Isolation of a Novel Microcystin-Degrading Bacterium and the Evolutionary Origin of mlr Gene Cluster


School of Resource and Environmental Engineering, Wuhan University of Technology, 122 Luoshi Road, Wuhan 430070, China
*
Author to whom correspondence should be addressed.
†
Lian Qin and Xiaoxing Zhang contributed equally to this work and are Co-Firstauthors.
Toxins 2019, 11(5), 269; https://doi.org/10.3390/toxins11050269
Submission received: 11 April 2019
/
Revised: 29 April 2019
/
Accepted: 9 May 2019
/
Published: 13 May 2019
(This article belongs to the Collection Toxicological Challenges of Aquatic Toxins)
Abstract
:The mlr-dependent biodegradation plays an essential role in the natural attenuation of microcystins (MCs) in eutrophic freshwater ecosystems. However, their evolutionary origin is still unclear due to the lack of mlr gene cluster sequences. In this study, a Sphingopyxis sp. strain X20 with high MC-degrading ability was isolated, and the mlrA gene activity was verified by heterologous expression. The whole sequence of the mlr gene cluster in strain X20 was obtained through PCR and thermal asymmetric interlaced (TAIL)-PCR, and then used for evolutionary origin analyses together with the sequences available in GenBank. Phylogenetic analyses of mlr gene clusters suggested that the four mlr genes had the same origin and evolutionary history. Genomic island analyses showed that there is a genomic island on the genome of sphingomonads that is capable of degrading MCs, on which the mlr gene cluster anchors. The concentrated distribution of the mlr gene cluster in sphingomonads implied that these genes have likely been present in the sphingomonads gene pool for a considerable time. Therefore, the mlr gene cluster may have initially entered into the genome of sphingomonads together with the genomic island by a horizontal gene transfer event, and then become inherited by some sphingomonads. The species other than sphingomonads have likely acquired mlr genes from sphingomonads by recently horizontal gene transfer due to the sporadic distribution of MC-degrading species and the mlr genes in them. Our results shed new light on the evolutionary origin of the mlr cluster and thus facilitate the interpretation of characteristic distribution of the mlr gene in bacteria and the understanding of whole mlr pathway.
Key Contribution: A genomic island containing an mlr gene cluster was found in sphingomonads MC-degraders for the first time. A novel evolutionary origin scheme of the mlr gene cluster was proposed.
1. Introduction
Harmful cyanobacterial blooms (HCBs) in freshwater bodies have become a global environmental problem, and are receiving growing attention with their increase in magnitude, frequency, and duration [1]. Microcystins (MCs) are a group of cyclic heptapeptide hepatotoxins produced by HCBs, among which microcystin-LR (MCLR) is the most widespread and best studied [2,3]. Due to their hepatotoxicity and potential carcinogenic activity, the World Health Organization (WHO) has suggested a guideline value of 1 µg·L−1 MCLR equivalents for drinking water [4].
MCs are resistant to traditional water treatment processes due to their chemically stable structure [5]. Biodegradation is one of the major pathways for MCs’ attenuation in the natural environment, and thus may be used in the removal of MCs [3,6]. Although a variety of MC-degrading pathways have been proposed, only the mlr-dependent pathway is confirmed and elucidated in detail. In this pathway, four genes—mlrA, mlrB, mlrC and mlrD—are involved, which form an mlr gene cluster and encode three hydrolysis enzymes (MlrA, MlrB, and MlrC) and one oligopeptide transporter-like protein (MlrD), respectively [7]. MlrA is the first enzyme involved in MCLR degradation, which hydrolyzes the circular MCLR into linear MCLR. Then, the MlrB enzyme hydrolyzes the linear MCLR into a tetrapeptide. MlrC is responsible for the further hydrolysis of the tetrapeptide to amino acids and smaller peptides [8]. Later studies showed that MlrC can also decompose linear MCLR to Adda directly [9,10]. This is the most well-characterized pathway for MC degradation thus far. However, there are still some questions to be answered about this process. Currently, only the part of the process from MCLR to Adda has been elucidated [11]; how the Adda residue is decomposed is still unclear, and the gene involved in Adda degradation is also unknown. The Adda residue is a specific amino that is only found in MCs and nodularins to date. Besides, it is a main building block for the synthesis of MCs, and is crucial to the toxicity of MCs [12]. Understanding the metabolism of Adda is essential for the ecological risk assessment of MCs. In addition, the function of MlrD has not been determined, although it is deduced to be responsible for the transport of MCLR or its products [13].
Up to now, over 70 strains of MC-degrading bacteria have been isolated from various environmental habitats, and the majority of them are from phylum proteobacteria, especially from the class α-proteobacteria [13,14,15,16,17]. Most of these α-proteobacteria are proved to harbor the mlrA gene, suggesting that they may degrade MCs through the mlr-dependent mechanism [18,19]. In addition, these strains have been found in many places over the world, and most of them possess strong MC-degrading activity [18,19,20,21,22]. These findings imply that MC-degrading bacteria harboring mlr genes may play a significant role in the diminishment of MCs in the natural environment, and may be applied to MCs pollution treatment. However, by now, we know little about the diversity of mlr genes. Moreover, acknowledgement of the distribution of the mlr-dependent pathway in bacteria is also deficient. These deficiencies affect the assessment of MCs pollution negatively, and impede the application of biodegradation process to MCs pollution treatment.
Understanding the evolutionary origin of mlr genes will help to infer the diversity of mlr genes and the distribution of the mlr-dependent pathway among bacteria. It also conduces determining the degradation mechanism adopted by novel MC-degrading isolates [13]. However, very few studies have been performed on the evolutionary origin of mlr genes, and the existing results are controversial. The sporadic distribution of mlrA genes in Sphingomonas supported that Sphingomonas may acquire the mlrA gene by horizontal gene transfer [23], whereas phylogenetic analyses argued that mlrA genes are likely as conserved and ancient as the 16S rRNA gene [20]. More recently, Zhu [13] investigated the evolutionary origin of mlrA through comparing the mlrA tree with the 16S rDNA tree. The congruent topologies in both trees for α-proteobacteria and the incongruent topologies for other proeobacteria indicated that a-proteobacteria is likely to have acquired the mlrA gene by vertical evolution, and other proeobacteria possibly have acquired the mlrA gene by horizontal gene transfer [13]. Although this thesis can explain the distribution and diversity of mlrA among MC-degrading bacteria, it is inferred based on limited sequences [13]. The confirmation of this will have to await additional MC-degrading isolates and their mlr gene sequences. Furthermore, only the evolutionary origin of the mlA gene has been investigated currently. The origin of the other three genes involved in this process—mlrB, mlrC and mlrD—is still unknown, and whether these genes had the same origin and evolutionary history with mlrA is unclear.
In this study, a novel Sphingopyxis sp. strain X20 with high MCs degradability was isolated from Dianchi Lake sediment. The whole mlr gene cluster was sequenced by PCR and TAIL-PCR, and the activity of the mlrA gene was verified by heterologous expression. To clarify the evolutionary origin of the mlr gene cluster, a spliced sequences dataset was constructed based on the sequences of four mlr genes from each isolate available in GenBank. The evolutionary origin of the mlr gene cluster was deduced through phylogenetic analyses of the spliced sequences, mlrA sequences, and the related 16S rDNA sequences. Genomic island (GI) analyses of sphingomonads and the strain X20 were also conducted to further verify the origin of the mlr gene cluster.
2. Results and Discussion
2.1. Isolation and Degradation Activity of MC-Degrading Bacterium
An MC-degrading bacterium, which was designated as strain X20, was isolated from the sediments of Dianchi Lake. It was a gram-negative aerobic bacterium, and formed bright yellow, round colonies on solid yeast extract-peptone medium (YPM). To identify it, a 16S rDNA tree was constructed using sequences of strain X20 and the related type strains (Figure 1). Strain X20 had the highest homology with Sphingopyxis sp. BZ30 in the tree, with 100% bootstrap support. Moreover, they clustered with five other types of Sphingopyxis to form a clade, which was clearly separated from other genera (Figure 1). These results suggested that strain X20 belongs to the genus Sphingopyxis.
Strain X20 degraded 5 mg·L−1 of MCLR to below the detection limit without a lag phase within 10 h (Figure 2). The pseudo-first order rate constant was up to 0.22 h−1, which was higher than that of most MC-degraders currently isolated [13,14,16]. This high rate suggested that strain X20 may be one of the main degraders involved in MCs degradation in Dianchi Lake. Their appearance may explain why the concentration of MCs in Dianchi Lake has been maintained at a relatively lower level, although the toxic cyanobacterial bloom frequently occurs [24]. The high rate also implied that these indigenous bacteria have the potential to be used for the treatment of MCs pollution.
Besides strain X20, many other species of sphingomonads, which comprises five closely related genera—Sphingopyxis, Sphingomonas, Sphingosinicella, Novosphingobium and Sphingobium—have also been found to degrade MCs [20,21,22,25,26,27]. These species were isolated from many different environmental habitats around the world, and usually have strong MC-degrading ability, suggesting that they may play an important role in the natural attenuation of MCs. Sphingomonads are a versatile bacteria group previously classified as Sphingomonas [28]. They are widely distributed in both polluted and unpolluted environments. In addition to MCs, sphingomonads can decompose a variety of hazardous organic compounds, such as polycyclic aromatic hydrocarbons, dioxins, herbicides, and pesticides [14,28]. Catabolic diversity may provide them a competitive advantage over other bacteria and help them acclimate to diverse environments. This may explain the frequent appearance of sphingomonads that are capable of degrading MCs in eutrophic waterbodies [29].
2.2. Whole mlr Gene Cluster in Strain X20
The whole sequence of the mlr gene cluster in strain X20 was successfully amplified and sequenced by using traditional PCR and thermal asymmetric interlaced (TAIL)-PCR approaches. The activity of the mlr gene cluster obtained was verified through the heterologous expression of mlrA gene in E. coli. Biodegradation experiments showed that both the recombinant strains and the recombinant enzyme can degrade MCLR (data not shown). During the degradation of MCLR, a linear MCLR, the same intermediate product as that produced by ACM-3962 [8], occurred and accumulated in culture media. These results demonstrated that the same mlr-dependent pathway is adopted in strain X20 as other Sphingopyxis MC-degraders [9,21]. Previous results indicated that MlrD may be responsible for the transportation of MCs during degradation, whereas our result showed that MlrD is not essential in the first step of MCLR degradation, because the recombinant strain without the mlrD gene can also degrade MCLR and excrete the intermediate product (linear MCLR) out of cells. This phenomenon has also been found in other studies [13,30]. Hence, the function of mlrD in MCs degradation needs further research.
The mlr cluster in strain X20 had a total length of 5575 bp and a G + C content of 59.05%, in which mlrA, mlrB, mlrC, and mlrD were contained with the full length of 1011 bp, 1626 bp, 1587 bp, and 1272 bp, respectively. The four mlr genes in strain X20 have the same order and translation orientation as that in strain ACM-3962 [7]. Up to date, three full sequences of the mlr gene cluster and one partial sequence have been reported, which are from Sphingopyxis sp. C-1, Sphingosinicella sp. B-9, Novosphingobium sp. THN1, and Sphingomonas sp. ACM-3962, respectively. The four mlr cluster sequences have high similarity to that in strain X20 (86.8–98.1%), suggesting that they may come from a similar ancestor gene. It is noteworthy that most of the mlr sequences reported so far are from α-proteobacteria, especially from sphingomonads. Except for sphingomonads, no MC-degrading ability or mlr genes have been identified in other Sphingomonadaceae to date, although mlr genes have been found in a species of Rhizobiales [13] and two species of β-proteobacteria [17] and γ-proteobacteria [31]. The reason for the concentrated distribution of MC-degrading ability and mlr genes in sphingomonads is still unclear. The information on the evolutionary origin of mlr genes may facilitate the clarification of this phenomenon.
2.3. Evolutionary Origin of mlr Gene Cluster
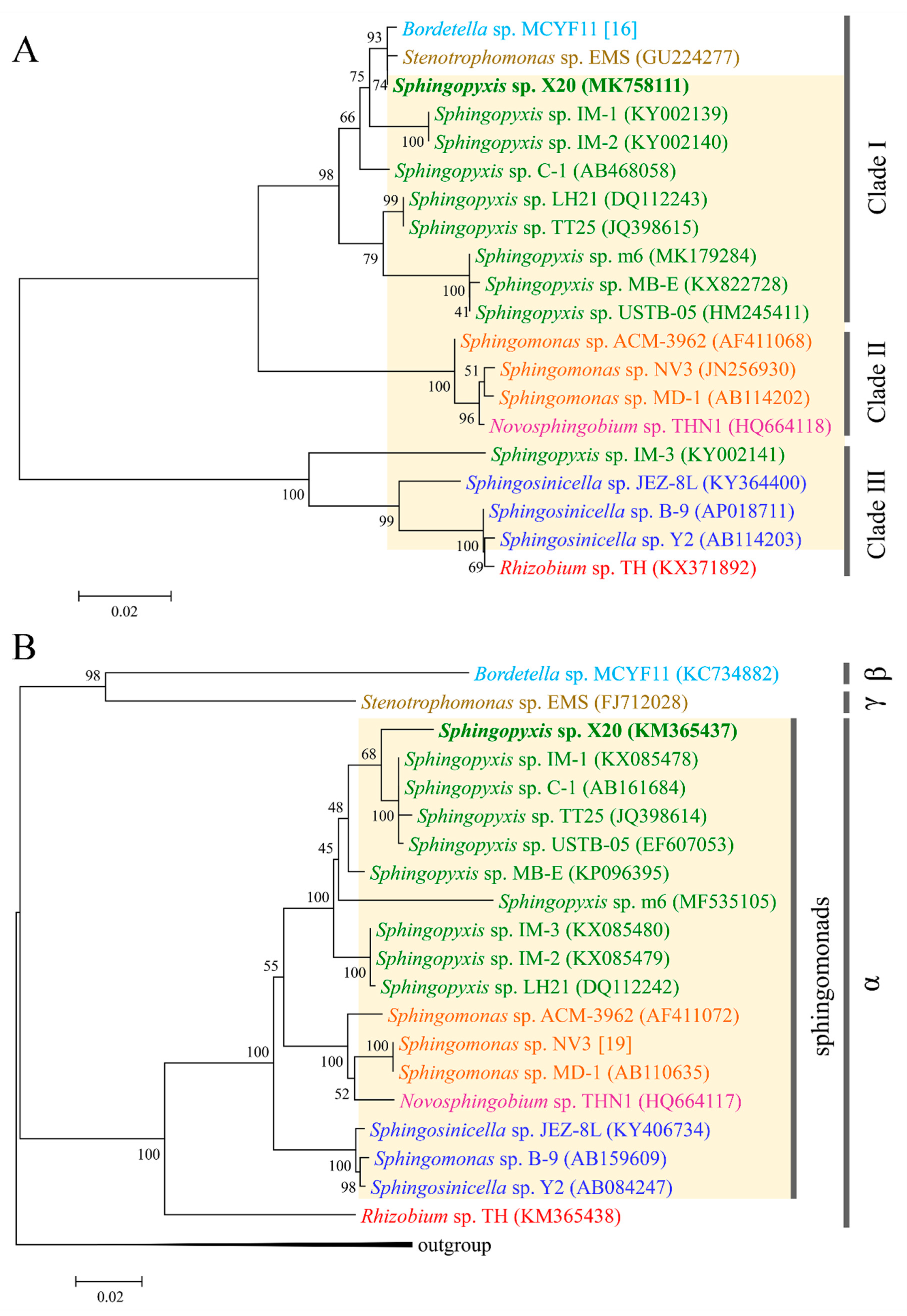
Although the evolutionary origin of mlrA has been elucidated in some detail, the origin of three other mlr genes, especially mlrB and mlrC, remains unknown [13,14]. Since the enzymes encoded by the two genes are also critical for MCs decomposition, it is necessary to understand their evolutionary origin [7]. To verify whether these genes co-evolved with mlrA, phylogenetic trees were constructed using the spliced mlr sequences by ML, NJ, and ME methods, respectively. The same topology was found in the trees constructed by the NJ and ME methods (Figure 3A). As shown in Figure 3A, all the species were divided into three clades. The Sphingopyxis species formed a clade (clade I), while the Sphingomonas species formed another tight clade (clade II) with a Novosphingobium species. The two subclades composed a major clade, and another main clade consisted of the Sphingosinicella and Rhizobium species (Figure 3A). In a phylogenetic tree based on protein sequences translated from the nucleotide sequences of the spliced mlr, all the species occupied the same phylogenetic positions as those in the mlr tree except for Sphingopyxis sp. LH21, which did not cluster with the other Sphingopyxis species (Figure S1). Moreover, a similar topology was also observed for analyses performed separately with the four mlr genes (data not shown). The topological congruence indicated that the four mlr genes might have the same origin and evolutionary history.
GI analyses provided further evidences for the co-evolution of the four mlr genes. Only three genomes of MC-degrading bacteria (Sphingopyxis sp. C-1, NZ_BBRO00000000; Sphingosinicella sp. B-9, AP018711; and Novosphingobium sp. THN1, CP028347) have been sequenced so far, and they are all from sphingomonads. GI analyses showed that there was a GI of 60.3 kb within the strain C-1 genome, on which the mlr gene cluster anchored. A similar GI of 130.0 kb was also found on the strain B-9 genome, inside which a shorter GI (18.2 kb) with the mlr gene cluster was nested. Although the similar GI was not found in strain THN1, a highly similar DNA region to the GI (about 33 kb, with similarities of 88.7% and 86.4% to strain C-1 and B-9) was found on the genome, with mlr genes on it. Therefore, it is likely that strain THN1 also possessed a similar GI, and the failure of detection might be due to the loss or rearrangement of some sequence regions during its evolution. Further BLAST analyses found no similar GI or sequence on the genomes of other sphingomonads without MC-degrading ability. Moreover, many genes on the GI were specific to the three strains, whereas the genes adjacent to the GI were also found on the genomes of other sphingomonads currently available, independent of their MC-degradation ability. These results implied that the GI is likely unique to the MC-degrading bacteria with the mlr-dependent pathway. To test the hypothesis, four genes (mlrE, mlrF, GI1, and GI2) on the GI, which were unique to the three MC-degrading bacteria, were selected as the marker genes to determine whether strain X20 possessed the same GI (Figure 4). Three of the four genes—mlrE, GI1, and GI2—were successfully amplified and sequenced. The three genes in strain X20 share very high similarity (86.0–98.6%) with those from strains C-1, B-9, and THN1, suggesting that similar GI may also be present in strain X20. The reason for failure in mlrF amplification is still unclear. The mismatch between primers and template might be one of the possible reasons, since only three mlrF sequences were reported to date, which exacerbated the difficulty of designing appropriate primers.
Since the genomes have not been reported for most MC-degrading bacteria, it is still unknown whether the mlr genes anchor on the similar GIs or conserved regions in these species. However, our results suggested that mlr gene clusters have likely entered into the genome of sphingomonads by horizontal gene transfer of the GI, and then evolved together with it, since the G + C contents (60.8% and 60.1%) of the GIs were significantly lower than that (63.7% and 63.9%) of associated genomes, but near to that (59.0% and 59.1%) of the mlr gene clusters in Sphingopyxis sp. C-1 and Sphingosinicella sp. B-9. That the mlrE and GI2 trees have identical topological properties with the mlr tree for strains C-1, B-9, THN1, and X20 (data not shown) provided further evidence for this hypothesis. In addition, despite the relatively concentrated distribution of the mlr-dependent pathway in sphingomonads, not all species of sphingomonads possess these genes. This phenomenon also agrees well with the above deduction.
To further clarify the evolutionary origin of the mlr gene cluster, 16S rDNA trees were constructed using the dataset from the same strains. Our results showed that the trees constructed by the NJ, ML, and ME methods shared the same topology. Furthermore, the 16S rDNA tree had a similar pattern with the mlr tree for sphingomonads (Figure 3A,B). In the two trees, all the sphingomonads formed three clades with taxonomically closer species clustering together. The congruent topology indicated that the mlr gene clusters in various genera of sphingomonads may originate from a single ancestor gene, rather than from recent horizontal gene transfer. Considering that limited mlr clusters data may play a role in the congruence, a comparison between the mlrA tree and 16S rDNA tree was also conducted by using currently available mlrA genes and the associated 16S rRNA genes. The same topology was obtained for sphingomonads species in both mlrA trees and 16S rDNA trees constructed by the NJ, ML, and ME methods (Figure 5A,B). The same topology was also observed in the phylogenetic tree inferred from MlrA protein sequences (Figure S2). The topological congruence provided further support for the above proposition. Currently, most α-proteobacteria containing mlr genes were from sphingomonads, which is composed of closely related genera, and only a Rhizobium sp. strain TH was from the order Rhizobiales. The broad distribution of mlr gene cluster in sphingomonads degraders suggested that these genes have been present in the sphingomonads gene pool for a considerable time. These findings implied that the mlr gene cluster together with a GI probably has been acquired very early in the evolution of sphingomonads by a horizontal gene transfer event, and then some species of sphingomonads gained it through vertical inheritance. Therefore, the acquisition of GI with the mlr gene cluster is likely a key step in the evolution of the mlr-dependent pathway.
As for Rhizobium sp. strain TH, the origin of mlr is likely different from that of sphingomonads. Strain TH tightly clustered with Sphingosinicella species in both mlrA and mlr trees (Figure 5A and Figure 3A), rather than formed a distinct clade, although it belongs to the order Rhizobiales, which is taxonomically distinct from sphingomonads (Figure 5B and Figure 3B). This result disagreed with that reported previously [13]. The divergence might be due to the limited dataset in the reference [13]. Considering the concentrated distribution of mlr gene clusters among sphingomonads, the high homology of mlr gene clusters between strain TH and Sphingosinicella species suggested that strain TH possibly acquired the mlr gene cluster from Sphingosinicella by recent lateral gene transfer. Although the mlrA gene has been detected and sequenced in a β-proteobacterial isolate (Bordetella sp. MCYF11) and a γ-proteobacterial isolate (Stenotrophomonas sp. EMS) [17,31], the mlr gene clusters have not been sequenced to date. Hence, the evolutionary origin of the mlr cluster is still unknown for Proteobacteria other than α-proteobacteria. Nevertheless, the high homology of the mlrA gene with Sphingopyxis species (Figure 5A) and the sporadic distribution of mlr genes among these species indicated that these MC-degrading bacteria might obtain the mlr gene cluster from Sphingopyxis by recent lateral gene transfer.
Although GIs are widespread in bacterial genomes, it is the first time, to our knowledge, that a GI containing an mlr gene cluster was reported. Acquisition of the GI enabled sphingomonads to expand its genome to exploit new environmental niches and may provide them with a competitive advantage over other species during water blooms. This may be one of the reasons that most of the MC-degrading bacteria with an mlr-dependent pathway are from sphingomonads. Up to now, only part of the mlr pathway for MC degradation has been clarified; other major genes, especially those involved in Adda degradation, have not been elucidated. Since the mlr gene cluster may have co-evolved with the GI, it is likely that other main genes involved in MC degradation are present on it [32,33]. The analyses of the GI may help discover new genes within this pathway and contribute to the clarification of the whole MC-degrading process.
3. Conclusions
An MC-degrading bacterium strain X20 was isolated from Dianchi Lake and identified as Sphingopyxis sp. The complete mlr gene cluster sequence of strain X20 was obtained and the activity of mlrA gene was verified by heterologous expression. Phylogenetic analysis and genomic island analyses suggested that the four mlr genes had the same origin and evolutionary history. The mlr gene cluster may has initially entered into the genome of sphingomonads by the horizontal gene transfer of a genomic island and then was inherited by some sphingomonads. Thereafter, the species other than sphingomonads obtained it by recent horizontal gene transfer.
4. Materials and Methods
4.1. Materials and Reagents
The MCLR standard was purchased from Sigma-Aldrich (St. Louis, MO, USA). The MCLR for biodegradation experiments was extracted and purified from laboratory-cultured Microcystis aeruginosa PCC 7806, as described previously [34]. Methanol and trifluoroacetic acid (TFA) (Tedia Company, Inc., Fairfield, OH, USA) used as the mobile phase of high-performance liquid chromatography (HPLC) were of HPLC grade. Other chemicals were of analytical grade.
4.2. Isolation and Identification of MC-Degrading Bacterium
Surface sediment was sampled from Dianchi Lake in China. The isolation and identification of MC-degrading bacterium were performed as reported previously [13]. Briefly, after the enrichment with MCLR-containing mineral salt medium (MSM), individual colonies were isolated from sediment by serial dilution in MSM and subsequent isolation on solid media. The MC-degrading ability was detected in MCLR-containing MSM by high-performance liquid chromatography (HPLC). One isolate with high MC-degrading ability was isolated and named as X20. The 16S rDNA of strain X20 was amplified and sequenced, and the sequencing data have been deposited in GenBank under accession number KM365437. Based on the sequences of strain X20 and related type strains in GenBank, a phylogenetic tree was constructed using the neighbor-joining method in MEGA7.0.
4.3. MCLR Degradation Experiments
Strain X20 was incubated overnight in yeast extract-peptone medium (YPM) (containing yeast extract 3 g, peptone 3 g, MgSO4·7H2O 0.5 g, and CaCl2 0.3 g per liter) on a shaker (120 rpm) at 30 °C for enrichment. The enriched cells were centrifuged at 8000 rpm for 5 min and washed with MSM twice. The cells were resuspended in MSM containing MCLR (approximately 5 mg·L−1) and cultivated at 30 °C. The same culture medium without bacterial inoculum was used as a control. Samples were collected from the cultures at regular intervals. The concentration of MCLR was monitored by HPLC, and the bacterial growth was measured via detection of the absorbance at 600 nm (OD600). All the experiments were conducted in triplicate.
4.4. Sequencing of the mlr Gene Cluster
The partial sequence of the mlr gene cluster in strain X20 was obtained by amplification of regions spanning mlrC-mlrA, mlrA-mlrD, and mlrD-mlrB, as described previously [13]. The flanking regions of the mlr gene cluster were obtained by thermal asymmetric interlaced (TAIL)-PCR. Two groups of nested insertion-specific primers for TAIL-PCR (Table 1) were designed based on the mlrC and mlrB partial sequences of strain X20. TAIL-PCR was performed with the Genome Walking kit (Takara) according to the manufacturer’s instructions. All the PCR products above were sequenced and assembled to obtain a full-length mlr gene cluster, which has been deposited in the GenBank database (accession no. MK758111). Comparison with other mlr gene clusters were conducted by BLAST.
4.5. Heterogeneous Expression of the mlrA Gene
To verify the activity of the mlr gene cluster, primers were designed based on the mlrA sequence of strain X20, with BamHI and HindIII sites added to the forward and reverse primers (Table 1). The heterogeneous expression of mlrA gene was performed in E. coli BL21 (DE3) and the activity of recombinant strains and recombinant enzyme were detected as reported previously [13]. The concentrations of MCLR and its intermediate products were determined by comparing retention times under the same HPLC conditions with that of standard MCLR or intermediate products previously reported [13].
4.6. Phylogenetic Analyses
Due to the lack of a whole mlr gene cluster, a spliced sequences dataset was assembled by the sequences of four mlr genes from each isolate for the construction of a phylogenetic tree. Each of the four mlr genes was retrieved from GenBank (Table S1) and aligned and trimmed by clustalX 2.1 to the longest fragment available, respectively. The segments of mlrA, mlrB, mlrC, and mlrD with 700 bp, 335bp, 546 bp, and 539 bp, respectively, were obtained and assembled to form a set of 2120 bp-spliced sequences. Phylogenetic trees were inferred using both the segments and the spliced sequences datasets by the maximum-likelihood (ML), minimum-evolution (ME), and neighbor-joining (NJ) methods in MEGA7. A phylogenetic tree was also constructed based on 16S rDNA sequences from the same sets of taxa by using Desulfobacter halotolerans DSM 11383T (NR_026439) as an outgroup. In contrast to the limited number of sequences for three other genes, more mlrA sequences have been reported. Therefore, a phylogenetic tree was inferred using the mlrA sequences currently available to further clarify the evolutionary origin of mlr genes. A 16S rDNA tree was also constructed by using Bacillus cereus ATCC 14579T (MH281748) and Arthrobacter globiformis DSM 20124T (NR_026187) as outgroups. Phylogenetic trees were also constructed based on protein sequences translated from the nucleotide sequences of the spliced mlr or mlrA genes, respectively.
4.7. Genomic Island Analyses
The genome sequences of three MC-degrading bacteria—Sphingopyxis sp. C-1 (NZ_BBRO00000000), Sphingosinicella sp. B-9 (AP018711), and Novosphingobium sp. THN1 (CP028347)—were retrieved from GenBank. The genomic islands (GIs) on the genomes of these strains were screened by the IslandViewer 4 web server with default settings [35]. The islands detected were compared with each other by BLAST to identify the conserved region, which was then used to search the genomes of sphingomonads by BLAST for similar sequences. The genes on the GIs and adjacent to the GIs were also used to search the GenBank database by BLAST.
To verify the existence of a similar GI in strain X20, two genes (mlrE and mlrF) previously reported [33] and two conserved genes (named as GI1 and GI2) on the GIs were chosen as markers. The primers were designed according to the conserved regions in the three strains (strain C-1, B-9, and THN1) (Table 1). The PCR reaction was performed with the genomic DNA of strain X20 as a template, and the PCR conditions were as follows: 94 °C for 5 min followed by 30 cycles of 94 °C for 20 s, 55 °C for 15 s and 72 °C for 40 s; then, 72 °C for 10 min. The amplified products of three genes (mlrE, GI1, and GI2) were sequenced after purification, and the comparisons of them among various strains were performed by BLAST. The phylogenetic trees were also inferred by the NJ method using the three genes, respectively.
Supplementary Materials
The following are available online at https://www.mdpi.com/2072-6651/11/5/269/s1, Figure S1: Phylogenetic tree inferred from protein sequences of spliced mlr sequences. Evolutionary analysis was conducted by Neighbor-Joining method in MEGA7. The numbers at each node were the bootstrap values for the percentages of 1000 replicate trees, Figure S2: Phylogenetic tree based on MlrA protein sequences. Evolutionary analyses were conducted by the Neighbor-Joining method in MEGA7. The numbers at each node were the bootstrap values for the percentages of 1000 replicate trees. * represent the protein sequence translated from the nucleotide sequence of the mlrA gene, Table S1: Sequences used to construct assembled mlr gene cluster.
Author Contributions
Data curation, X.Z., K.W., Y.S. and D.L.; Funding acquisition, X.C.; Supervision, X.C.; Visualization, L.Q.; Writing – original draft, L.Q. and X.Z.; Writing – review & editing, X.C.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (21077083).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huisman, J.; Codd, G.A.; Paerl, H.W.; Ibelings, B.W.; Verspagen, J.M.H.; Visser, P.M. Cyanobacterial blooms. Nat. Rev. Microbiol. 2018, 16, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.P.; Sinha, R.P.; Incharoensakdi, A. The cyanotoxin-microcystins: Current overview. Rev. Environ. Sci. Biotechnol. 2014, 13, 215–249. [Google Scholar] [CrossRef]
- Li, J.; Li, R.; Li, J. Current research scenario for microcystins biodegradation—A review on fundamental knowledge, application prospects and challenges. Sci. Total Environ. 2017, 595, 615–632. [Google Scholar] [CrossRef]
- WHO. Guidelines for Drinking-Water Quality—Second Edition. In Addendum to Volume 2: Health Criteria and Other Supporting Information; World Health Organization: Geneva, Switzerland, 1998. [Google Scholar]
- Hall, T.; Hart, J.; Croll, B.; Gregory, R. Laboratory-scale investigations of algal toxin removal by water treatment. Water Environ. J. 2000, 14, 143–149. [Google Scholar] [CrossRef]
- Ho, L.; Sawade, E.; Newcombe, G. Biological treatment options for cyanobacteria metabolite removal—A review. Water Res. 2012, 46, 1536–1548. [Google Scholar] [CrossRef]
- Bourne, D.G.; Riddles, P.; Jones, G.J.; Smith, W.; Blakeley, R.L. Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin LR. Environ. Toxicol. 2001, 16, 523–534. [Google Scholar] [CrossRef]
- Bourne, D.G.; Jones, G.J.; Blakeley, R.L.; Jones, A.; Negri, A.P.; Riddles, P. Enzymatic pathway for the bacterial degradation of the cyanobacterial cyclic peptide toxin microcystin LR. Appl. Environ. Microbiol. 1996, 62, 4086–4094. [Google Scholar] [PubMed]
- Shimizu, K.; Maseda, H.; Okano, K.; Kurashima, T.; Kawauchi, Y.; Xue, Q.; Utsumi, M.; Zhang, Z.; Sugiura, N. Enzymatic pathway for biodegrading microcystin LR in Sphingopyxis sp. C-1. J. Biosci. Bioeng. 2012, 114, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Dziga, D.; Wasylewski, M.; Szetela, A.; Bochenska, O.; Wladyka, B. Verification of the Role of MlrC in Microcystin Biodegradation by Studies Using a Heterologously Expressed Enzyme. Chem. Res. Toxicol. 2012, 25, 1192–1194. [Google Scholar] [CrossRef]
- Dziga, D.; Zielinska, G.; Wladyka, B.; Bochenska, O.; Maksylewicz, A.; Strzalka, W.; Meriluoto, J. Characterization of Enzymatic Activity of MlrB and MlrC Proteins Involved in Bacterial Degradation of Cyanotoxins Microcystins. Toxins 2016, 8, 76. [Google Scholar] [CrossRef]
- Fontanillo, M.; Köhn, M. Microcystins: Synthesis and structure–activity relationship studies toward PP1 and PP2A. Bioorg. Med. Chem. 2018, 26, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, Y.; Chen, X.; Hu, Y.O.O.; Xiang, H.; Tao, J.; Ling, Y. Biodegradation mechanism of microcystin-LR by a novel isolate of Rhizobium sp. TH and the evolutionary origin of the mlrA gene. Int. Biodeterior. Biodegrad. 2016, 115, 17–25. [Google Scholar] [CrossRef]
- Kormas, K.A.; Lymperopoulou, D.S. Cyanobacterial toxin degrading bacteria: Who are they? Biomed. Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Thees, A.; Atari, E.; Birbeck, J.; Westrick, J.A.; Huntley, J.F. Isolation and characterization of Lake Erie bacteria that degrade the cyanobacterial microcystin toxin MC-LR. J. Great Lakes Res. 2019, 45, 138–149. [Google Scholar] [CrossRef]
- Krishnan, A.; Zhang, Y.Q.; Mou, X.Z. Isolation and Characterization of Microcystin-Degrading Bacteria from Lake Erie. Bull. Environ. Contam Toxicol. 2018, 101, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhou, Y.; Sun, R.; Wei, H.; Li, Y.; Yin, L.; Pu, Y. Biodegradation of microcystin-LR and-RR by a novel microcystin-degrading bacterium isolated from Lake Taihu. Biodegradation 2014, 25, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Maghsoudi, E.; Fortin, N.; Greer, C.; Maynard, C.; Page, A.; Vo Duy, S.; Sauve, S.; Prevost, M.; Dorner, S. Cyanotoxin degradation activity and mlr gene expression profiles of a Sphingopyxis sp. isolated from Lake Champlain, Canada. Environ. Sci. Process. Impacts 2016, 18, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Somdee, T.; Thunders, M.; Ruck, J.; Lys, I.; Allison, M.; Page, R. Degradation of [Dha(7)]MC-LR by a Microcystin Degrading Bacterium Isolated from Lake Rotoiti, New Zealand. ISRN Microbiol. 2013, 2013, 596429. [Google Scholar] [CrossRef]
- Jiang, Y.; Shao, J.; Wu, X.; Xu, Y.; Li, R. Active and silent members in the mlr gene cluster of a microcystin-degrading bacterium isolated from Lake Taihu, China. FEMS Microbiol. Lett. 2011, 322, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Hoefel, D.; Saint, C.P.; Newcombe, G. Isolation and identification of a novel microcystin-degrading bacterium from a biological sand filter. Water Res. 2007, 41, 4685–4695. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, G.; Yan, H. Microbial biodegradation of microcystin-RR by bacterium Sphingopyxis sp. USTB-05. J. Environ. Sci. 2010, 22, 168–175. [Google Scholar] [CrossRef]
- Saito, T.; Okano, K.; Park, H.D.; Itayama, T.; Inamori, Y.; Neilan, B.A.; Burns, B.P.; Sugiura, N. Detection and sequencing of the microcystin LR-degrading gene, mlrA, from new bacteria isolated from Japanese lakes. FEMS Microbiol. Lett. 2003, 229, 271–276. [Google Scholar] [CrossRef]
- Zhu, L.; Wu, Y.; Song, L.; Gan, N. Ecological dynamics of toxic Microcystis spp. and microcystin-degrading bacteria in Dianchi Lake, China. Appl. Environ. Microbiol. 2014, 80, 1874–1881. [Google Scholar] [CrossRef]
- Jones, G.J.; Bourne, D.G.; Blakeley, R.L.; Doelle, H. Degradation of the cyanobacterial hepatotoxin microcystin by aquatic bacteria. Nat. Toxins 1994, 2, 228–235. [Google Scholar] [CrossRef]
- Tsuji, K.; Asakawa, M.; Anzai, Y.; Sumino, T.; Harada, K.-I. Degradation of microcystins using immobilized microorganism isolated in an eutrophic lake. Chemosphere 2006, 65, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-D.; Sasaki, Y.; Maruyama, T.; Yanagisawa, E.; Hiraishi, A.; Kato, K. degradation of the cyanobacterial hepatotoxin microcystin by a new bacterium isolated from a hypertrophic lake. Environ. Toxicol. 2001, 16, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A. Molecular characteristics of xenobiotic-degrading sphingomonads. Appl. Microbiol. Biotechnol. 2009, 81, 793–811. [Google Scholar] [CrossRef]
- Lezcano, M.Á.; Velázquez, D.; Quesada, A.; El-Shehawy, R. Diversity and temporal shifts of the bacterial community associated with a toxic cyanobacterial bloom: An interplay between microcystin producers and degraders. Water Res. 2017, 125, 52–61. [Google Scholar] [CrossRef]
- Wang, R.P.; Li, J.M.; Jiang, Y.G.; Lu, Z.J.; Li, R.H.; Li, J. Heterologous expression of mlrA gene originated from Novosphingobium sp. THN1 to degrade microcystin-RR and identify the first step involved in degradation pathway. Chemosphere 2017, 184, 159–167. [Google Scholar] [CrossRef]
- Chen, J.; Hu, L.B.; Zhou, W.; Yan, S.H.; Yang, J.D.; Xue, Y.F.; Shi, Z.Q. Degradation of Microcystin-LR and RR by a Stenotrophomonas sp. Strain EMS Isolated from Lake Taihu, China. Int. J. Mol. Sci. 2010, 11, 896–911. [Google Scholar] [CrossRef]
- Jin, H.Y.; Hiraoka, Y.; Okuma, Y.; Hashimoto, E.H.; Kurita, M.; Anas, A.R.J.; Uemura, H.; Tsuji, K.; Harada, K.I. Microbial Degradation of Amino Acid-Containing Compounds Using the Microcystin-Degrading Bacterial Strain B-9. Mar. Drugs 2018, 16, 50. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Shimizu, K.; Maseda, H.; Kawauchi, Y.; Utsumi, M.; Itayama, T.; Zhang, Z.; Sugiura, N. Whole-genome sequence of the microcystin-degrading bacterium Sphingopyxis sp. strain C-1. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, X.; Yang, L.; Xiao, B.; Wu, X.; Wang, J.; Wan, H. An effective pathway for the removal of microcystin LR via anoxic biodegradation in lake sediments. Water Res. 2010, 44, 1884–1892. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Phylogenetic analysis of the 16S rDNA sequences from strain X20 and the related type strains by the neighbor-joining (NJ) method in MEGA7. Bootstrap values represent percentages from 1000 replicates of the data.
Figure 1.
Phylogenetic analysis of the 16S rDNA sequences from strain X20 and the related type strains by the neighbor-joining (NJ) method in MEGA7. Bootstrap values represent percentages from 1000 replicates of the data.

Figure 2.
Degradation of microcystin-LR (MCLR) by strain X20 at 30 °C. Error bars represent the standard deviations.
Figure 2.
Degradation of microcystin-LR (MCLR) by strain X20 at 30 °C. Error bars represent the standard deviations.

Figure 3.
Phylogenetic tree inferred from spliced mlr gene sequences (A) and 16S rDNA (B) from the same set of microcystin (MC)-degrading bacteria. Evolutionary analyses were conducted by the neighbor-joining method in MEGA7. The numbers at each node were the bootstrap values for the percentages of 1000 replicate trees.
Figure 3.
Phylogenetic tree inferred from spliced mlr gene sequences (A) and 16S rDNA (B) from the same set of microcystin (MC)-degrading bacteria. Evolutionary analyses were conducted by the neighbor-joining method in MEGA7. The numbers at each node were the bootstrap values for the percentages of 1000 replicate trees.

Figure 4.
Comparison of the conserved region on the genomic islands (GIs) of Sphingosinicella sp. B-9 (AP018711), Sphingopyxis sp. C-1 (NZ_BBRO00000000), and Novosphingobium sp. THN1 (CP028347). Genomic comparisons were performed using BLASTn, with a maximum e-value of 0.001 and a minimum hit length of 20 bp. The figure was produced using Easyfig v2.2.3. Predicted genes and the direction of transcription were notated by block arrows. The grey-black region indicates the sequence similarity (from 80% to 100%). The corresponding genes in strain X20 were also noted.
Figure 4.
Comparison of the conserved region on the genomic islands (GIs) of Sphingosinicella sp. B-9 (AP018711), Sphingopyxis sp. C-1 (NZ_BBRO00000000), and Novosphingobium sp. THN1 (CP028347). Genomic comparisons were performed using BLASTn, with a maximum e-value of 0.001 and a minimum hit length of 20 bp. The figure was produced using Easyfig v2.2.3. Predicted genes and the direction of transcription were notated by block arrows. The grey-black region indicates the sequence similarity (from 80% to 100%). The corresponding genes in strain X20 were also noted.

Figure 5.
Phylogenetic tree of the mlrA gene (A) and 16S rDNA (B) from the same set of MC-degrading bacteria. Evolutionary analyses were conducted by the neighbor-joining method in MEGA7. The numbers at each node were the bootstrap values for the percentages of 1000 replicate trees. The Greek letters denoted α-proteobacteria, β-proteobacteria, and γ-proteobacteria, respectively.
Figure 5.
Phylogenetic tree of the mlrA gene (A) and 16S rDNA (B) from the same set of MC-degrading bacteria. Evolutionary analyses were conducted by the neighbor-joining method in MEGA7. The numbers at each node were the bootstrap values for the percentages of 1000 replicate trees. The Greek letters denoted α-proteobacteria, β-proteobacteria, and γ-proteobacteria, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Primer sequences used in this study.
| Gene | Primer | Sequence (5′–3′) | Purpose | References |
|---|---|---|---|---|
| mlrC-mlrA | mlrCf1 | TCCCCGAAACCGATTCTCCA | Partial mlr | [21] |
| MR | CTCCTCCCACAAATCAGGAC | [23] | ||
| mlrA-mlrD | MF | GACCCGATGTTCAAGATACT | Partial mlr | [23] |
| mlrDr1 | ACAGTGTTGCCGAGCTGCTCA | [21] | ||
| mlrD-mlrB | mlrDf1 | GCTGGCTGCGACGGAAATG | Partial mlr | [21] |
| mlrBr1 | CGTGCGGACTACTGTTGG | |||
| mlrB | mlrBf2 | ATGACTGCAACAAAGCTTTT | Partial mlr | This study |
| mlrBr2 | TTATCCACGAACAACCCACC | |||
| mlrC | CR1 | CCCTGGCAGTACAATTGGGCTTTGA | Flanking region | This study |
| CR2 | CACAGGGCTTGCCGAGAATGTCA | |||
| CR3 | CGTCAGCGAAATTCGCGACCAGT | |||
| mlrB | BF1 | AGGTAGGTCAGGCAGATAGGTG | Flanking region | This study |
| BF2 | AAGATCAGGATGAGAACGGCCG | |||
| BF3 | AGATCAGCAAGTCCAAAGCCGC | |||
| mlrA | MlrAxf | GACGGATCCATGCGGGAGTTTGTCAAAC | Expression | This study |
| MlrAxr | TATAAGCTTCGCGTTCGCGCCGGACTTG | |||
| mlrE | mlrEf | TTCGGTAGACGGAACACA | GI verification | This study |
| mlrEr | ACACGGCATTGATCTGAAT | |||
| mlrF | mlrFf | GATGGAAGAGGTGATGGCAATT | GI verification | This study |
| mlrFr | AGGACGAATACTGGTGGTAGTC | |||
| GI1 | G1f | ACTCTGGACCAGCGGCTAA | GI verification | This study |
| G1r | CAAGCGGACTGACAAGTTCTG | |||
| GI2 | G2f | GCAACCGTCATCAGTGGATC | GI verification | This study |
| G2r | CCGCCGTAGTATTCGTGAATG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Qin, L.; Zhang, X.; Chen, X.; Wang, K.; Shen, Y.; Li, D. Isolation of a Novel Microcystin-Degrading Bacterium and the Evolutionary Origin of mlr Gene Cluster. Toxins 2019, 11, 269. https://doi.org/10.3390/toxins11050269
AMA Style
Qin L, Zhang X, Chen X, Wang K, Shen Y, Li D. Isolation of a Novel Microcystin-Degrading Bacterium and the Evolutionary Origin of mlr Gene Cluster. Toxins. 2019; 11(5):269. https://doi.org/10.3390/toxins11050269
Chicago/Turabian StyleQin, Lian, Xiaoxing Zhang, Xiaoguo Chen, Ke Wang, Yitian Shen, and Dan Li. 2019. "Isolation of a Novel Microcystin-Degrading Bacterium and the Evolutionary Origin of mlr Gene Cluster" Toxins 11, no. 5: 269. https://doi.org/10.3390/toxins11050269
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.