More Than a Pore: The Cellular Response to Cholesterol-Dependent Cytolysins

Abstract
:1. Introduction
2. Physical Properties of the CDCs
2.1. Structure and Membrane Binding
2.2. Oligomerization and Pore Formation

{kind=link}
{kind=link}
| Response | Outcome | Toxin | Reference |
|---|---|---|---|
| Translocation of a bacterial effector | Hydrolysis of NAD+ | SLO | [2] |
| Lipid raft fusion | Tyrosine phosphorylation | LLO | [25] |
| IFNy secretion | Inflammation | PLY | [5] |
| iNOS production | Anti-microbial | PLY | [5] |
| Histone dephosphorylation | Transcriptional modification | LLO | [71] |
| Vaccine adjuvant | Improved tumor clearance | LLO | [97] |
| MHC presentation | Adaptive immunity | LLO | [96] |
3. Membrane Response to CDC Intoxication
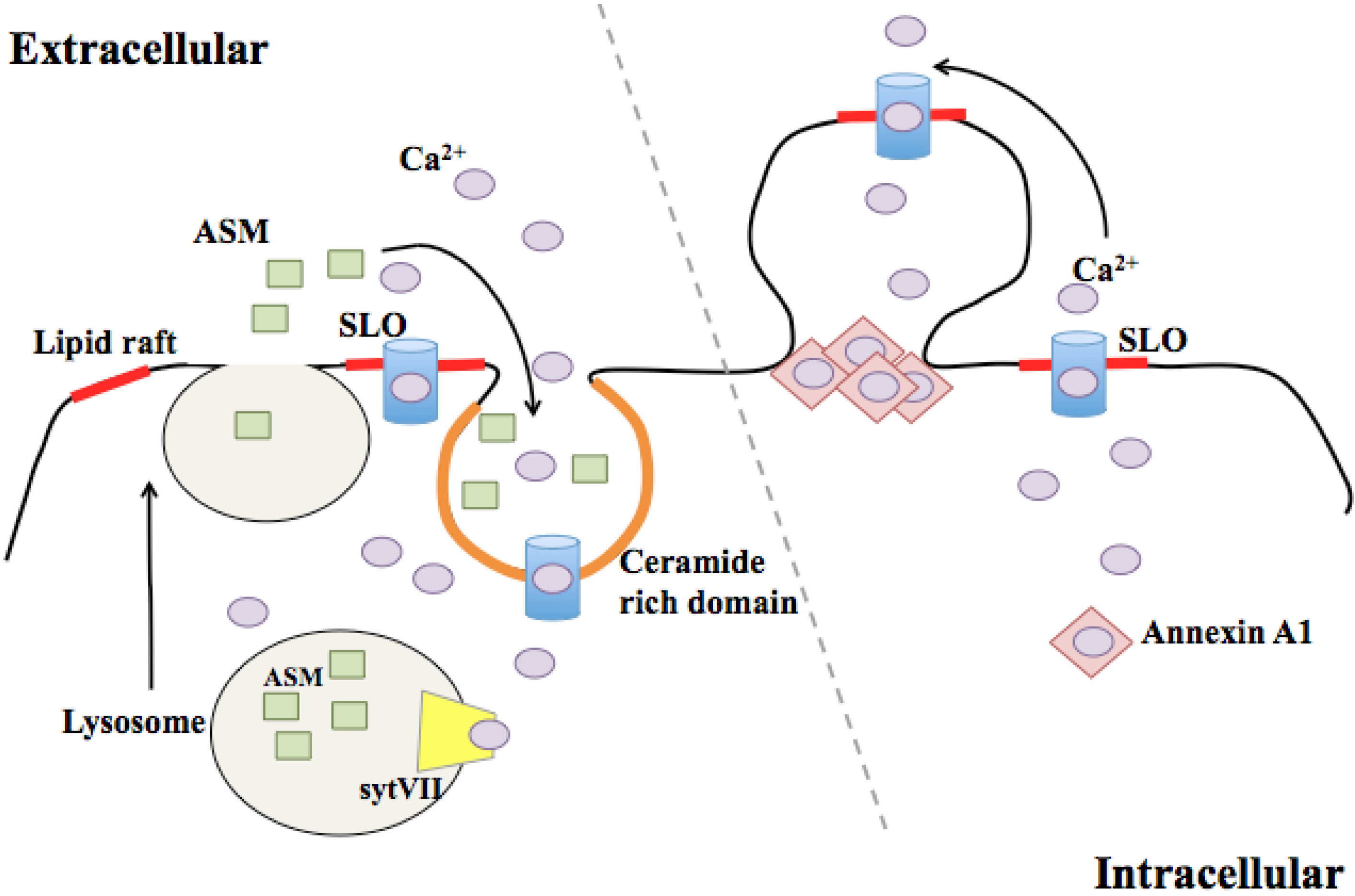
3.1. Endocytosis of Pores
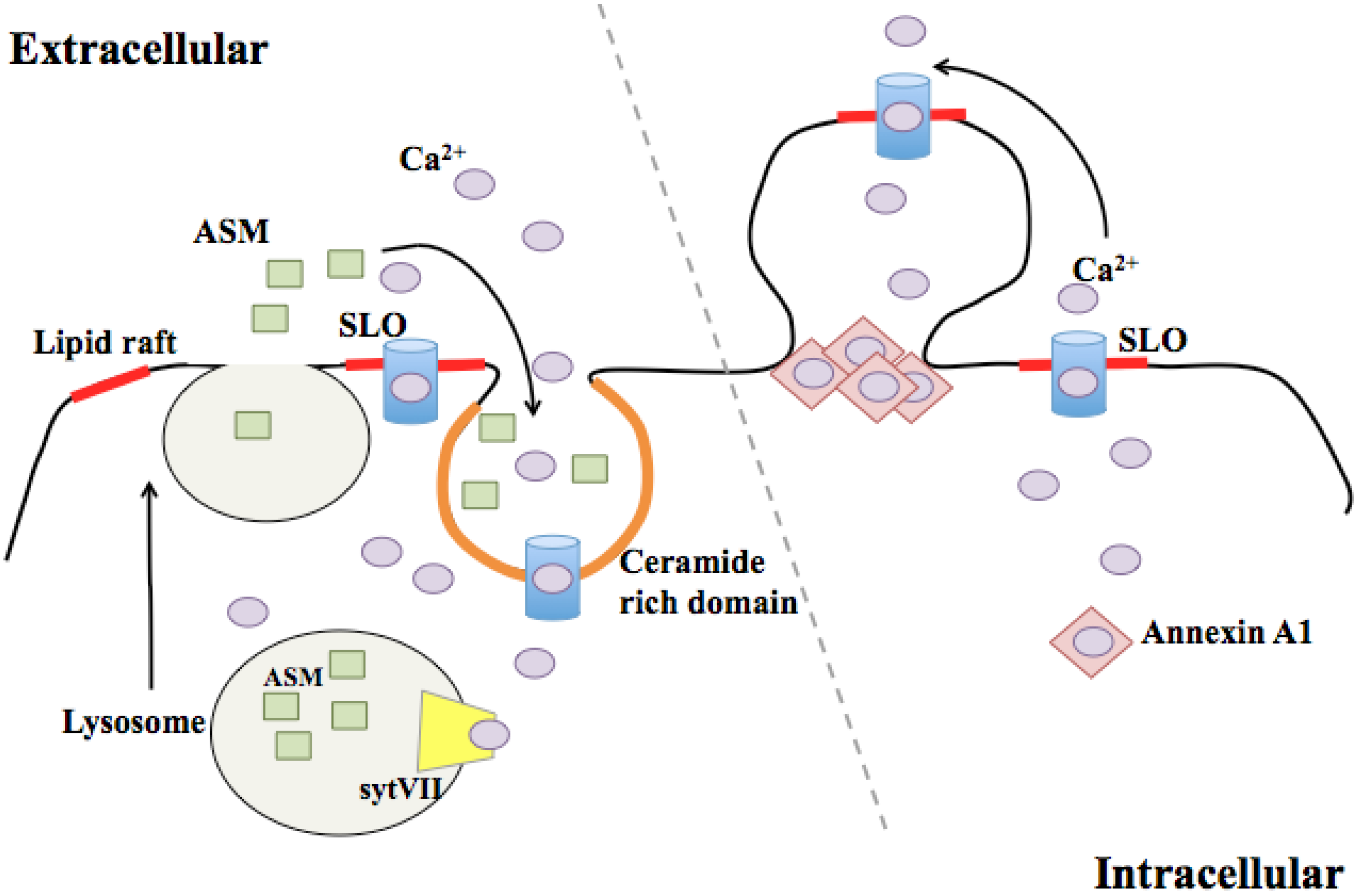
3.2. Membrane Blebbing
4. Intracellular Effects of CDC Intoxication
4.1. Effects on the Endoplasmic Reticulum and Golgi
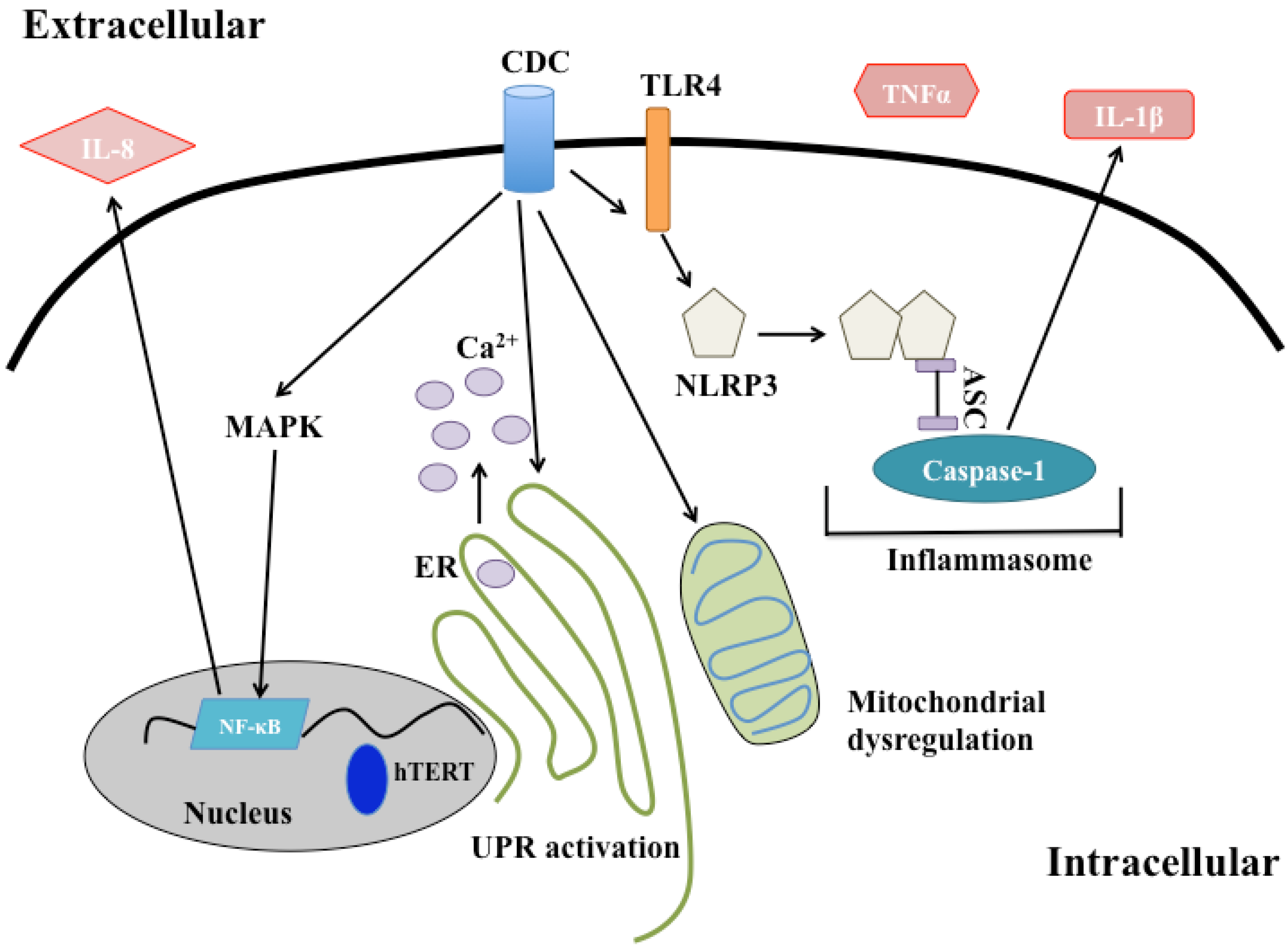
4.2. Effects on the Mitochondria and Nucleus
4.3. Endolysosomal Network
4.4. Immune Signaling
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Sierig, G.; Cywes, C.; Wessels, M.R.; Ashbaugh, C.D. Cytotoxic effects of streptolysin O and streptolysin S enhance the virulence of poorly encapsulated group A streptococci. Infect. Immun. 2003, 71, 446–455. [Google Scholar] [CrossRef]
- Magassa, N.A.G.; Chandrasekaran, S.; Caparon, M.G. Streptococcus pyogenes cytolysin-mediated translocation does not require pore formation by streptolysin O. EMBO Rep. 2010, 11, 400–405. [Google Scholar] [CrossRef]
- Portnoy, D.A.; Jacks, P.S.; Hinrichs, D.J. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 1988, 167, 1459–1471. [Google Scholar] [CrossRef]
- Chu, J.; Thomas, L.M.; Watkins, S.C.; Franchi, L.; Núñez, G.; Salter, R.D. Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J. Leukoc. Biol. 2009, 86, 1227–1238. [Google Scholar] [CrossRef]
- Baba, H.; Kawamura, I.; Kohda, C.; Nomura, T.; Ito, Y.; Kimoto, T.; Watanabe, I.; Ichiyama, S.; Mitsuyama, M. Induction of gamma interferon and nitric oxide by truncated pneumolysin that lacks pore-forming activity. Infect. Immun. 2002, 70, 107–113. [Google Scholar] [CrossRef]
- Walev, I.; Palmer, M.; Martin, E.; Jonas, D.; Weller, U.; Höhn-Bentz, H.; Husmann, M.; Bhakdi, S. Recovery of human fibroblasts from attack by the pore-forming alpha-toxin of Staphylococcus aureus. Microb. Pathog. 1994, 17, 187–201. [Google Scholar] [CrossRef]
- Jacobs, T.; Darji, A.; Frahm, N.; Rohde, M.; Wehland, J.; Chakraborty, T.; Weiss, S. Listeriolysin O: Cholesterol inhibits cytolysis but not binding to cellular membranes. Mol. Microbiol. 1998, 28, 1081–1089. [Google Scholar] [CrossRef]
- Hamon, M.A.; Cossart, P. K+ efflux is necessary for histone H3 dephosphorylation by Listeria monocytogenes listeriolysin O and other pore forming toxins. Infect. Immun. 2011, 79, 2839–2846. [Google Scholar] [CrossRef]
- Jones, S.; Prieter, K.; Portnoy, D.A. Conversion of an extracellular cytolysin into a phagosome-specific lysin which supports the growth of an intracellaulr pathogen. Mol. Micro. 1996, 21, 1219–1225. [Google Scholar]
- Dunstone, M.A.; Tweten, R.K. Packing a punch: the mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Curr. Opin. Struct. Biol. 2012, 22, 342–349. [Google Scholar] [CrossRef]
- Kafsack, B.F.C.; Pena, J.D.O.; Coppens, I.; Ravindran, S.; Boothroyd, J.C.; Carruthers, V.B. Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science 2009, 323, 530–533. [Google Scholar] [CrossRef]
- Sousa, M.V.; Richardson, M.; Fontes, W.; Morhy, L. Homology between the seed cytolysin enterolobin and bacterial aerolysins. J. Protein Chem. 1994, 13, 659–667. [Google Scholar] [CrossRef]
- Walev, I.; Bhakdi, S.C.; Hofmann, F.; Djonder, N.; Valeva, A.; Aktories, K.; Bhakdi, S. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. USA 2001, 98, 3185–3190. [Google Scholar]
- Lopez, J.A.; Susanto, O.; Jenkins, M.R.; Lukoyanova, N.; Sutton, V.R.; Law, R.H.P.; Johnston, A.; Bird, C.H.; Bird, P.I.; Whisstock, J.C.; et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 2013. [Google Scholar] [CrossRef]
- Heuck, A.P.; Moe, P.C.; Johnson, B.B. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell. Biochem. 2010, 51, 551–577. [Google Scholar]
- Rossjohn, J.; Feil, S.C.; McKinstry, W.J.; Tweten, R.K.; Parker, M.W. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell 1997, 89, 685–692. [Google Scholar] [CrossRef]
- Polekhina, G.; Giddings, K.S.; Tweten, R.K.; Parker, M.W. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc. Natl. Acad. Sci. USA 2005, 102, 600–605. [Google Scholar] [CrossRef]
- Bourdeau, R.W.; Malito, E.; Chenal, A.; Bishop, B.L.; Musch, M.W.; Villereal, M.L.; Chang, E.B.; Mosser, E.M.; Rest, R.F.; Tang, W.-J. Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J. Biol. Chem. 2009, 284, 14645–14656. [Google Scholar] [CrossRef]
- Xu, L.; Huang, B.; Du, H.; Zhang, X.C.; Xu, J.; Li, X.; Rao, Z. Crystal structure of cytotoxin protein suilysin from Streptococcus suis. Protein Cell 2010, 1, 96–105. [Google Scholar] [CrossRef]
- Hotze, E.M.; Tweten, R.K. Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. Acta 2012, 1818, 1028–1038. [Google Scholar] [CrossRef]
- Soltani, C.E.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20226–20231. [Google Scholar]
- Dowd, K.J.; Tweten, R.K. The cholesterol-dependent cytolysin signature motif: A critical element in the allosteric pathway that couples membrane binding to pore assembly. PLoS Pathog. 2012, 8, e1002787. [Google Scholar] [CrossRef]
- Giddings, K.S.; Zhao, J.; Sims, P.J.; Tweten, R.K. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 2004, 11, 1173–1178. [Google Scholar] [CrossRef]
- Gelber, S.E.; Aguilar, J.L.; Lewis, K.L.T.; Ratner, A.J. Functional and phylogenetic characterization of Vaginolysin, the human-specific cytolysin from Gardnerella vaginalis. J. Bacteriol. 2008, 190, 3896–3903. [Google Scholar] [CrossRef]
- Gekara, N.O.; Weiss, S. Lipid rafts clustering and signalling by listeriolysin O. Biochem. Soc. Trans. 2004, 32, 712–714. [Google Scholar] [CrossRef]
- Gekara, N.O.; Jacobs, T.; Chakraborty, T.; Weiss, S. The cholesterol-dependent cytolysin listeriolysin O aggregates rafts via oligomerization. Cell. Microbiol. 2005, 7, 1345–1356. [Google Scholar] [CrossRef]
- Nelson, L.D.; Chiantia, S.; London, E. Perfringolysin O association with ordered lipid domains: Implications for transmembrane protein raft affinity. Biophys. J. 2010, 99, 3255–3263. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Zitzer, A.; Bittman, R.; Verbicky, C.A.; Erukulla, R.K.; Bhakdi, S.; Weis, S.; Valeva, A.; Palmer, M. Coupling of cholesterol and cone-shaped lipids in bilayers augments membrane permeabilization by the cholesterol-specific toxins streptolysin O and Vibrio cholerae cytolysin. J. Biol. Chem. 2001, 276, 14628–14633. [Google Scholar]
- Kouzel, I.U.; Pohlentz, G.; Storck, W.; Radamm, L.; Hoffmann, P.; Bielaszewska, M.; Bauwens, A.; Cichon, C.; Schmidt, M.A.; Mormann, M.; Karch, H.; Müthing, J. Association of Shiga toxin glycosphingolipid receptors with membrane microdomains of toxin-sensitive lymphoid and myeloid cells. J. Lipid Res. 2013, 54, 692–710. [Google Scholar]
- Tilley, S.J.; Orlova, E.V.; Gilbert, R.J.C.; Andrew, P.W.; Saibil, H.R. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 2005, 121, 247–256. [Google Scholar]
- Ramachandran, R.; Heuck, A.P.; Tweten, R.K.; Johnson, A.E. Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat. Struct. Biol. 2002, 9, 823–827. [Google Scholar]
- Tweten, R.K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 2005, 73, 6199–6209. [Google Scholar] [CrossRef]
- Bhakdi, S.; Tranum-Jensen, J.; Sziegoleit, A. Mechanism of membrane damage by streptolysin-O. Infect. Immun. 1985, 47, 52–60. [Google Scholar]
- Schuerch, D.W.; Wilson-Kubalek, E.M.; Tweten, R.K. Molecular basis of listeriolysin O pH dependence. Proc. Natl. Acad. Sci. USA 2005, 102, 12537–12542. [Google Scholar]
- Czajkowsky, D.M.; Hotze, E.M.; Shao, Z.; Tweten, R.K. Vertical collapse of a cytolysin prepore moves its transmembrane beta-hairpins to the membrane. EMBO J. 2004, 23, 3206–3215. [Google Scholar] [CrossRef]
- Shaughnessy, L.M.; Hoppe, A.D.; Christensen, K.A.; Swanson, J.A. Membrane perforations inhibit lysosome fusion by altering pH and calcium in Listeria monocytogenes vacuoles. Cell. Microbiol. 2006, 8, 781–792. [Google Scholar] [CrossRef]
- Palmer, M.; Harris, R.; Freytag, C.; Kehoe, M.; Tranum-Jensen, J.; Bhakdi, S. Assembly mechanism of the oligomeric streptolysin O pore: The early membrane lesion is lined by a free edge of the lipid membrane and is extended gradually during oligomerization. EMBO J. 1998, 17, 1598–1605. [Google Scholar] [CrossRef]
- Gilbert, R.J.C.; Mikelj, M.; Dalla Serra, M.; Froelich, C.J.; Anderluh, G. Effects of MACPF/CDC proteins on lipid membranes. Cell. Mol. Life Sci. 2012. [Google Scholar] [CrossRef]
- Seong, S.-Y.; Matzinger, P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004, 4, 469–478. [Google Scholar] [CrossRef]
- Bhakdi, S.; Weller, U.; Walev, I.; Martin, E.; Jonas, D.; Palmer, M. A guide to the use of pore-forming toxins for controlled permeabilization of cell membranes. Med. Microbiol. Immunol. 1993, 182, 167–175. [Google Scholar]
- Gonzalez, M.R.; Bischofberger, M.; Frêche, B.; Ho, S.; Parton, R.G.; van der Goot, F.G. Pore-forming toxins induce multiple cellular responses promoting survival. Cell. Microbiol. 2011, 13, 1026–1043. [Google Scholar]
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 2004, 279, 20471–20479. [Google Scholar]
- Rodríguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997, 137, 93–104. [Google Scholar] [CrossRef]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 2010, 189, 1027–1038. [Google Scholar]
- Truman, J.-P.; Al Gadban, M.M.; Smith, K.J.; Hammad, S.M. Acid sphingomyelinase in macrophage biology. Cell. Mol. Life Sci. 2011, 68, 3293–3305. [Google Scholar] [CrossRef]
- Grassme, H.; Jendrossek, V.; Bock, J.; Riehle, A.; Gulbins, E. Ceramide-rich membrane rafts mediate CD40 clustering. J. Immunol. 2002, 168, 298–307. [Google Scholar]
- Corrotte, M.; Fernandes, M.C.; Tam, C.; Andrews, N.W. Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic 2012, 13, 483–494. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef]
- Lesieur, C.; Frutiger, S.; Hughes, G.; Kellner, R.; Pattus, F.; van der Goot, F.G. Increased stability upon heptamerization of the pore-forming toxin aerolysin. J. Biol. Chem. 1999, 274, 36722–36728. [Google Scholar]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar]
- Charras, G.; Paluch, E. Blebs lead the way: How to migrate without lamellipodia. Nat. Rev. Mol. Cell Biol. 2008, 9, 730–736. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2011, 18, 80–89. [Google Scholar] [CrossRef]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef]
- Cassidy, S.K.B.; Hagar, J.A.; Kanneganti, T.-D.; Franchi, L.; Núñez, G.; O’Riordan, M.X.D. Membrane damage during Listeria monocytogenes infection triggers a caspase-7 dependent cytoprotective response. PLoS Pathog. 2012, 8, e1002628. [Google Scholar] [CrossRef]
- Sebbagh, M.; Renvoizé, C.; Hamelin, J.; Riché, N.; Bertoglio, J.; Bréard, J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 2001, 3, 346–352. [Google Scholar] [CrossRef]
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 2001, 3, 339–345. [Google Scholar]
- Pillich, H.; Loose, M.; Zimmer, K.-P.; Chakraborty, T. Activation of the unfolded protein response by Listeria monocytogenes. Cell. Microbiol. 2012, 14, 949–964. [Google Scholar] [CrossRef]
- Todd, D.J.; Lee, A.-H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Bischof, L.J.; Kao, C.-Y.; Los, F.C.O.; Gonzalez, M.R.; Shen, Z.; Briggs, S.P.; van der Goot, F.G.; Aroian, R.V. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008, 4, e1000176. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Jäättelä, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Tweten, R.K.; Johnson, A.E.; Heuck, A.P. Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 2009, 48, 3977–3987. [Google Scholar]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Gekara, N.O.; Westphal, K.; Ma, B.; Rohde, M.; Groebe, L.; Weiss, S. The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell. Microbiol. 2007, 9, 2008–2021. [Google Scholar] [CrossRef]
- Kobayashi, T.; Pimplikar, S.W.; Parton, R.G.; Bhakdi, S.; Simons, K. Sphingolipid transport from the trans-Golgi network to the apical surface in permeabilized MDCK cells. FEBS Lett. 1992, 300, 227–231. [Google Scholar] [CrossRef]
- Bankaitis, V.A.; Garcia-Mata, R.; Mousley, C.J. Golgi membrane dynamics and lipid metabolism. Curr. Biol. 2012, 22, R414–R424. [Google Scholar] [CrossRef]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef]
- Goldmann, O.; Sastalla, I.; Wos-Oxley, M.; Rohde, M.; Medina, E. Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell. Microbiol. 2009, 11, 138–155. [Google Scholar] [CrossRef]
- Braun, J.S.; Hoffmann, O.; Schickhaus, M.; Freyer, D.; Dagand, E.; Bermpohl, D.; Mitchell, T.J.; Bechmann, I.; Weber, J.R. Pneumolysin causes neuronal cell death through mitochondrial damage. Infect Immun. 2007, 75, 4245–4254. [Google Scholar] [CrossRef]
- Stavru, F.; Bouillaud, F.; Sartori, A.; Ricquier, D.; Cossart, P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 2011, 108, 3612–3617. [Google Scholar] [CrossRef]
- Hamon, M.A.; Batsché, E.; Régnault, B.; Tham, T.N.; Seveau, S.; Muchardt, C.; Cossart, P. Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. USA 2007, 104, 13467–13472. [Google Scholar]
- Kayal, S.; Lilienbaum, A.; Poyart, C.; Memet, S.; Israel, A.; Berche, P. Listeriolysin O-dependent activation of endothelial cells during infection with Listeria monocytogenes: Activation of NF-kappa B and upregulation of adhesion molecules and chemokines. Mol. Microbiol. 1999, 31, 1709–1722. [Google Scholar] [CrossRef]
- Tang, P.; Rosenshine, I.; Cossart, P.; Finlay, B.B. Listeriolysin O activates mitogen-activated protein kinase in eucaryotic cells. Infect. Immun. 1996, 64, 2359–2361. [Google Scholar]
- Samba-Louaka, A.; Stavru, F.; Cossart, P. Role for telomerase in Listeria monocytogenes infection. Infect. Immun. 2012, 80, 4257–4263. [Google Scholar] [CrossRef]
- Schnupf, P.; Portnoy, D.A. Listeriolysin O: A phagosome-specific lysin. Microbes Infect. 2007, 9, 1176–1187. [Google Scholar] [CrossRef]
- Shaughnessy, L.M.; Lipp, P.; Lee, K.-D.; Swanson, J.A. Localization of protein kinase C epsilon to macrophage vacuoles perforated by Listeria monocytogenes cytolysin. Cell. Microbiol. 2007, 9, 1695–1704. [Google Scholar] [CrossRef]
- Henry, R.; Shaughnessy, L.; Loessner, M.J.; Alberti-Segui, C.; Higgins, D.E.; Swanson, J.A. Cytolysin-dependent delay of vacuole maturation in macrophages infected with Listeria monocytogenes. Cell. Microbiol. 2006, 8, 107–119. [Google Scholar] [CrossRef]
- Marquis, H.; Goldfine, H.; Portnoy, D.A. Proteolytic pathways of activation and degradation of a bacterial phospholipase C during intracellular infection by Listeria monocytogenes. J. Cell Biol. 1997, 137, 1381–1392. [Google Scholar] [CrossRef]
- Wadsworth, S.J.; Goldfine, H. Mobilization of protein kinase C in macrophages induced by Listeria monocytogenes affects its internalization and escape from the phagosome. Infect. Immun. 2002, 70, 4650–4660. [Google Scholar] [CrossRef]
- Birmingham, C.L.; Canadien, V.; Gouin, E.; Troy, E.B.; Yoshimori, T.; Cossart, P.; Higgins, D.E.; Brumell, J.H. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 2007, 3, 442–451. [Google Scholar]
- Meyer-Morse, N.; Robbins, J.R.; Rae, C.S.; Mochegova, S.N.; Swanson, M.S.; Zhao, Z.; Virgin, H.W.; Portnoy, D. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS One 2010, 5, e8610. [Google Scholar]
- Logsdon, L.K.; Håkansson, A.P.; Cortés, G.; Wessels, M.R. Streptolysin O inhibits clathrin-dependent internalization of group A Streptococcus. MBio 2011, 2, e00332-10. [Google Scholar]
- Sakurai, A.; Maruyama, F.; Funao, J.; Nozawa, T.; Aikawa, C.; Okahashi, N.; Shintani, S.; Hamada, S.; Ooshima, T.; Nakagawa, I. Specific behavior of intracellular Streptococcus pyogenes that has undergone autophagic degradation is associated with bacterial streptolysin O and host small G proteins Rab5 and Rab7. J. Biol. Chem. 2010, 285, 22666–22675. [Google Scholar]
- Thiery, J.; Keefe, D.; Boulant, S.; Boucrot, E.; Walch, M.; Martinvalet, D.; Goping, I.S.; Bleackley, R.C.; Kirchhausen, T.; Lieberman, J. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat. Immunol. 2011, 12, 770–777. [Google Scholar]
- Davis, M.J.; Swanson, J.A. Technical advance: Caspase-1 activation and IL-1β release correlate with the degree of lysosome damage, as illustrated by a novel imaging method to quantify phagolysosome damage. J. Leukoc. Biol. 2010, 88, 813–822. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar]
- Dong, C.; Davis, R.J.; Flavell, R.A. MAP kinases in the immune response. Annu. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [CrossRef]
- Dogan, S.; Zhang, Q.; Pridmore, A.C.; Mitchell, T.J.; Finn, A.; Murdoch, C. Pneumolysin-induced CXCL8 production by nasopharyngeal epithelial cells is dependent on calcium flux and MAPK activation by Toll-like receptor 4. Microbes Infect. 2011, 13, 65–75. [Google Scholar] [CrossRef]
- Nilsson, M.; Sørensen, O.E.; Mörgelin, M.; Weineisen, M.; Sjöbring, U.; Herwald, H. Activation of human polymorphonuclear neutrophils by streptolysin O from Streptococcus pyogenes leads to the release of proinflammatory mediators. Thromb. Haemost. 2006, 95, 982–990. [Google Scholar]
- Bebien, M.; Hensler, M.E.; Davanture, S.; Hsu, L.-C.; Karin, M.; Park, J.M.; Alexopoulou, L.; Liu, G.Y.; Nizet, V.; Lawrence, T. The pore-forming toxin β hemolysin/cytolysin triggers p38 MAPK-dependent IL-10 production in macrophages and inhibits innate immunity. PLoS Pathog. 2012, 8, e1002812. [Google Scholar] [CrossRef]
- Hackett, S.P.; Stevens, D.L. Streptococcal toxic shock syndrome: synthesis of tumor necrosis factor and interleukin-1 by monocytes stimulated with pyrogenic exotoxin A and streptolysin O. J. Infect. Dis. 1992, 165, 879–885. [Google Scholar] [CrossRef]
- Houldsworth, S.; Andrew, P.W.; Mitchell, T.J. Pneumolysin stimulates production of tumor necrosis factor alpha and interleukin-1 beta by human mononuclear phagocytes. Infect. Immun. 1994, 62, 1501–1503. [Google Scholar]
- Malley, R.; Henneke, P.; Morse, S.C.; Cieslewicz, M.J.; Lipsitch, M.; Thompson, C.M.; Kurt-Jones, E.; Paton, J.C.; Wessels, M.R.; Golenbock, D.T. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. USA 2003, 100, 1966–1971. [Google Scholar] [CrossRef]
- Park, J.M.; Ng, V.H.; Maeda, S.; Rest, R.F.; Karin, M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J. Exp. Med. 2004, 200, 1647–1655. [Google Scholar]
- Zhang, H.; Tay, P.N.; Cao, W.; Li, W.; Lu, J. Integrin-nucleated Toll-like receptor (TLR) dimerization reveals subcellular targeting of TLRs and distinct mechanisms of TLR4 activation and signaling. FEBS Lett. 2002, 532, 171–176. [Google Scholar] [CrossRef]
- Harder, J.; Franchi, L.; Muñoz-Planillo, R.; Park, J.-H.; Reimer, T.; Núñez, G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J. Immunol. 2009, 183, 5823–5829. [Google Scholar] [CrossRef]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Keyel, P.A.; Heid, M.E.; Salter, R.D. Macrophage responses to bacterial toxins: A balance between activation and suppression. Immunol. Res. 2011, 50, 118–123. [Google Scholar] [CrossRef]
- Carrero, J.A.; Vivanco-Cid, H.; Unanue, E.R. Listeriolysin O is strongly immunogenic independently of its cytotoxic activity. PLoS One 2012, 7, e32310. [Google Scholar]
- Wallecha, A.; Wood, L.; Pan, Z.-K.; Maciag, P.C.; Shahabi, V.; Paterson, Y. Listeria monocytogenes-derived listeriolysin O has pathogen-associated molecular pattern-like properties independent of its hemolytic ability. Clin. Vaccine Immunol. 2013, 20, 77–84. [Google Scholar] [CrossRef]
- Harty, J.T.; Schreiber, R.D.; Bevan, M.J. CD8 T cells can protect against an intracellular bacterium in an interferon gamma-independent fashion. Proc. Natl. Acad. Sci. USA 1992, 89, 11612–11616. [Google Scholar] [CrossRef]
- Ladel, C.H.; Flesch, I.E.; Arnoldi, J.; Kaufmann, S.H. Studies with MHC-deficient knock-out mice reveal impact of both MHC I- and MHC II-dependent T cell responses on Listeria monocytogenes infection. J. Immunol. 1994, 153, 3116–3122. [Google Scholar]
- Gekara, N.O.; Zietara, N.; Geffers, R.; Weiss, S. Listeria monocytogenes induces T cell receptor unresponsiveness through pore-forming toxin listeriolysin O. J. Infect. Dis. 2010, 202, 1698–1707. [Google Scholar] [CrossRef]
- Carrero, J.A.; Calderon, B.; Unanue, E.R. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J. Immunol. 2004, 172, 4866–4874. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cassidy, S.K.B.; O'Riordan, M.X.D. More Than a Pore: The Cellular Response to Cholesterol-Dependent Cytolysins. Toxins 2013, 5, 618-636. https://doi.org/10.3390/toxins5040618
Cassidy SKB, O'Riordan MXD. More Than a Pore: The Cellular Response to Cholesterol-Dependent Cytolysins. Toxins. 2013; 5(4):618-636. https://doi.org/10.3390/toxins5040618
Chicago/Turabian StyleCassidy, Sara K. B., and Mary X. D. O'Riordan. 2013. "More Than a Pore: The Cellular Response to Cholesterol-Dependent Cytolysins" Toxins 5, no. 4: 618-636. https://doi.org/10.3390/toxins5040618