
Immunization of Mice with Anthrax Protective Antigen Limits Cardiotoxicity but Not Hepatotoxicity Following Lethal Toxin Challenge

Abstract
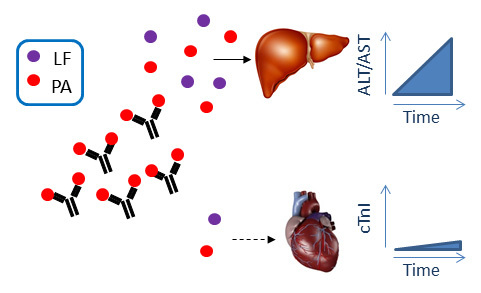
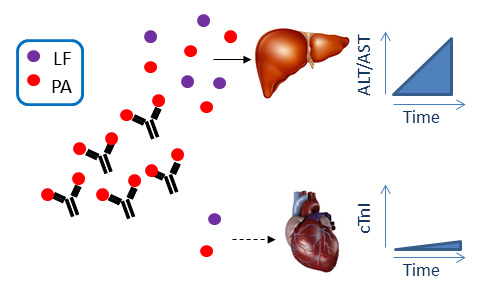
:
{kind=link}
{kind=link}
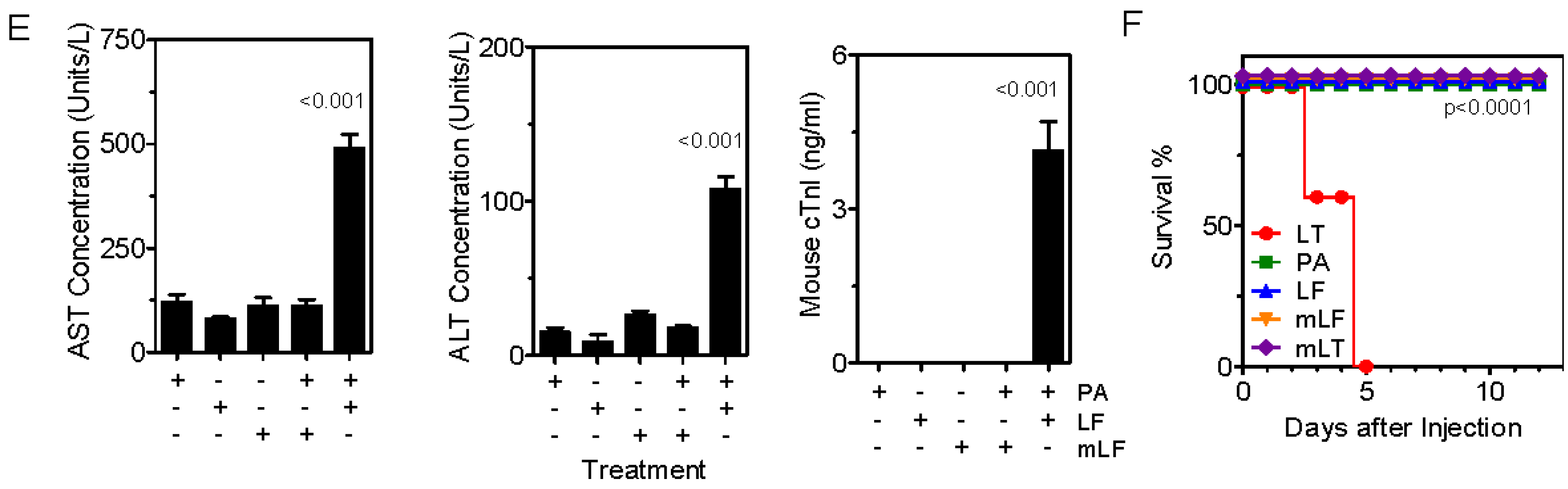{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
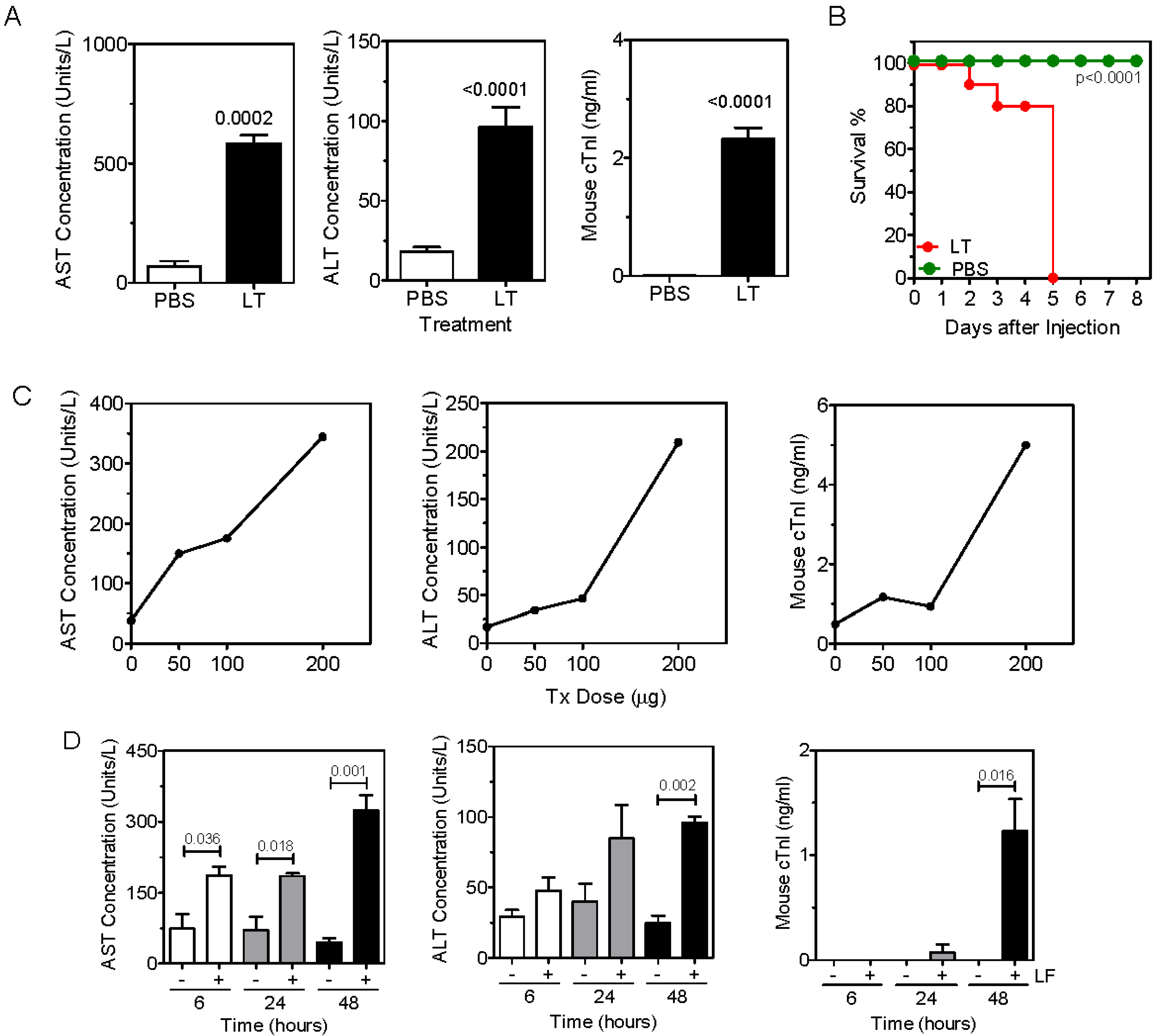
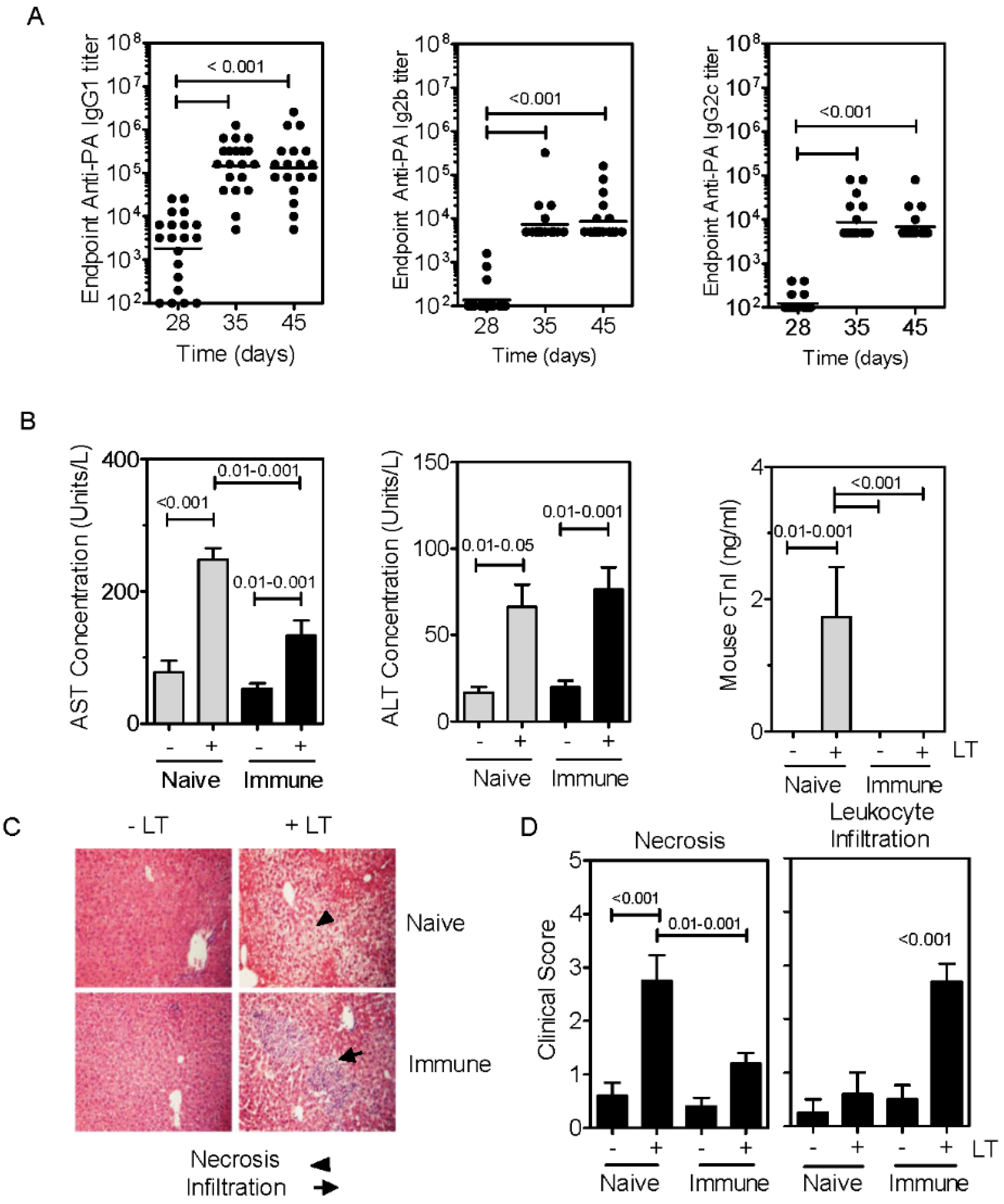
2.1. Anthrax Toxin Leads to Elevation of Serum Hepatic AST and ALT and Cardiac Troponin I in Naïve Mice


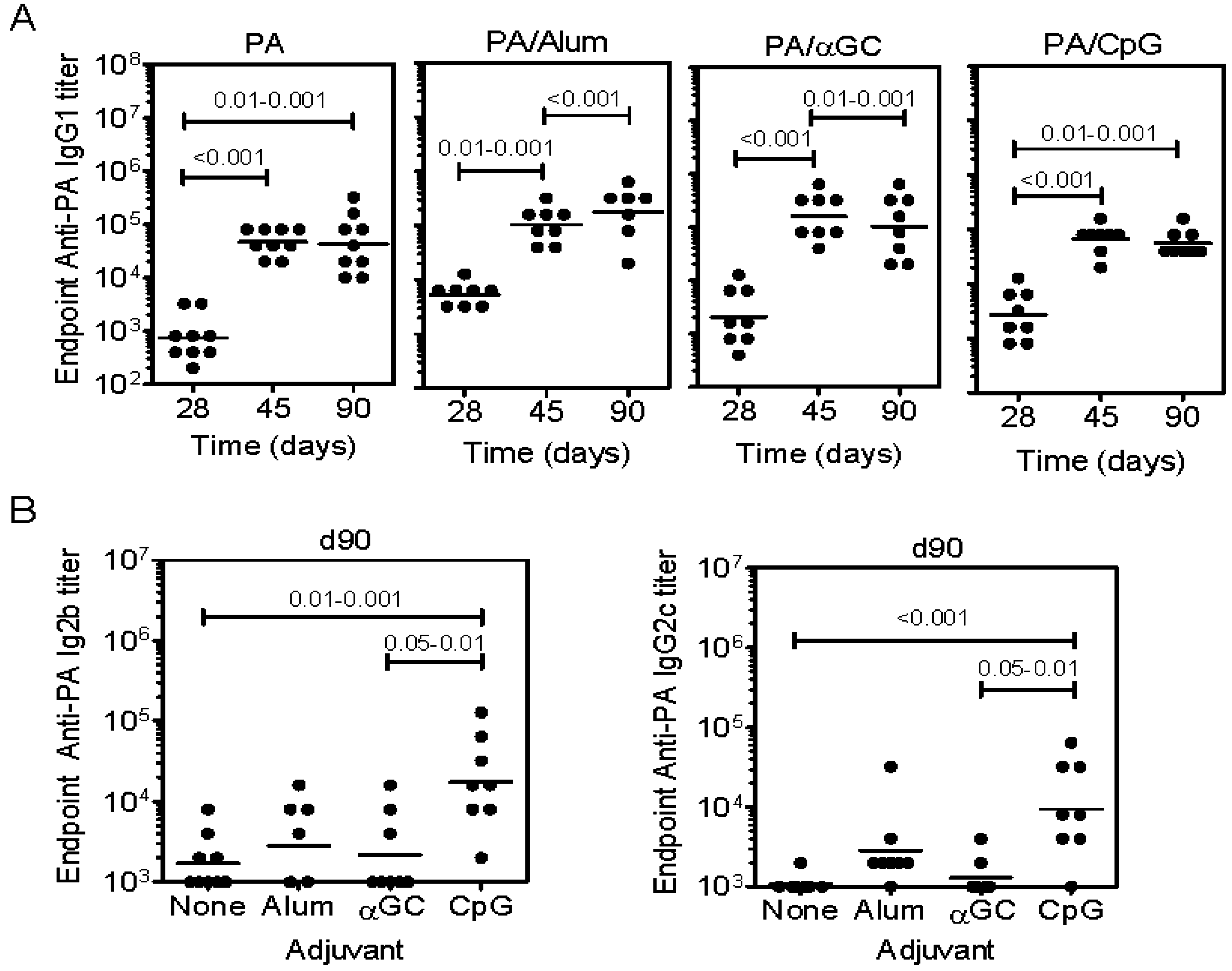
2.2. Immunization with PA Induces Ab and Inhibits Cardiotoxicity but not Hepatotoxicity

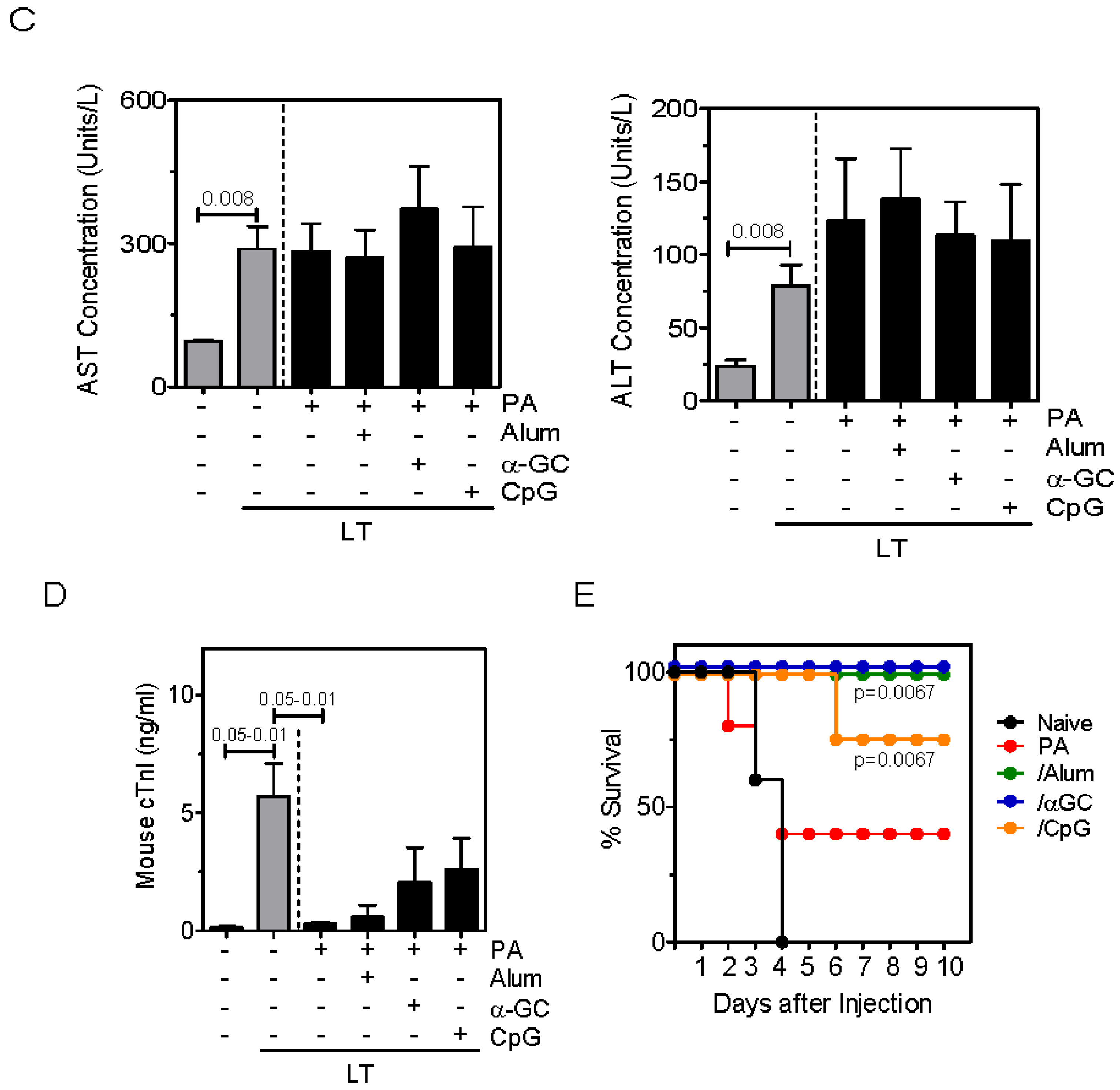
2.3. Inclusion of Adjuvants in the PA Vaccine Does not Affect LT-Induced Hepatotoxicity


3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Toxin Expression and Purification
4.3. Mice
4.4. Ethics Statement
4.5. Retro-Orbital Eye Bleed and Serum Collection
4.6. Immunizations and Experimental Schedule
4.7. In Vivo Toxin Challenge
4.8. ELISA for Serum Ig
4.9. AST, ALT, and cTn1 Measurement
4.10. Histological Analysis of Liver
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J.; Young, J.A. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 2003, 19, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Bergsbaken, T.; Cookson, B.T. Anthrax lethal toxin and salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 4312–4317. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H. Bacillus anthracis calmodulin-dependent adenylate cyclase: Chemical and enzymatic properties and interactions with eucaryotic cells. Adv. Cyclic Nucleotide Protein Phosphorylation Res. 1984, 17, 189–198. [Google Scholar] [PubMed]
- Agrawal, A.; Lingappa, J.; Leppla, S.H.; Agrawal, S.; Jabbar, A.; Quinn, C.; Pulendran, B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 2003, 424, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Xu, L.; Chen, T.Y.; Cyr, J.M.; Frucht, D.M. Anthrax lethal toxin has direct and potent inhibitory effects on b cell proliferation and immunoglobulin production. J. Immunol. 2006, 176, 6155–6161. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.K.; Lang, G.A.; Larabee, J.L.; Devera, T.S.; Aye, L.M.; Shah, H.B.; Ballard, J.D.; Lang, M.L. Bacillus anthracis lethal toxin disrupts tcr signaling in cd1d-restricted nkt cells leading to functional anergy. PLoS Pathog. 2009, 5, e1000588. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D'Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Mock, M.; Mignot, T. Anthrax toxins and the host: A story of intimacy. Cell. Microbiol. 2003, 5, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Moayeri, M.; Liu, J.; Crown, D.; Fattah, R.J.; Wein, A.N.; Yu, Z.X.; Finkel, T.; Leppla, S.H. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013, 501, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Abboud, N.; Casadevall, A. Immunogenicity of Bacillus anthracis protective antigen domains and efficacy of elicited antibody responses depend on host genetic background. Clin. Vaccine Immunol. 2008, 15, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Boyaka, P.N.; Tafaro, A.; Fischer, R.; Leppla, S.H.; Fujihashi, K.; McGhee, J.R. Effective mucosal immunity to anthrax: Neutralizing antibodies and th cell responses following nasal immunization with protective antigen. J. Immunol. 2003, 170, 5636–5643. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Lee, J.H.; Hung, C.F.; Wu, T.C.; Kim, T.W. Enhancement of antibody responses to Bacillus anthracis protective antigen domain iv by use of calreticulin as a chimeric molecular adjuvant. Infect. Immun. 2008, 76, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Peachman, K.K.; Rao, M.; Alving, C.R.; Burge, R.; Leppla, S.H.; Rao, V.B.; Matyas, G.R. Correlation between lethal toxin-neutralizing antibody titers and protection from intranasal challenge with bacillus anthracis ames strain spores in mice after transcutaneous immunization with recombinant anthrax protective antigen. Infect. Immun. 2006, 74, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Nakouzi, A.; Abboud, N.; Revskaya, E.; Goldman, D.; Collier, R.J.; Dadachova, E.; Casadevall, A. A monoclonal antibody to bacillus anthracis protective antigen defines a neutralizing epitope in domain 1. Infect. Immun. 2006, 74, 4149–4156. [Google Scholar] [CrossRef] [PubMed]
- Welkos, S.; Little, S.; Friedlander, A.; Fritz, D.; Fellows, P. The role of antibodies to Bacillus anthracis and anthrax toxin components in inhibiting the early stages of infection by anthrax spores. Microbiology 2001, 147, 1677–1685. [Google Scholar] [PubMed]
- Quinn, C.P.; Dull, P.M.; Semenova, V.; Li, H.; Crotty, S.; Taylor, T.H.; Steward-Clark, E.; Stamey, K.L.; Schmidt, D.S.; Stinson, K.W.; et al. Immune responses to Bacillus anthracis protective antigen in patients with bioterrorism-related cutaneous or inhalation anthrax. J. Infect. Dis. 2004, 190, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Pittman, P.R.; Leitman, S.F.; Oro, J.G.; Norris, S.L.; Marano, N.M.; Ranadive, M.V.; Sink, B.S.; McKee, K.T., Jr. Protective antigen and toxin neutralization antibody patterns in anthrax vaccinees undergoing serial plasmapheresis. Clin. Diagn. Lab. Immunol. 2005, 12, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Ribot, W.J.; Powell, B.S.; Ivins, B.E.; Little, S.F.; Johnson, W.M.; Hoover, T.A.; Norris, S.L.; Adamovicz, J.J.; Friedlander, A.M.; Andrews, G.P. Comparative vaccine efficacy of different isoforms of recombinant protective antigen against Bacillus anthracis spore challenge in rabbits. Vaccine 2006, 24, 3469–3476. [Google Scholar] [CrossRef] [PubMed]
- Devera, T.S.; Aye, L.M.; Lang, G.A.; Joshi, S.K.; Ballard, J.D.; Lang, M.L. Cd1d-dependent B-cell help by NK-like T cells leads to enhanced and sustained production of bacillus anthracis lethal toxin-neutralizing antibodies. Infect. Immun. 2010, 78, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Devera, T.S.; Joshi, S.K.; Aye, L.M.; Lang, G.A.; Ballard, J.D.; Lang, M.L. Regulation of anthrax toxin-specific antibody titers by natural killer T cell-derived IL-4 and IFN-gamma. PLoS ONE 2011, 6, e23817. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Xu, Q.; Hesek, E.D.; Pichichero, M.E. N-fragment of edema factor as a candidate antigen for immunization against anthrax. Vaccine 2006, 24, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Pelat, T.; Hust, M.; Laffly, E.; Condemine, F.; Bottex, C.; Vidal, D.; Lefranc, M.P.; Dubel, S.; Thullier, P. High-affinity, human antibody-like antibody fragment (single-chain variable fragment) neutralizing the lethal factor (LF) of Bacillus anthracis by inhibiting protective antigen-LF complex formation. Antimicrob. Agents Chemother. 2007, 51, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Little, S.F.; Leppla, S.H.; Friedlander, A.M. Production and characterization of monoclonal antibodies against the lethal factor component of Bacillus anthracis lethal toxin. Infect. Immun. 1990, 58, 1606–1613. [Google Scholar] [PubMed]
- Abboud, N.; Chow, S.K.; Saylor, C.; Janda, A.; Ravetch, J.V.; Scharff, M.D.; Casadevall, A. A requirement for fcgammar in antibody-mediated bacterial toxin neutralization. J. Exp. Med. 2010, 207, 2395–2405. [Google Scholar] [CrossRef] [PubMed]
- Barochia, A.V.; Cui, X.Z.; Sun, J.F.; Li, Y.; Solomon, S.B.; Migone, T.S.; Subramanian, G.M.; Bolmer, S.D.; Eichacker, P.Q. Protective antigen antibody augments hemodynamic support in anthrax lethal toxin shock in canines. J. Infect. Dis. 2012, 205, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Boyer, J.L.; Hackett, N.R.; Wilson, J.M.; Crystal, R.G. Induction of protective immunity to anthrax lethal toxin with a nonhuman primate adenovirus-based vaccine in the presence of preexisting anti-human adenovirus immunity. Infect. Immun. 2005, 73, 6885–6891. [Google Scholar] [CrossRef] [PubMed]
- Aulinger, B.A.; Roehrl, M.H.; Mekalanos, J.J.; Collier, R.J.; Wang, J.Y. Combining anthrax vaccine and therapy: A dominant-negative inhibitor of anthrax toxin is also a potent and safe immunogen for vaccines. Infect. Immun. 2005, 73, 3408–3414. [Google Scholar] [CrossRef] [PubMed]
- Dumas, E.K.; Cox, P.M.; Fullenwider, C.O.; Nguyen, M.; Centola, M.; Frank, M.B.; Dozmorov, I.; James, J.A.; Farris, A.D. Anthrax lethal toxin-induced gene expression changes in mouse lung. Toxins 2011, 3, 1111–1130. [Google Scholar] [CrossRef] [PubMed]
- Mabry, R.; Brasky, K.; Geiger, R.; Carrion, R., Jr.; Hubbard, G.B.; Leppla, S.; Patterson, J.L.; Georgiou, G.; Iverson, B.L. Detection of anthrax toxin in the serum of animals infected with Bacillus anthracis by using engineered immunoassays. Clin. Vaccine Immunol. 2006, 13, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Moayeri, M.; Chen, Z.; Harma, H.; Zhao, J.; Hu, H.; Purcell, R.H.; Leppla, S.H.; Hewlett, I.K. Detection of anthrax toxin by an ultrasensitive immunoassay using europium nanoparticles. Clin. Vaccine Immunol. 2009, 16, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, N.; Howard, J.C.; Williams, B.D. Demonstration of hepatic fc receptor function in vivo. Clin. Exp. Immunol. 1989, 78, 278–284. [Google Scholar] [PubMed]
- Skogh, T.; Blomhoff, R.; Eskild, W.; Berg, T. Hepatic uptake of circulating IgG immune complexes. Immunology 1985, 55, 585–594. [Google Scholar] [PubMed]
- Salles, II; Voth, D.E.; Ward, S.C.; Averette, K.M.; Tweten, R.K.; Bradley, K.A.; Ballard, J.D. Cytotoxic activity of Bacillus anthracis protective antigen observed in a macrophage cell line overexpressing antxr1. Cell. Microbiol. 2006, 8, 1272–1281. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devera, T.S.; Prusator, D.K.; Joshi, S.K.; Ballard, J.D.; Lang, M.L. Immunization of Mice with Anthrax Protective Antigen Limits Cardiotoxicity but Not Hepatotoxicity Following Lethal Toxin Challenge. Toxins 2015, 7, 2371-2384. https://doi.org/10.3390/toxins7072371
Devera TS, Prusator DK, Joshi SK, Ballard JD, Lang ML. Immunization of Mice with Anthrax Protective Antigen Limits Cardiotoxicity but Not Hepatotoxicity Following Lethal Toxin Challenge. Toxins. 2015; 7(7):2371-2384. https://doi.org/10.3390/toxins7072371
Chicago/Turabian StyleDevera, T. Scott, Dawn K. Prusator, Sunil K. Joshi, Jimmy D. Ballard, and Mark L. Lang. 2015. "Immunization of Mice with Anthrax Protective Antigen Limits Cardiotoxicity but Not Hepatotoxicity Following Lethal Toxin Challenge" Toxins 7, no. 7: 2371-2384. https://doi.org/10.3390/toxins7072371