
Determination of Ochratoxin A in Wheat and Maize by Solid Bar Microextraction with Liquid Chromatography and Fluorescence Detection



Abstract
:1. Introduction
2. Results and Discussion
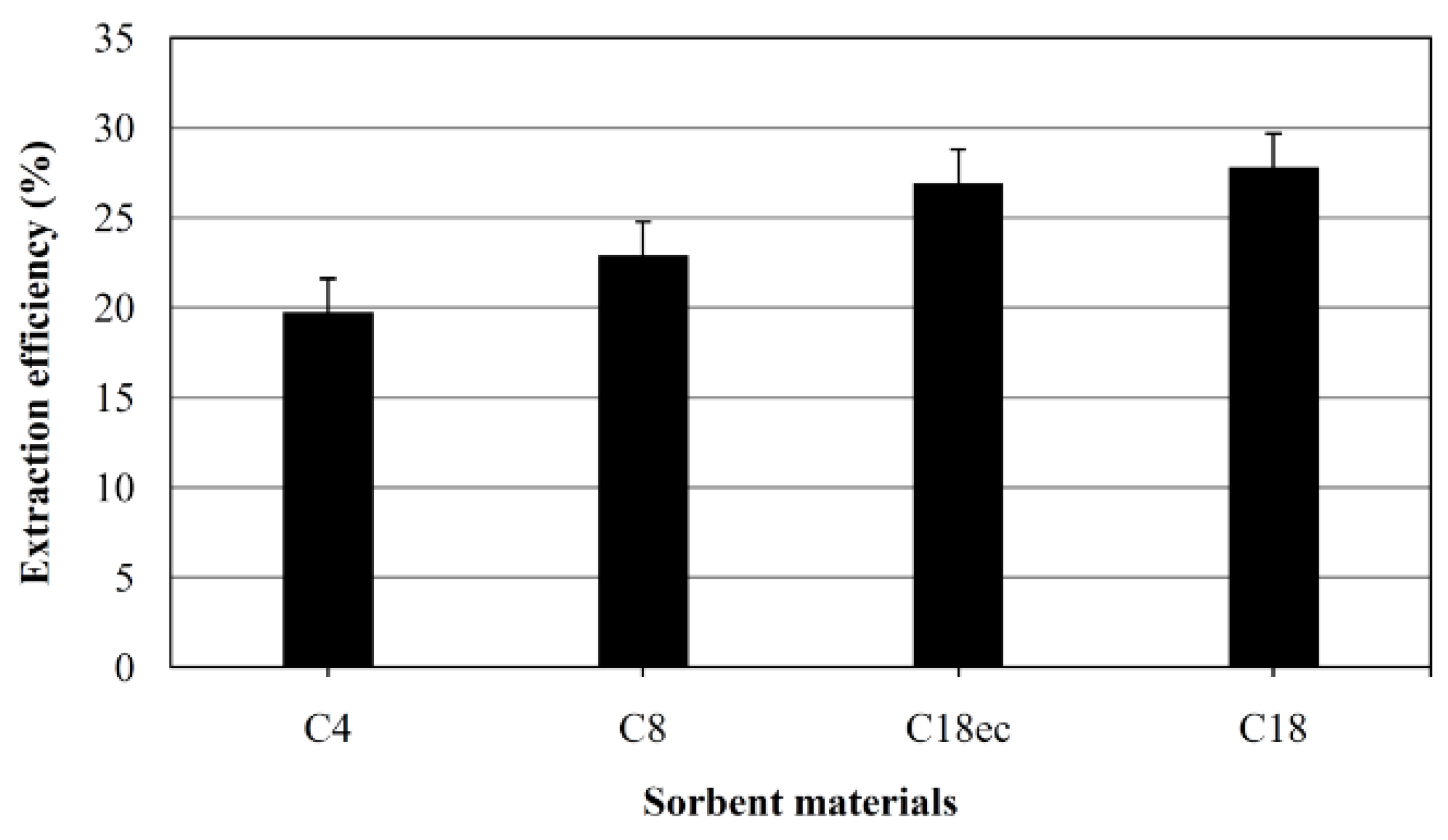
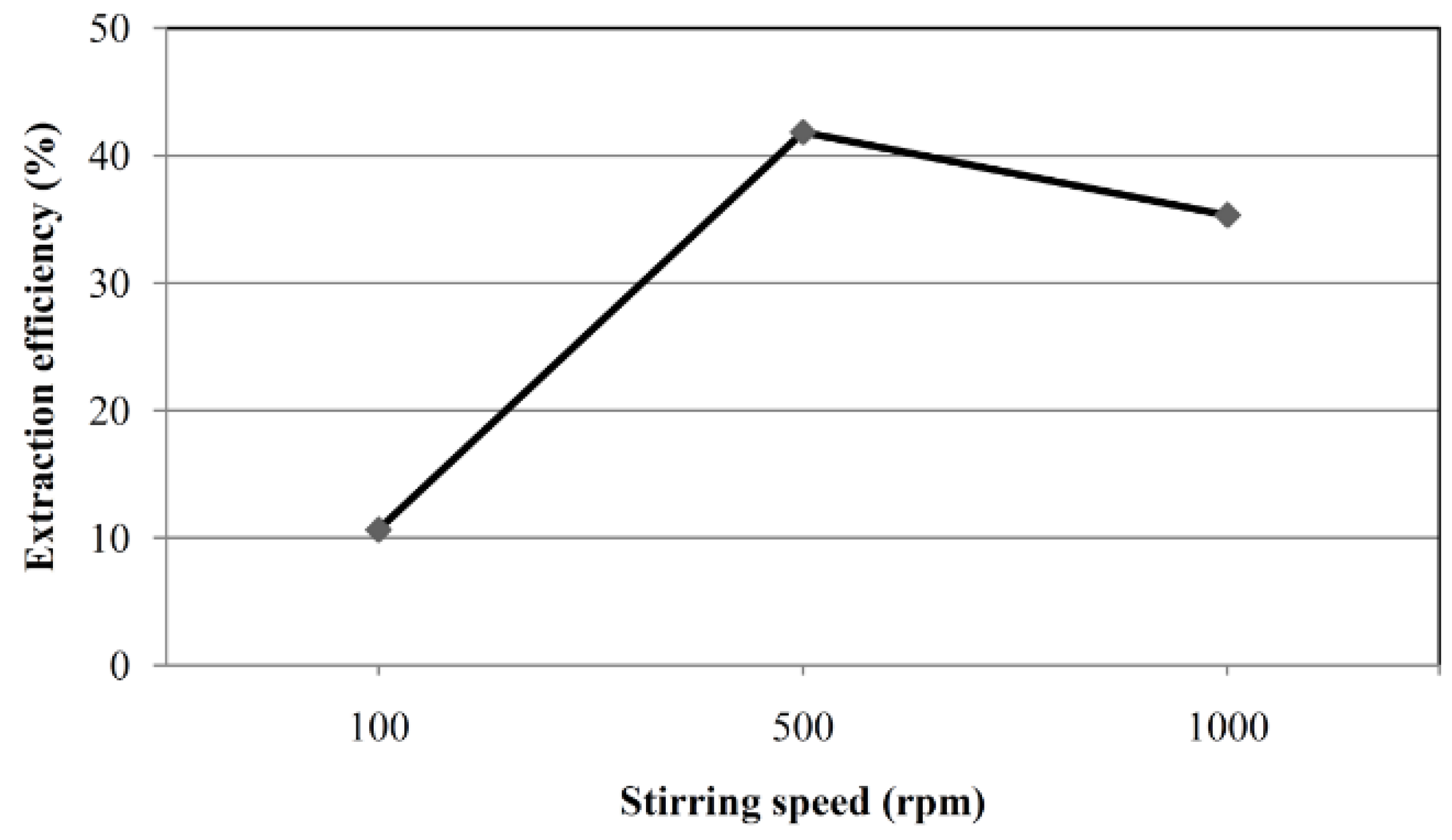
2.1. Optimization of SBME Conditions





2.2. Analysis of Maize and Wheat Samples


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Performance parameter | Wheat | Maize |
|---|---|---|
| LOD | 2.5 μg·kg−1 | 0.9 μg·kg−1 |
| LOQ | 8.7 μg·kg−1 | 3.4 μg·kg−1 |
| Correlation coefficient ( r) * | 0.995 | 0.998 |
| Repeatability as RSD ( n = 5) | 5.0% | 3.4% |
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Sample Preparation
3.3. Solid Bar Microextraction Procedure
3.4 Chromatography
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Davolos, D.; Pietrangeli, B.A. Molecular and bioinformatic study on the ochratoxin A (OTA)-producing Aspergillus affinis (section Circumdati). Mycotoxin Res. 2014, 30, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, K. Occurrence of ochratoxin A in commodities and processed food—A review of EU occurrence data. Food Addit. Contam. 2005, 22 (Suppl. 1), 26–30. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.C.; Pena, A.; Lino, C.M. A review on ochratoxin A occurrence and effects of processing of cereal and cereal derived food products. Food Microbiol. 2010, 27, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Streit, E.; Schatzmayr, G.; Tassis, P.; Tzika, E.; Marin, D.; Taranu, I.; Tabuc, C.; Nicolau, A.; Aprodu, I.; Puel, O.; et al. Current situation of mycotoxin contamination and co-occurrence in animal feed—Focus on Europe. Toxins 2012, 4, 788–809. [Google Scholar] [CrossRef] [PubMed]
- Barberis, C.L.; Pena, G.; Carranza, C.; Magnoli, C.E. Effect of indigenous mycobiota on ochratoxin A production by Aspergillus carbonarius isolated from soil: Ochratoxin in mixed cultures. Mycotoxin Res. 2014, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Dietrich, D.R. Ochratoxin A: The continuing enigma. Crit. Rev. Toxicol. 2005, 35, 33–60. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, V.; Di Giacomo, C.; Acquaviva, R.; Barbagallo, I.; Bognanno, M.; Galvano, F. Toxicity of ochratoxin A and its modulation by antioxidants: A review. Toxins 2013, 5, 1742–1766. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-Y.; Balch, C.G.; Cliff, R.L.; Sharma, B.S.; Boermans, H.J.; Swamy, H.V.L.N.; Quinton, V.M.; Karrow, N.A. Exposure to Penicillium mycotoxins alters gene expression of enzymes involved in the epigenetic regulation of bovine macrophages (BoMacs). Mycotoxin Res. 2013, 29, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Berrada, H.; Soriano, J.M.; Moltó, J.C.; Mañes, J. Rapid determination of ochratoxin A in cereals and cereal products by liquid chromatography. J. Chromatogr. A 2004, 1046, 127–131. [Google Scholar] [PubMed]
- Dall’asta, C.; Galaverna, G.; De Dea Lindner, J.; Virgili, R.; Neviani, E.; Dossena, A.A. New validated HPLC-FLD method for detecting ochratoxin A in dry-cured meat and in blue cheese. Mycotoxin Res. 2007, 23, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Lindenmeier, M.; Schieberle, P.; Rychlik, M. Determination of ochratoxin A in food: comparison of a stable isotope dilution assay, liquid chromatography-fluorescence detection and an enzyme-linked immunosorbent assay. Mycotoxin Res. 2011, 27, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Medina, A.; Valle-Algarra, F.M.; Gimeno-Adelantado, J.V.; Mateo, R.; Mateo, F.; Jiménez, M. New method for determination of ochratoxin A in beer using zinc acetate and solid-phase extraction silica cartridges. J. Chromatogr. A 2006, 1121, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.V.; Cichna-Markl, M.; Chung, D.H.; Shim, W.B.; Zentek, J.; Razzazi-Fazeli, E. Determination of ochratoxin A in grains by immuno-ultrafiltration and HPLC-fluorescence detection after postcolumn derivatisation in an electrochemical cell. Anal. Bioanal. Chem. 2011, 400, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Meulenberg, E.P. Immunochemical methods for ochratoxin A detection: A review. Toxins 2012, 4, 244–266. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Anaya, I.; Broto-Puig, F.; Agut, M.; Comellas, L. Two-dimensional thin-layer chromatographic method for the analysis of ochratoxin A in green coffee. J. Food Prot. 2005, 68, 1920–1922. [Google Scholar] [PubMed]
- Schenzel, J.; Forrer, H.R.; Vogelgsang, S.; Bucheli, T.D. Development, validation and application of a multi-mycotoxin method for the analysis of whole wheat plants. Mycotoxin Res. 2012, 28, 135–147. [Google Scholar] [PubMed]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Manzanares, N.; García-Campaña, A.M.; Gámiz-Gracia, L. Comparison of different sample treatments for the analysis of ochratoxin A in wine by capillary HPLC with laser-induced fluorescence detection. Anal. Bioanal. Chem. 2011, 401, 2987–2994. [Google Scholar] [CrossRef] [PubMed]
- Monaci, L.; Palmisano, F. Determination of ochratoxin A in foods: State-of-the-art and analytical challenges. Anal. Bioanal. Chem. 2004, 378, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, A.M.; Scherpenisse, P.; Bergwerff, A.A. Ochratoxin a contents in wine: comparison of organically and conventionally produced products. J. Agric. Food Chem. 2006, 54, 7399–7404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.N.; Lai, E.P.C.; Miller, J.D. Analysis of wheat extracts for ochratoxin A by molecularly imprinted solid-phase extraction and pulsed elution. Anal. Bioanal. Chem. 2004, 378, 1903–1906. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, A.-M.; Scott, P.M.; Tague, B. Analysis of cocoa products for ochratoxin A and aflatoxins. Mycotoxin Res. 2013, 29, 193–201. [Google Scholar] [CrossRef] [PubMed]
- García-Fonseca, S.; Ballesteros-Gómez, A.; Rubio, S.; Pérez-Bendito, D. Coacervative extraction of Ochratoxin A in wines prior to liquid chromatography/fluorescence determination. Anal. Chim. Acta 2008, 617, 3–10. [Google Scholar] [CrossRef] [PubMed]
- González-Peñas, E.; Leache, C.; Viscarret, M.; Pérez de Obanos, A.; Araguás, C.; López de Cerain, A. Determination of ochratoxin A in wine using liquid-phase microextraction combined with liquid chromatography with fluorescence detection. J. Chromatogr. A. 2004, 1025, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lee, H.K. Solvent bar microextraction. Anal. Chem. 2004, 76, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Aresta, A.; Palmisano, F.; Vatinno, R.; Zambonin, C.G. Ochratoxin A determination in beer by solid-phase microextraction coupled to liquid chromatography with fluorescence detection: A fast and sensitive method for assessment of noncompliance to legal limits. J. Agric. Food Chem. 2006, 54, 1594–1598. [Google Scholar] [CrossRef] [PubMed]
- Vatinno, R.; Aresta, A.; Zambonin, C.G.; Palmisano, F. Determination of ochratoxin A in green coffee beans by solid-phase microextraction and liquid chromatography with fluorescence detection. J. Chromatogr. A. 2008, 1187, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Aresta, A.; Cioffi, N.; Palmisano, F.; Zambonin, C.G. Simultaneous determination of ochratoxin A and cyclopiazonic, mycophenolic, and tenuazonic acids in cornflakes by solid-phase microextraction coupled to high-performance liquid chromatography. J. Agric. Food Chem. 2003, 51, 5232–5237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cudjoe, E.; Vuckovic, D.; Pawliszyn, J. Direct monitoring of ochratoxin A in cheese with solid-phase microextraction coupled to liquid chromatography-tandem mass spectrometry. J. Chromatogr. A. 2009, 1216, 7505–7509. [Google Scholar] [CrossRef] [PubMed]
- Zambonin, C.G. Coupling solid-phase microextraction to liquid chromatography. A review. Anal. Bioanal. Chem. 2003, 375, 73–80. [Google Scholar] [PubMed]
- Basheer, C.; Chong, H.G.; Hii, T.M.; Lee, H.K. Application of porous membrane-protected micro-solid-phase extraction combined with HPLC for the analysis of acidic drugs in wastewater. Anal. Chem. 2007, 79, 6845–6850. [Google Scholar]
- AL-Hadithi, N.; Saad, B.; Grote, M. A solid bar microextraction method for the liquid chromatographic determination of trace diclofenac, ibuprofen and carbamazepine in river water. Microchim. Acta 2011, 172, 31–37. [Google Scholar] [CrossRef]
- Lee, T.P.; Saad, B.; Khayoon, W.S.; Salleh, B. Molecularly imprinted polymer as sorbent in micro-solid phase extraction of ochratoxin A in coffee, grape juice and urine. Talanta 2012, 88, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Rhouati, A.; Yang, C.; Hayat, A.; Marty, J.-L. Aptamers: A promising tool for ochratoxin A detection in food analysis. Toxins 2013, 5, 1988–2008. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, J.; Wang, X.; Duan, Y. Amplified fluorescent aptasensor through catalytic recycling for highly sensitive detection of ochratoxin A. Biosens. Bioelectron. 2014, 65, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Sanzani, S.M.; Reverberi, M.; Fanelli, C.; Ippolito, A. Detection of ochratoxin a using molecular beacons and real-time PCR thermal cycler. Toxins 2015, 7, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Tittlemier, S.A.; Roscoe, M.; Drul, D.; Blagden, R.; Kobialka, C.; Chan, J.; Gaba, D. Single laboratory evaluation of a planar waveguide-based system for a simple simultaneous analysis of four mycotoxins in wheat. Mycotoxin Res. 2013, 29, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.T.; Danzer, K.; Townshend, A. Use of the term “recovery” and “apparent recovery” in analytical procedures. (IUPAC Recommendations 2002). Pure Appl. Chem. 2002, 74, 2201–2205. [Google Scholar] [CrossRef]
- Kaus, R. Detection limits and quantitation limits in the view of international harmonization and the consequences for analytical laboratories. Accred. Qual. Assur. 1998, 3, 150–154. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Hadithi, N.; Kössler, P.; Karlovsky, P. Determination of Ochratoxin A in Wheat and Maize by Solid Bar Microextraction with Liquid Chromatography and Fluorescence Detection. Toxins 2015, 7, 3000-3011. https://doi.org/10.3390/toxins7083000
Al-Hadithi N, Kössler P, Karlovsky P. Determination of Ochratoxin A in Wheat and Maize by Solid Bar Microextraction with Liquid Chromatography and Fluorescence Detection. Toxins. 2015; 7(8):3000-3011. https://doi.org/10.3390/toxins7083000
Chicago/Turabian StyleAl-Hadithi, Nabil, Philip Kössler, and Petr Karlovsky. 2015. "Determination of Ochratoxin A in Wheat and Maize by Solid Bar Microextraction with Liquid Chromatography and Fluorescence Detection" Toxins 7, no. 8: 3000-3011. https://doi.org/10.3390/toxins7083000
APA StyleAl-Hadithi, N., Kössler, P., & Karlovsky, P. (2015). Determination of Ochratoxin A in Wheat and Maize by Solid Bar Microextraction with Liquid Chromatography and Fluorescence Detection. Toxins, 7(8), 3000-3011. https://doi.org/10.3390/toxins7083000