
Shiga Toxin 2-Induced Endoplasmic Reticulum Stress Is Minimized by Activated Protein C but Does Not Correlate with Lethal Kidney Injury

Abstract
:1. Introduction
2. Results
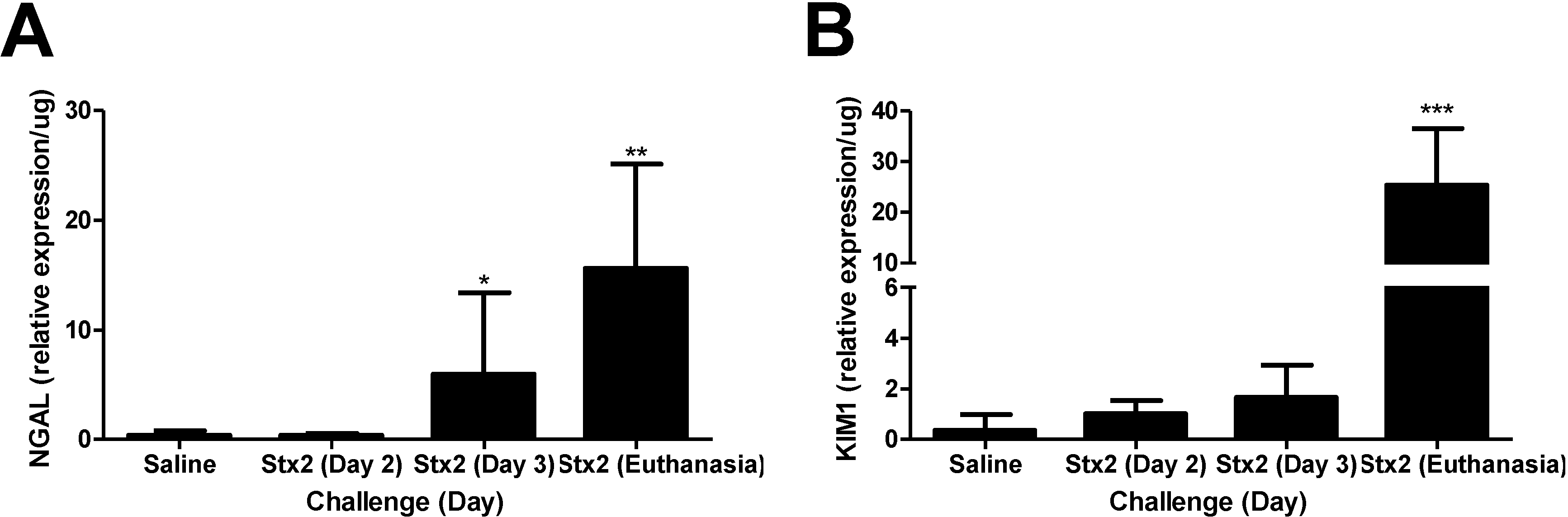
2.1. Murine Responses in Two Injection Stx2 Model
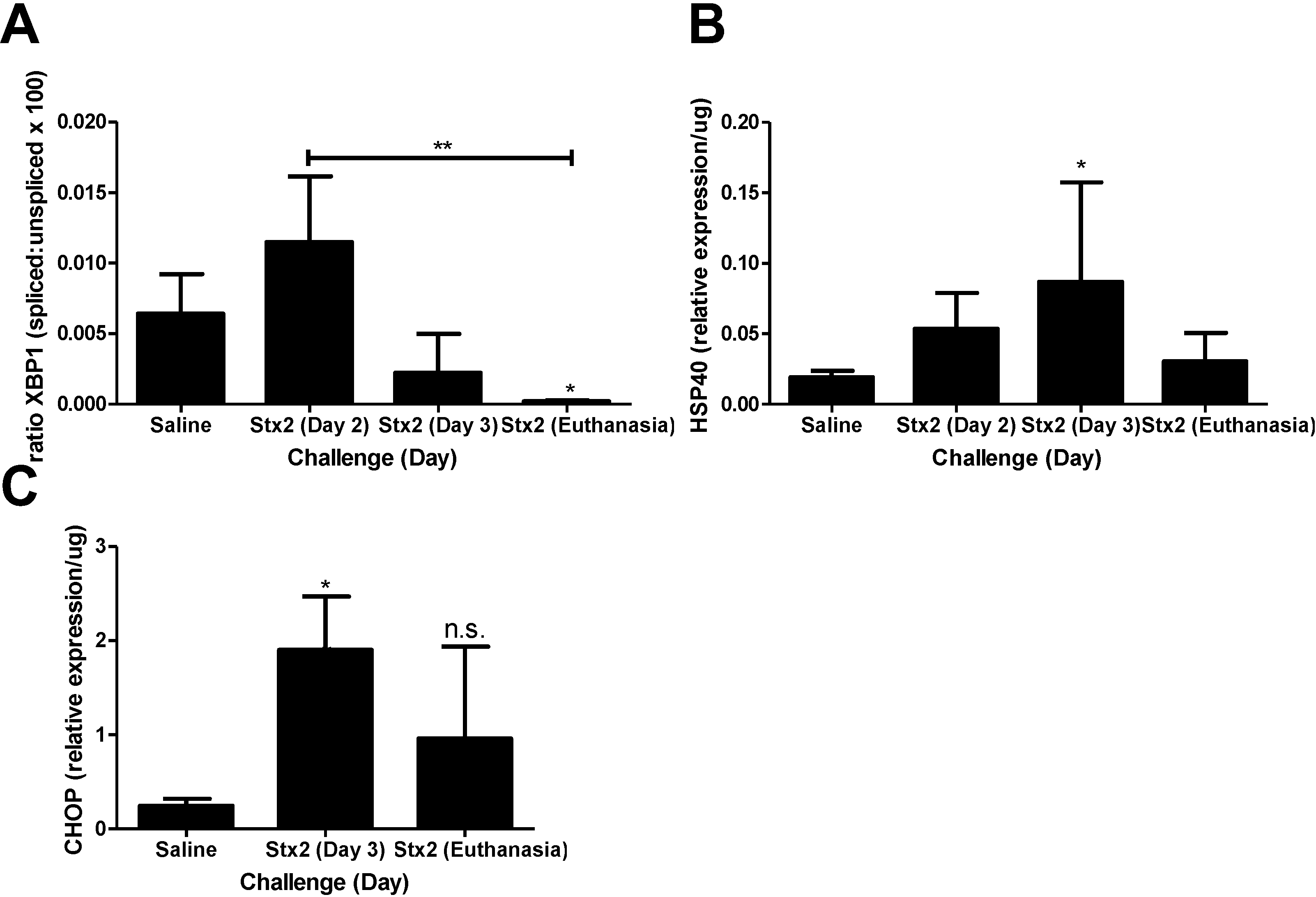
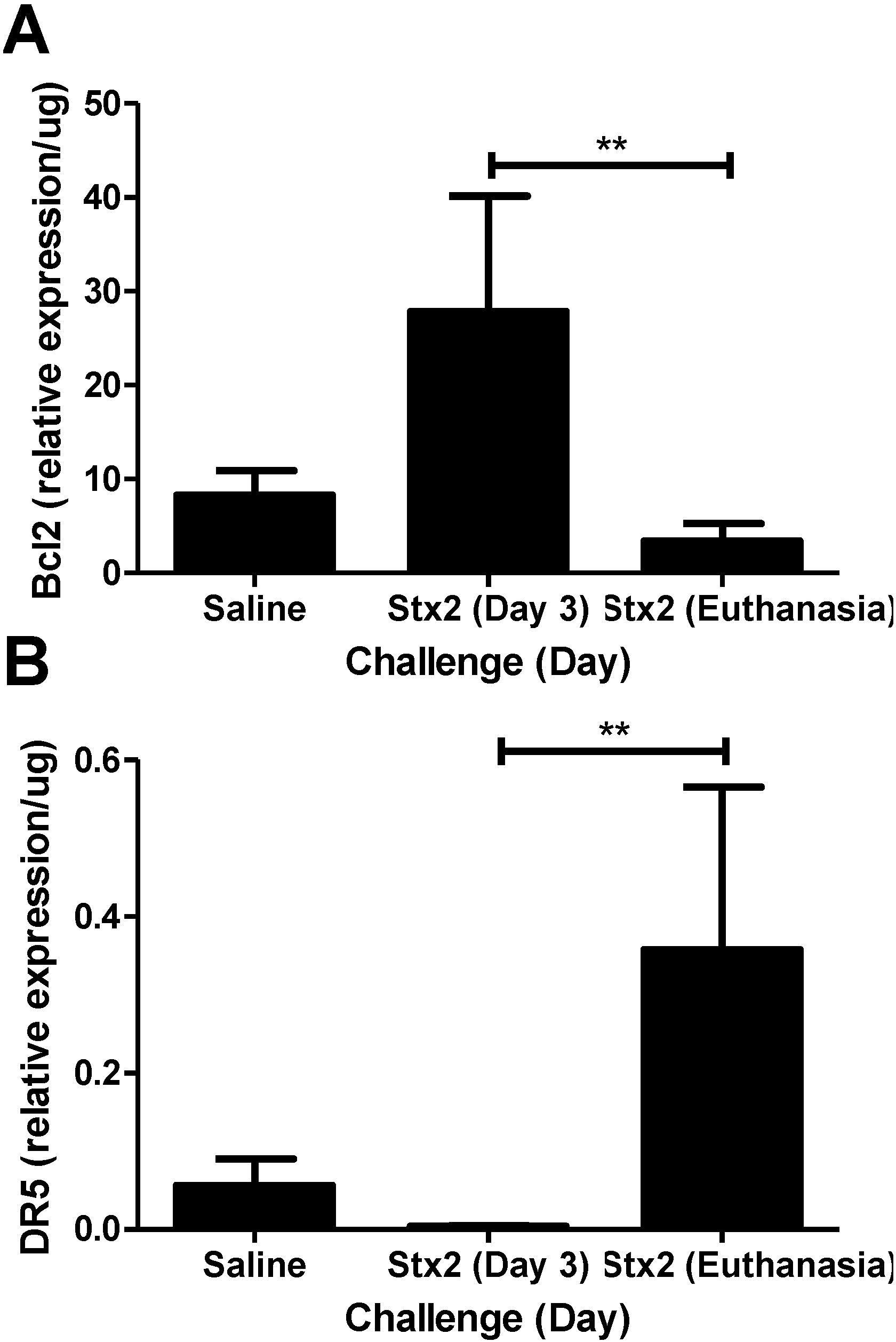
2.2. ER Stress and Apoptosis

2.3. Activated Protein C and Z-VAD-FMK Treatment



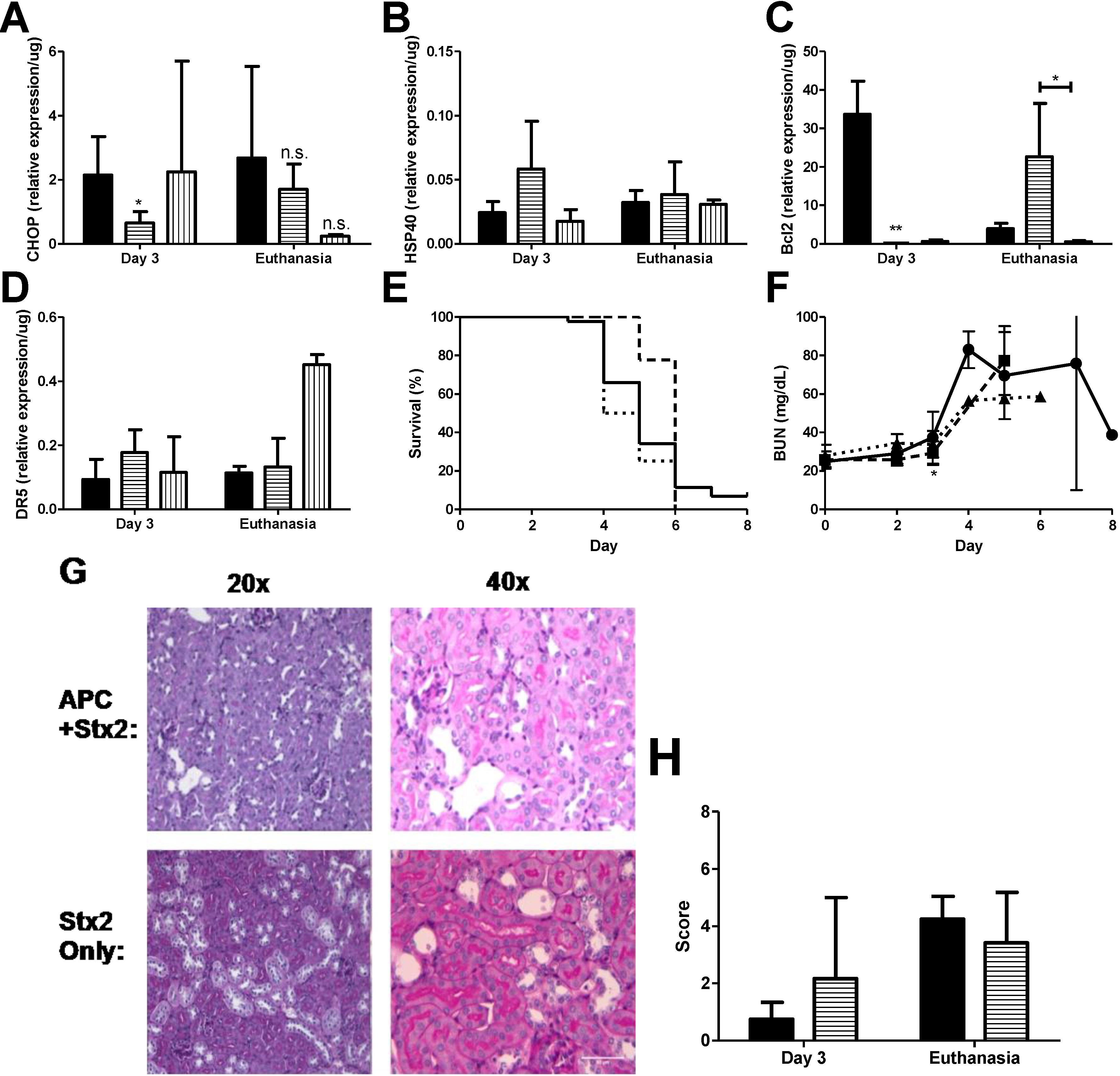
2.4. ER Stress, Apoptosis, and APC Treatment in Stx2-Producing Citrobacter rodentium Model

3. Discussion
3.1. Stx2-Induced Murine Lethal Kidney Injury Is Accompanied by Transcriptional Evidence of ER Stress and Apoptosis
3.2. APC Treatment of Stx2 and Stx2 Producing C. rodentium Challenged Mice Down-Regulates ER Stress but Does Not Reverse Lethal Kidney Injury
3.3. Z-VAD-FMK Treatment of Stx2 Challenged Mice Is Ineffective
3.4. Targeting Intracellular Stx2 Activities Remains a Viable Therapeutic Opportunity
4. Materials and Methods
4.1. Reagents
4.2. Citrobacter rodentium Culture
4.3. Animal Experiments
4.4. RNA Isolation and qPCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence |
|---|---|
| Lcn1 (NGAL) | F: 5'CCCTGTATGGAAGAACCAAGGA3' |
| R: 5'CGGTGGGGACAGAGAAGATG3' | |
| Kim1 | F: 5'GGAGATACCTGGAGTAATCACACTG3' |
| R: 5'TAGCCACGGTGCTCACAAGC3' | |
| Xbp1 | F: 5'AAACAGAGTAGCAGCTCAGACTGC3' |
| R: 3'ATCTCTAAGACTAGGGGCTTGGT3' | |
| ERdj4 (HSP40) | F: 5'AGGAACCTGGGAGCTTGACTA3' |
| R: 5'ACACATGACGTGCTTGGAATG3' | |
| Bcl2 | F: 5'TTCTTTGAGTTCGGTGGGGTC3' |
| R: 5'TGCATATTTGTTTGGGGCAGG3' | |
| Dr5 | F: 5'TTCCAGTAGTGCTGCTGATTGG3' |
| R: 5'CAAACGCACTGAGATCCTCCT3' | |
| Ddit3 (CHOP) | F: 5'AGTTATCTTGAGCCTAACACGTCG3' |
| R: 5'CACTTCCTTCTGGAACACTCTCTC3' | |
| Hprt | F: 5'TGGGCTTACCTCACTGCTTTC3' |
| R: 5'CCTGGTTCATCATCGCTAATCAC3' |
4.5. Spliced XBP1 Assay
4.6. Histology
| Criteria (6 fields analyzed per animal) | Score | |
|---|---|---|
| Injury foci in low magnification field (20×) | 0 | 0 |
| 1 per field | 1 | |
| 2 per field | 1 | |
| 3 per field | 1 | |
| 4+ per field | 1 | |
| Tubule dilation | 0 per field | 0 |
| 1–4 per field | 1 | |
| 4+ per field | 1 | |
| Epithelial cell shedding | 1 | |
| Pyknotic bodies | 1 | |
| Total possible Score | 8 | |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Frenzen, P.D.; Drake, A.; Angulo, F.J. Economic cost of illness due to Escherichia coli O157 infections in the United States. J. Food Prot. 2005, 68, 2623–2630. [Google Scholar]
- Riley, L.W.; Remis, R.S.; Helgerson, S.D.; McGee, H.B.; Wells, J.G.; Davis, B.R.; Hebert, R.J.; Olcott, E.S.; Johnson, L.M.; Hargrett, N.T.; et al. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 1983, 308, 681–685. [Google Scholar] [CrossRef]
- Manning, S.D.; Motiwala, A.S.; Springman, A.C.; Qi, W.; Lacher, D.W.; Ouellette, L.M.; Mladonicky, J.M.; Somsel, P.; Rudrik, J.T.; Dietrich, S.E.; et al. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. USA 2008, 105, 4868–4873. [Google Scholar] [CrossRef]
- Rangel, J.M.; Sparling, P.H.; Crowe, C.; Griffin, P.M.; Swerdlow, D.L. Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982–2002. Emerg. Infect. Dis. 2005, 11, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Paton, J.C.; Paton, A.W. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 1998, 11, 450–479. [Google Scholar] [PubMed]
- Karch, H.; Tarr, P.I.; Bielaszewska, M. Enterohaemorrhagic Escherichia coli in human medicine. Int. J. Med. Microbiol. 2005, 295, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Gould, L.H.; Demma, L.; Jones, T.F.; Hurd, S.; Vugia, D.J.; Smith, K.; Shiferaw, B.; Segler, S.; Palmer, A.; Zansky, S.; et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000–2006. Clin. Infect. Dis. 2009, 49, 1480–1485. [Google Scholar] [CrossRef]
- Rowe, P.C.; Orrbine, E.; Lior, H.; Wells, G.A.; Yetisir, E.; Clulow, M.; McLaine, P.N. Risk of hemolytic uremic syndrome after sporadic Escherichia coli O157:H7 infection: Results of a Canadian collaborative study. Investigators of the Canadian Pediatric Kidney Disease Research Center. J. Pediatr. 1998, 132, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Oakes, R.S.; Kirkham, J.K.; Nelson, R.D.; Siegler, R.L. Duration of oliguria and anuria as predictors of chronic renal-related sequelae in post-diarrheal hemolytic uremic syndrome. Pediatr. Nephrol. 2008, 23, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.S.; Mooney, J.C.; Brandt, J.R.; Staples, A.O.; Jelacic, S.; Boster, D.R.; Watkins, S.L.; Tarr, P.I. Risk factors for the hemolytic uremic syndrome in children infected with Escherichia coli O157:H7: A multivariable analysis. Clin. Infect. Dis. 2012, 55, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin gene expression by Shiga toxin-producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Geitz, C.; Tarr, P.I.; Frosch, M.; Karch, H. Non-O157:H7 pathogenic Shiga toxin-producing Escherichia coli: Phenotypic and genetic profiling of virulence traits and evidence for clonality. J. Infect. Dis. 1999, 179, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Boerlin, P.; McEwen, S.A.; Boerlin-Petzold, F.; Wilson, J.B.; Johnson, R.P.; Gyles, C.L. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [PubMed]
- Sandvig, K.; van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 1996, 76, 949–966. [Google Scholar] [PubMed]
- Sandvig, K.; Olsnes, S.; Brown, J.E.; Petersen, O.W.; van Deurs, B. Endocytosis from coated pits of Shiga toxin: A glycolipid-binding protein from Shigella dysenteriae 1. J. Cell. Biol. 1989, 108, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Garred, O.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, N.; Taguchi, T.; Mori, T.; Uchida, H.; Sato, N.; Takeda, T.; Fujimoto, J. Induction of apoptosis in normal human renal tubular epithelial cells by Escherichia coli Shiga toxins 1 and 2. J. Infect. Dis. 1998, 178, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Cherla, R.P.; Lee, S.Y.; Tesh, V.L. Shiga toxins and apoptosis. FEMS Microbiol. Lett. 2003, 228, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Hakansson, A.; Perez, M.T.; Isaksson, C.; Carlemalm, E.; Caprioli, A.; Svanborg, C. Apoptosis of renal cortical cells in the hemolytic-uremic syndrome: In vivo and in vitro studies. Infect. Immunity 1998, 66, 636–644. [Google Scholar]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Mallick, E.M.; McBee, M.E.; Vanguri, V.K.; Melton-Celsa, A.R.; Schlieper, K.; Karalius, B.J.; O'Brien, A.D.; Butterton, J.R.; Leong, J.M.; Schauer, D.B. A novel murine infection model for Shiga toxin-producing Escherichia coli. J. Clin. Investig. 2012, 122, 4012–4024. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. The cytoprotective protein C pathway. Blood 2007, 109, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Toltl, L.J.; Austin, R.C.; Liaw, P.C. Activated protein C modulates inflammation, apoptosis and tissue factor procoagulant activity by regulating endoplasmic reticulum calcium depletion in blood monocytes. J. Thromb. Haemost. 2011, 9, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Calvo, M.; Peterson, E.P.; Leiting, B.; Ruel, R.; Nicholson, D.W.; Thornberry, N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998, 273, 32608–32613. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P. Neutrophil gelatinase-associated lipocalin: A promising biomarker for human acute kidney injury. Biomark. Med. 2010, 4, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Liangos, O.; Perianayagam, M.C.; Vaidya, V.S.; Han, W.K.; Wald, R.; Tighiouart, H.; MacKinnon, R.W.; Li, L.; Balakrishnan, V.S.; Pereira, B.J.; et al. Urinary N-acetyl-β-(d)-glucosaminidase activity and kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J. Am. Soc. Nephrol. 2007, 18, 904–912. [Google Scholar] [CrossRef]
- Donohue-Rolfe, A.; Kondova, I.; Oswald, S.; Hutto, D.; Tzipori, S. Escherichia coli O157:H7 strains that express Shiga toxin (Stx) 2 alone are more neurotropic for gnotobiotic piglets than are isotypes producing only Stx1 or both Stx1 and Stx2. J. Infect. Dis. 2000, 181, 1825–1829. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.W.; Otero, J.H.; Hendershot, L.M.; Snapp, E. ERdj4 protein is a soluble endoplasmic reticulum (ER) DnaJ family protein that interacts with ER-associated degradation machinery. J. Biol. Chem. 2012, 287, 7969–7978. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Habener, J.F. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992, 6, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; He, Y.; Jing, Y.; Li, K.; Zhang, J. Albumin overload induces apoptosis in renal tubular epithelial cells through a CHOP-dependent pathway. OMICS 2010, 14, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Collins, V.; Freeman, S.; Tesh, V.L.; Kurosawa, S. Distinct physiologic and inflammatory responses elicited in baboons after challenge with Shiga toxin type 1 or 2 from enterohemorrhagic Escherichia coli. Infect. Immunity 2010, 78, 2497–2504. [Google Scholar] [CrossRef]
- Huang, D.C.; Hahne, M.; Schroeter, M.; Frei, K.; Fontana, A.; Villunger, A.; Newton, K.; Tschopp, J.; Strasser, A. Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-x(l). Proc. Natl. Acad. Sci. USA 1999, 96, 14871–14876. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-κB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Cherla, R.P.; Caliskan, I.; Tesh, V.L. Shiga toxin 1 induces apoptosis in the human myelogenous leukemia cell line THP-1 by a caspase-8-dependent, tumor necrosis factor receptor-independent mechanism. Infect. Immunity 2005, 73, 5115–5126. [Google Scholar] [CrossRef]
- Yoshida, T.; Chen, C.C.; Zhang, M.S.; Wu, H.C. Disruption of the Golgi apparatus by brefeldin A inhibits the cytotoxicity of ricin, modeccin, and Pseudomonas toxin. Exp. Cell Res. 1991, 192, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.P.; Oda, T.; Chen, C.; Kuwazuru, Y.; Wu, H.C. Involvement of the Golgi region in the intracellular trafficking of cholera toxin. J. Cell. Physiol. 1993, 154, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Kojio, S.; Zhang, H.; Ohmura, M.; Gondaira, F.; Kobayashi, N.; Yamamoto, T. Caspase-3 activation and apoptosis induction coupled with the retrograde transport of Shiga toxin: Inhibition by brefeldin A. FEMS Immunol. Med. Microbiol. 2000, 29, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Watanabe, M.; Kita, E.; Igai, K.; Omata, K.; Yaffe, M.B.; Natori, Y. A multivalent peptide library approach identifies a novel Shiga toxin inhibitor that induces aberrant cellular transport of the toxin. FASEB J. 2006, 20, 2597–2599. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Collins, V.; Freeman, S.; Debord, D.; Nishikawa, K.; Oh, S.Y.; Leibowitz, C.S.; Kurosawa, S. Rescue from lethal Shiga toxin 2-induced renal failure with a cell-permeable peptide. Pediatr. Nephrol. 2011, 26, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Sillence, D.J.; Falguieres, T.; Jarvis, R.M.; Johannes, L.; Lord, J.M.; Platt, F.M.; Roberts, L.M. The association of Shiga-like toxin with detergent-resistant membranes is modulated by glucosylceramide and is an essential requirement in the endoplasmic reticulum for a cytotoxic effect. Mol. Biol. Cell 2006, 17, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parello, C.S.L.; Mayer, C.L.; Lee, B.C.; Motomochi, A.; Kurosawa, S.; Stearns-Kurosawa, D.J. Shiga Toxin 2-Induced Endoplasmic Reticulum Stress Is Minimized by Activated Protein C but Does Not Correlate with Lethal Kidney Injury. Toxins 2015, 7, 170-186. https://doi.org/10.3390/toxins7010170
Parello CSL, Mayer CL, Lee BC, Motomochi A, Kurosawa S, Stearns-Kurosawa DJ. Shiga Toxin 2-Induced Endoplasmic Reticulum Stress Is Minimized by Activated Protein C but Does Not Correlate with Lethal Kidney Injury. Toxins. 2015; 7(1):170-186. https://doi.org/10.3390/toxins7010170
Chicago/Turabian StyleParello, Caitlin S. L., Chad L. Mayer, Benjamin C. Lee, Amanda Motomochi, Shinichiro Kurosawa, and Deborah J. Stearns-Kurosawa. 2015. "Shiga Toxin 2-Induced Endoplasmic Reticulum Stress Is Minimized by Activated Protein C but Does Not Correlate with Lethal Kidney Injury" Toxins 7, no. 1: 170-186. https://doi.org/10.3390/toxins7010170