
The Anti-Cancer Potency and Mechanism of a Novel Tumor-Activated Fused Toxin, DLM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
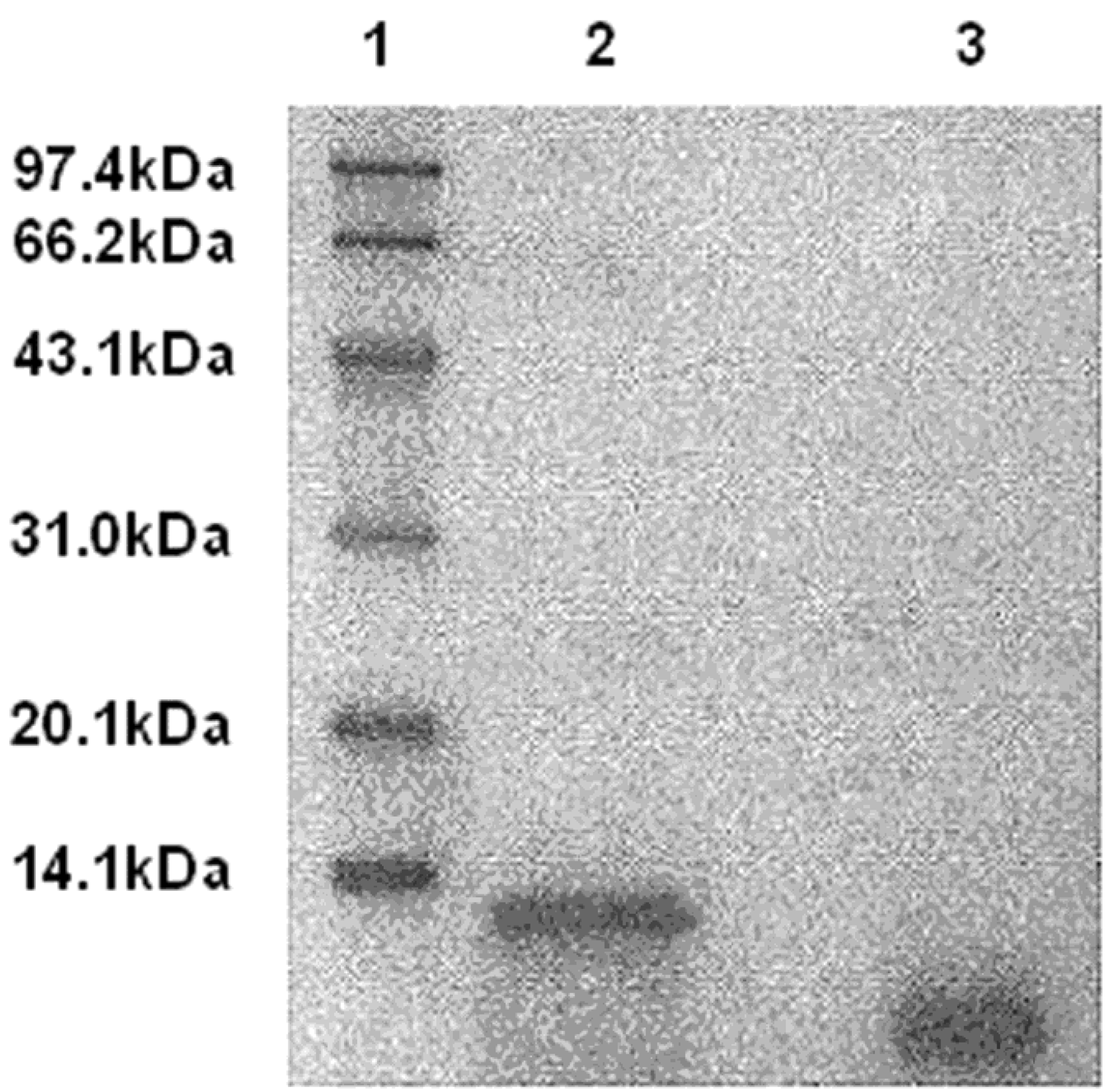
2.1. Disintegrin Is Released from DLM by uPA Cleavage

2.2. Hemolysis Bioactivity of the DLM

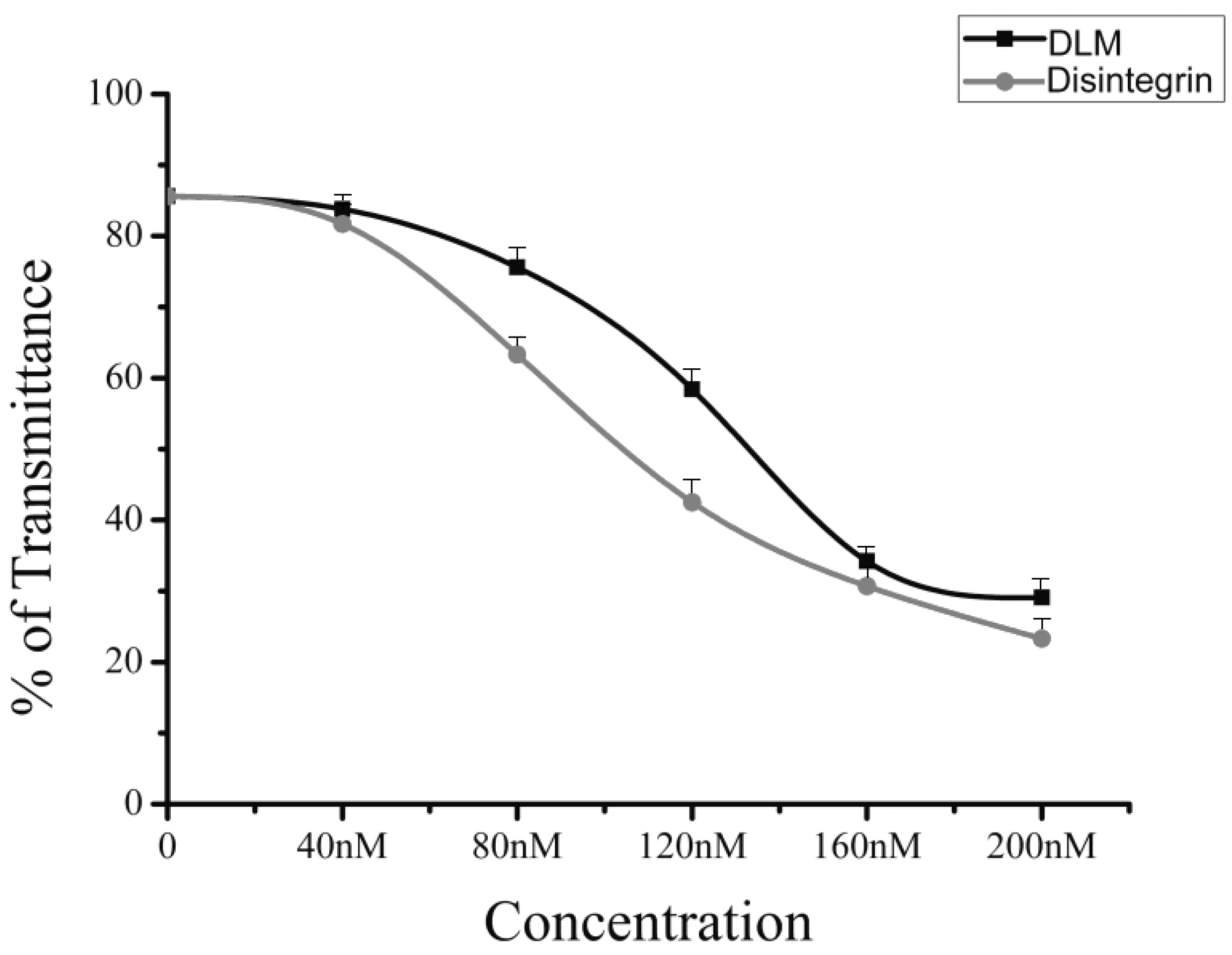
2.3. Native Disintegrin and DLM Activity

2.4. Binding of Disintegrin to Inactive Platelets

2.5. MTT Assay

2.6. DNA Ladder

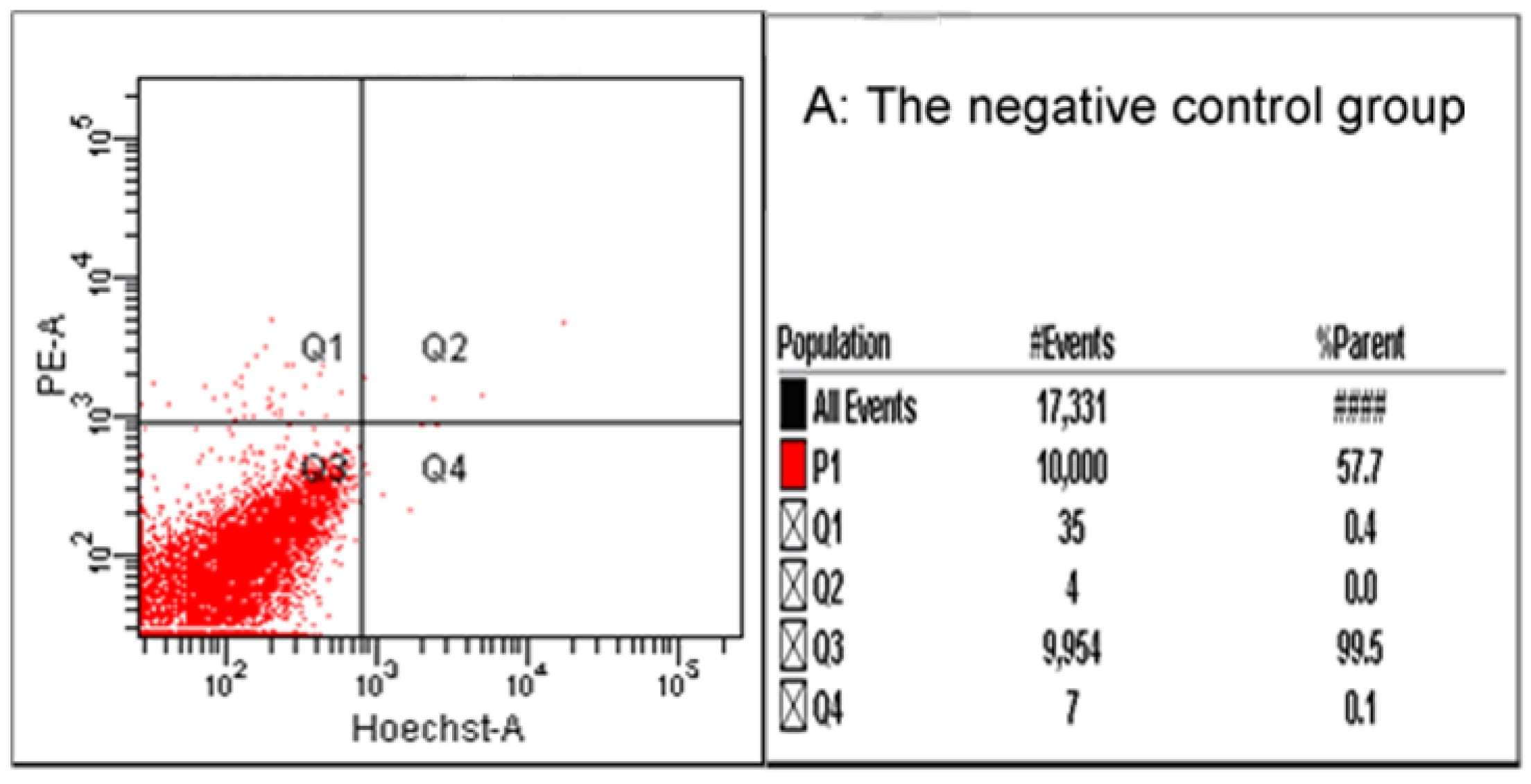
2.7. Flow Cytometry and Fluorescence Microscope




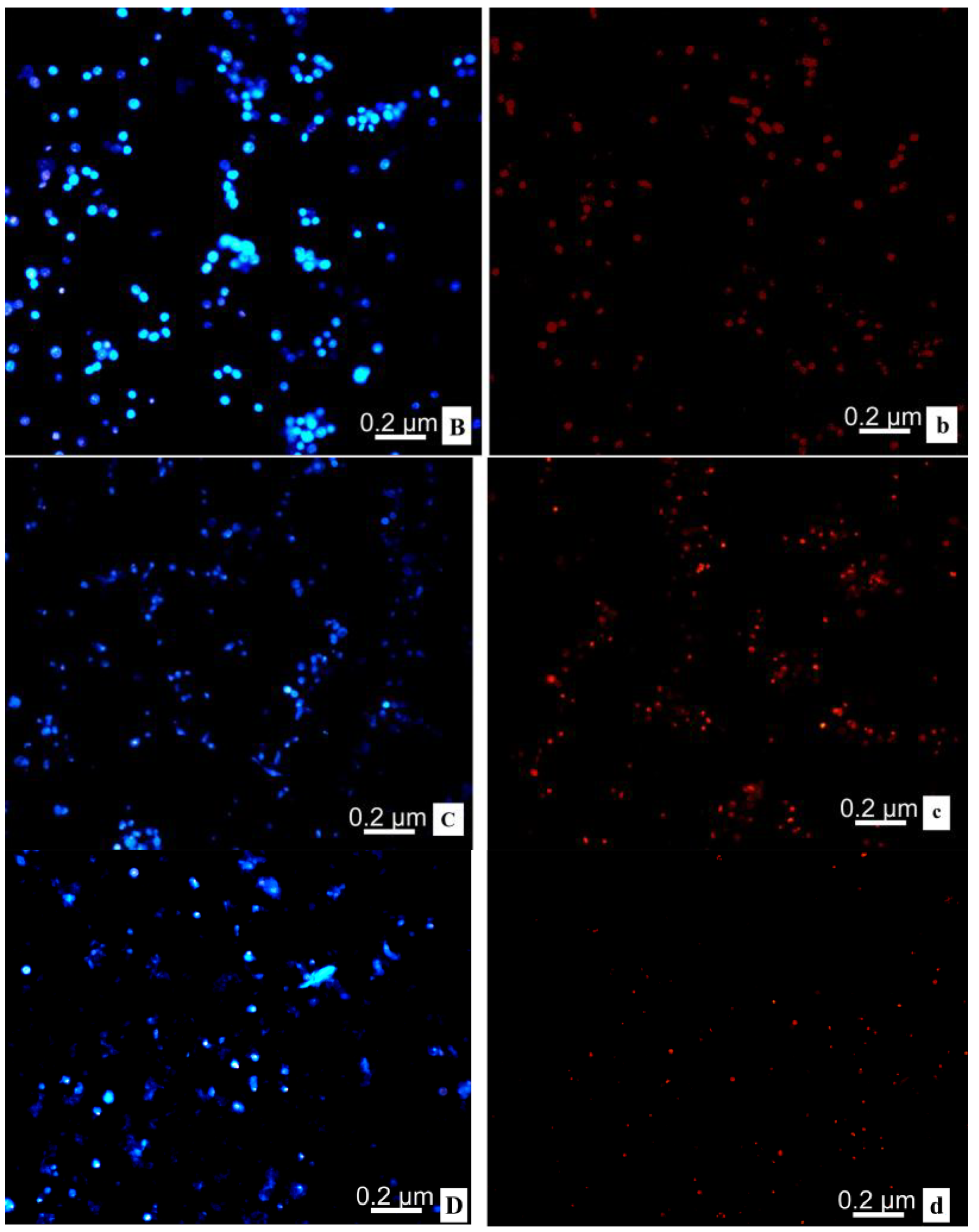
2.8. Transmission Electron Microscope; TEM


3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
- (1)
- SMMC-7721, a human hepatoma cell line,
- (2)
- MCF-7, a human breast carcinoma cell line,
- (3)
- BT-549, a human breast carcinoma cell line,
- (4)
- MDA-MB-231, a human breast carcinoma cell line,
- (5)
- SKOV-3, a human ovarian neoplasm cell line,
- (6)
- MCF-10, a human normal breast cell,
- (7)
- L-02, a human normal liver cell, and
- (8)
- HEK293, a human normal embryonic kidney cell.
4.2. DLM
4.3. Hemolysis Assay of DLM
4.4. Disintegrin Released from DLM by uPA in Vitro
4.5. Assay DLM Activity
4.6. Binding of Disintegrin and DLM to Inactive Platelets
4.7. MTT Assay
4.8. Flow Cytometry and Fluorescent Microscopy
4.9. DNA Ladder Assay
4.10. TEM
Supplementary Materials
Acknowledgments
Author Contributions
Abbreviations
| DLM | disintegrin-linker-melittin | TEM | transmission electron microscope |
| RGD | Arg-Gly-Asp | PRP | platelet rich plasma |
| PPP | platelet poor plasma | ADP | adenosine diphosphate |
| DMSO | dimethyl sulfoxide | MTT | methyl thiazolyl tetrazolium |
| PMA | phorbol ester | PGE1 | prostaglandin E1 |
| OD | optical density | FITC | fluorescein isothiocyanate |
| uPA | urokinase-type plasminogen activator | uPAR | Receptor of the urokinase-type plasminogen activator |
| BMMY | Basic medium with 2% peptone, 1% yeast extracts, 1.34% Yeast Nitrogen Base without Amino Acids, 1% methanol, 100 mM Potassium phosphate and 4 × 10−5 biotin | BMGY | Basic medium with 2% peptone, 1% yeast extracts, 1.34% Yeast Nitrogen Base without Amino Acids, 1% Glycerol, 100 mM Potassium phosphate and 4 × 10−5 biotin |
| RPMI | Roswell Park Memorial Institute |
Conflicts of Interest
References
- Skeel, R.T. Handbook of Cancer Chemotherapy, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2003. [Google Scholar]
- Takimoto, C.H.; Calvo, E. Principles of oncologic pharmacotherapy. In Cancer Management: A Multidisciplinary Approach, 11th ed.; Pazdur, R., Wagman, L.D., Camphausen, K.A., Hoskins, W.J., Eds.; CMP United Business Media: Manhasset, NY, USA, 2009. [Google Scholar]
- Moertel, C.G.; Fleming, T.R.; Macdonald, J.S.; Haller, D.G.; Laurie, J.A.; Goodman, P.J.; Ungerleider, J.S.; Emerson, W.A.; Tormey, D.C.; Glick, J.H.; et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N. Engl. J. Med. 1990, 322, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Sikora, K.; Advani, S.; Koroltchouk, V.; Magrath, I.; Levy, L.; Pinedo, H.; Schwartsmann, G.; Tattersall, M.; Yan, S. Essential drugs for cancer therapy: A world health organization consultation. Ann. Oncol. 1999, 10, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.; González-Bártulos, M.; Soriano-Castell, D.; Ribas, X.; Costas, M.; Tebar, F.; Massaguer, A.; Feliu, L.; Planas, M. Identification of BP16 as a non-toxic cell-penetrating peptide with highly efficient drug delivery properties. Org. Biomol. Chem. 2014, 12, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I.; Zuniga, E.S.; Berger, W.T.; Seitz, J.D. Tumor-targeting drug delivery of new-generation taxoids. Future Med. Chem. 2012, 4, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V. Targeted delivery of chemotherapeutics: Tumor-activated prodrug therapy. Adv. Drug Deliv. Rev. 1998, 31, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody-drug conjugates: Basic concepts, examples and future perspectives. J. Control. Release 2012, 161, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Newell, D.R. How to develop a successful cancer drug—Molecules to medicines or targets to treatments? Eur. J. Cancer 2005, 41, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Jaracz, S.; Chen, J.; Kuznetsova, L.V.; Ojima, I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg. Med. Chem. 2005, 13, 5043–5054. [Google Scholar] [CrossRef] [PubMed]
- Iyer, U.; Kadambi, V.J. Antibody drug conjugates—Trojan horses in the war on cancer. J. Pharmacol. Toxicol. Methods 2011, 64, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Miranti, C.K.; Brugge, J.S. Sensing the environment: A historical perspective on integrin signal transduction. Nat. Cell Biol. 2002, 4, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, F.; Heisenberg, C.P. Trafficking and cell migration. Traffic 2009, 10, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, R.; Alahari, S.K. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010, 29, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.R.; Peeper, D.S. Metastasis mechanisms. Biochim. Biophys. Acta 2009, 1796, 293–308. [Google Scholar] [PubMed]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Ramos, O.H.; Kauskot, A.; Cominetti, M.R.; Bechyne, I.; Salla Pontes, C.L.; Chareyre, F.; Manent, J.; Vassy, R.; Giovannini, M.; Legrand, C.; et al. A novel αvβ3-blocking disintegrin containing the RGD motive, DisBa-01, inhibits bFGF-induced angiogenesis and melanoma metastasis. Clin. Exp. Metastasis 2008, 25, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Swenson, S.; Ramu, S.; Markland, F.S. Anti-angiogenesis and RGD-containing snake venom disintegrins. Curr. Pharm. Des. 2007, 13, 2860–2871. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Brahmbhatt, K.; Bhatt, H.; Parmar, U. Therapeutic potential of snake venom in cancer therapy: Current perspectives. Asian Pac. J. Trop. Biomed. 2013, 3, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.S.; Tang, C.H.; Chuang, W.J.; Huang, T.H.; Peng, H.C.; Huang, T.F.; Fu, W.M. Inhibition of tumor formation by snake venom disintegrin. Toxicon 2005, 45, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Swenson, S.; Costa, F.; Ernst, W.; Fujii, G.; Markland, F.S. Contortrostatin, a snake venom disintegrin with anti-angiogenic and anti-tumor activity. Pathophysiol. Haemost. Thromb. 2005, 34, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Selistre-de-Araujo, H.S.; Pontes, C.L.S.; Montenegro, C.F.; Martin, A.C. Snake venom disintegrins and cell migration. Toxins 2010, 2, 2606–2621. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sherwin, R.P.; Parrish, C.; Richters, V.; Groshen, S.G.; Tsao-Wei, D.; Markland, F.S. Contortrostatin, a dimeric disintegrin from Agkistrodon contortrix contortrix, inhibits breast cancer progression. Breast Cancer Res. Treat. 2000, 61, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadoon, M.K.; Rabah, D.M.; Badr, G. Enhanced anticancer efficacy of snake venom combined with silica nanoparticles in a murine model of human multiple myeloma: Molecular targets for cell cycle arrest and apoptosis induction. Cell. Immunol. 2013, 284, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Tomoyoshi, T.; Fumimasa, N.; Yasunori, Y.; Tanaka-Takiguchi, Y.; Homma, M.; Takiguchi, K. Multiple membrane interactions and versatile vesicle deformations elicited by melittin. Toxins 2013, 5, 637–664. [Google Scholar] [CrossRef] [PubMed]
- Oren, Z.; Shai, Y. Selective lysis of bacteria but not mammalian cell, by diasteromers of melittin: Structure-function study. Biochemistry 1997, 36, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Gevod, V.S.; Birdi, K.S. Melittin and the 8–26 franent. Differences in ionophofic properties as measured by monolayer method. Biophys. J. 1984, 45, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Russell, P.J.; Hewish, D.; Carter, T.; Sterling-Levis, K.; Ow, K.; Hattarki, M.; Doughty, L.; Guthrie, R.; Shapira, D.; Molloy, P.L.; et al. Cytotoxic properties of immunoconjugates containing melittin-like peptide 101 against prostate cancer: In vitro and in vivo studies. Cancer Immunol. Immunther. 2004, 53, 411–421. [Google Scholar] [CrossRef]
- Reiter, Y.; Ciobotariu, A.; Jones, J.; Morgan, B.P.; Fishelson, Z. Complement membrane attack complex, perforin, and bacterial exotoxins induce in K562 cell calcium-dependent cross-protection from lysis. J. Immunol. 1995, 155, 2203–2210. [Google Scholar] [PubMed]
- Soman, N.R.; Baldwin, S.L.; Hu, G. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J. Clin. Investig. 2009, 9, 2830–2842. [Google Scholar] [CrossRef]
- Jo, M.; Park, M.H.; Kollipara, P.S.; An, B.J.; Song, H.S.; Han, S.B.; Kim, J.H.; Song, M.J.; Hong, J.T. Anti-cancer effect of bee venom toxin and melittin in ovarian cancer cells through induction of death receptors and inhibition of JAK2/STAT3 pathway. Toxicol. Appl. Pharmacol. 2012, 258, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.G.; Tayeh, I.; Israel, L.; Castagna, M. Different susceptibility of lung cell lines to inhibitors of tumor promotion and inducers of differentiation. J. Biol. Regul. Homeost. Agents 1991, 5, 52–58. [Google Scholar] [PubMed]
- Lee, M.T.; Hung, W.C.; Chen, F.Y.; Huang, H.W. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 5087–5092. [Google Scholar] [CrossRef] [PubMed]
- Perez-Paya, E.; Dufourcq, J.; Braco, L.; Abad, C. Structural characterisation of the natural membrane-bound state of melittin: A fluorescence study of a dansylated analogue. Biochemistry 1997, 1329, 223–236. [Google Scholar]
- Holle, L.; Song, W.; Holle, E.; Huang, H.W. A matrix metalloproteinase 2 cleavable melittin/avidin conjugate specifically targets tumor cells in vitro and in vivo. Int. J. Oncol. 2003, 22, 93–98. [Google Scholar] [PubMed]
- Ling, C.Q.; Li, B.; Zhang, C.; Zhu, D.Z.; Huang, X.Q.; Gu, W.; Li, S.X. Inhibitory effect of recombinant adenovirus carrying melittin gene on hepatocellular carcinoma. Ann. Oncol. 2005, 16, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Sun, M.; Wang, Y.; Sun, D.J.; Zhang, S. Expression and functional characterization of a recombinant targeted toxin with an uPA cleavable linker in Pichia pastoris. Protein Expr. Purif. 2011, 76, 184–189. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, D.; Sun, M.; Zhu, W.; Wang, Z.; Li, Y.; Ma, J. The Anti-Cancer Potency and Mechanism of a Novel Tumor-Activated Fused Toxin, DLM. Toxins 2015, 7, 423-438. https://doi.org/10.3390/toxins7020423
Sun D, Sun M, Zhu W, Wang Z, Li Y, Ma J. The Anti-Cancer Potency and Mechanism of a Novel Tumor-Activated Fused Toxin, DLM. Toxins. 2015; 7(2):423-438. https://doi.org/10.3390/toxins7020423
Chicago/Turabian StyleSun, Dejun, Miaonan Sun, Wenhe Zhu, Zhiding Wang, Yuefei Li, and Jie Ma. 2015. "The Anti-Cancer Potency and Mechanism of a Novel Tumor-Activated Fused Toxin, DLM" Toxins 7, no. 2: 423-438. https://doi.org/10.3390/toxins7020423
APA StyleSun, D., Sun, M., Zhu, W., Wang, Z., Li, Y., & Ma, J. (2015). The Anti-Cancer Potency and Mechanism of a Novel Tumor-Activated Fused Toxin, DLM. Toxins, 7(2), 423-438. https://doi.org/10.3390/toxins7020423