
Sensitive, Rapid, Quantitative and in Vitro Method for the Detection of Biologically Active Staphylococcal Enterotoxin Type E


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
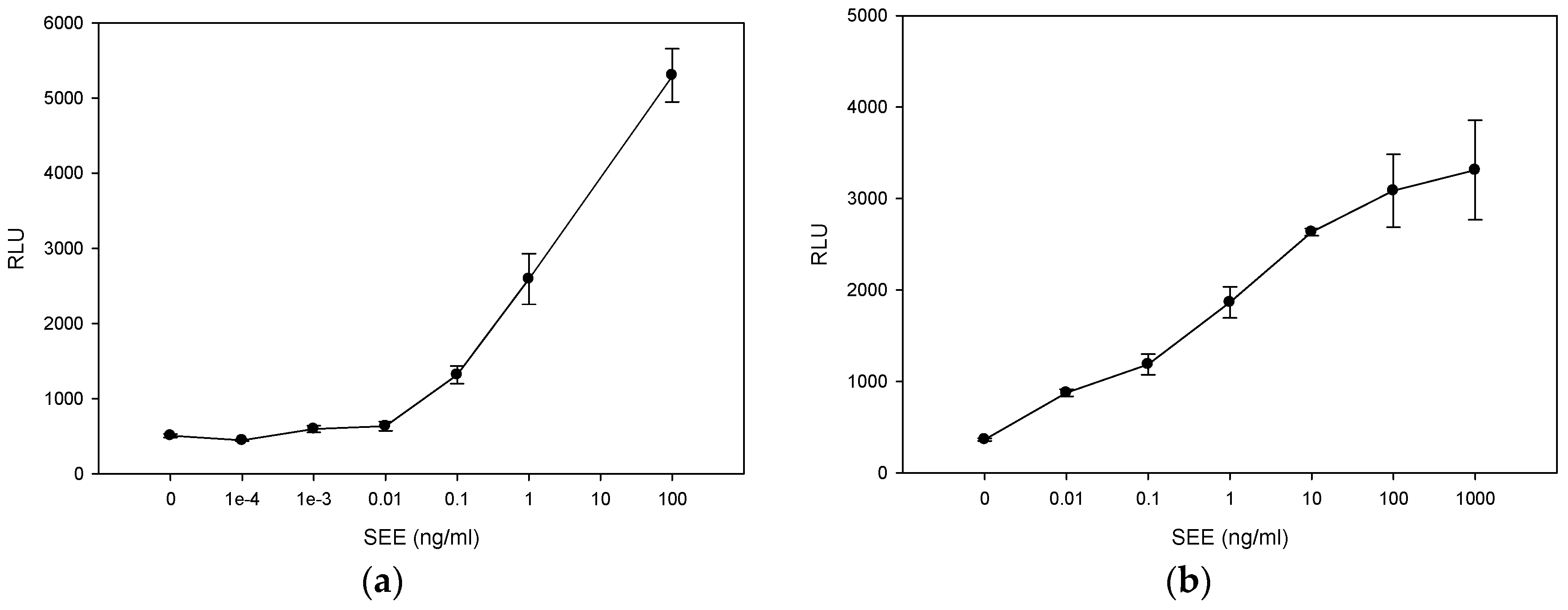{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Quantitative Splenocyte Proliferation Bioassay for Measuring Biologically Active SEE
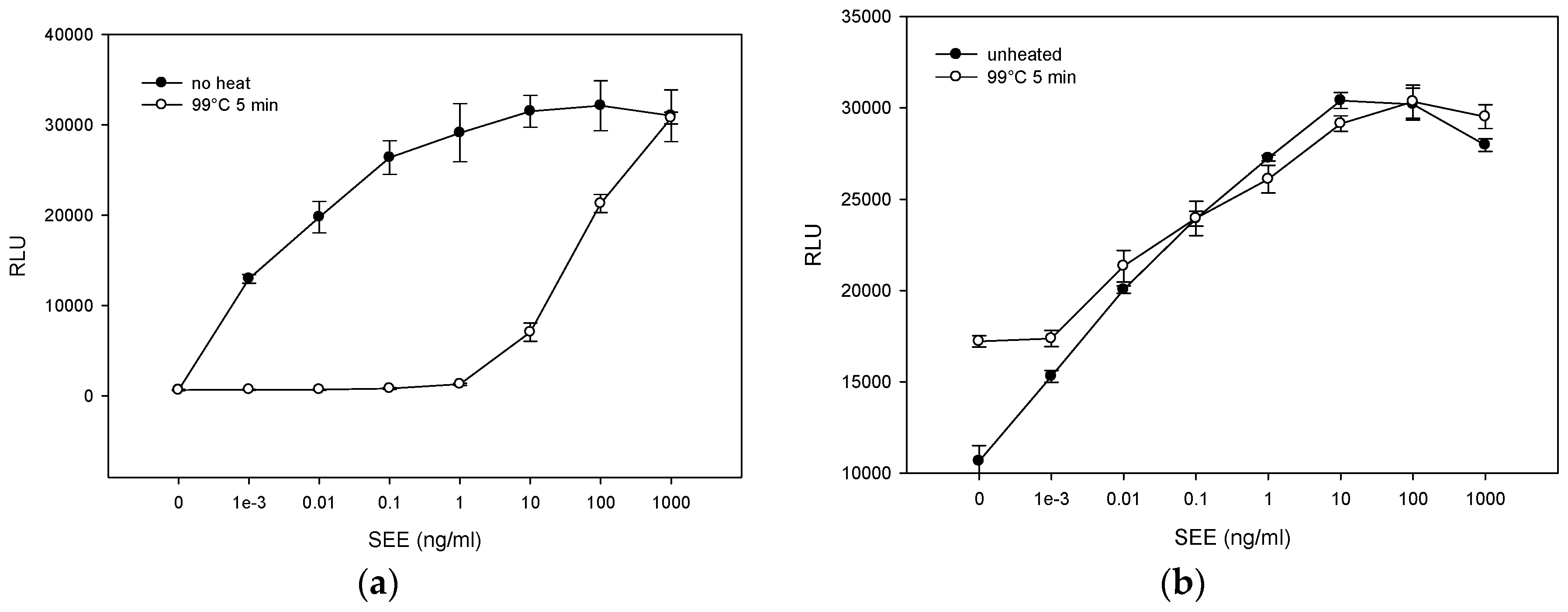
2.2. Quantitative Bioluminescence—Fast in Vitro Assay for Measuring Biologically Active SEE
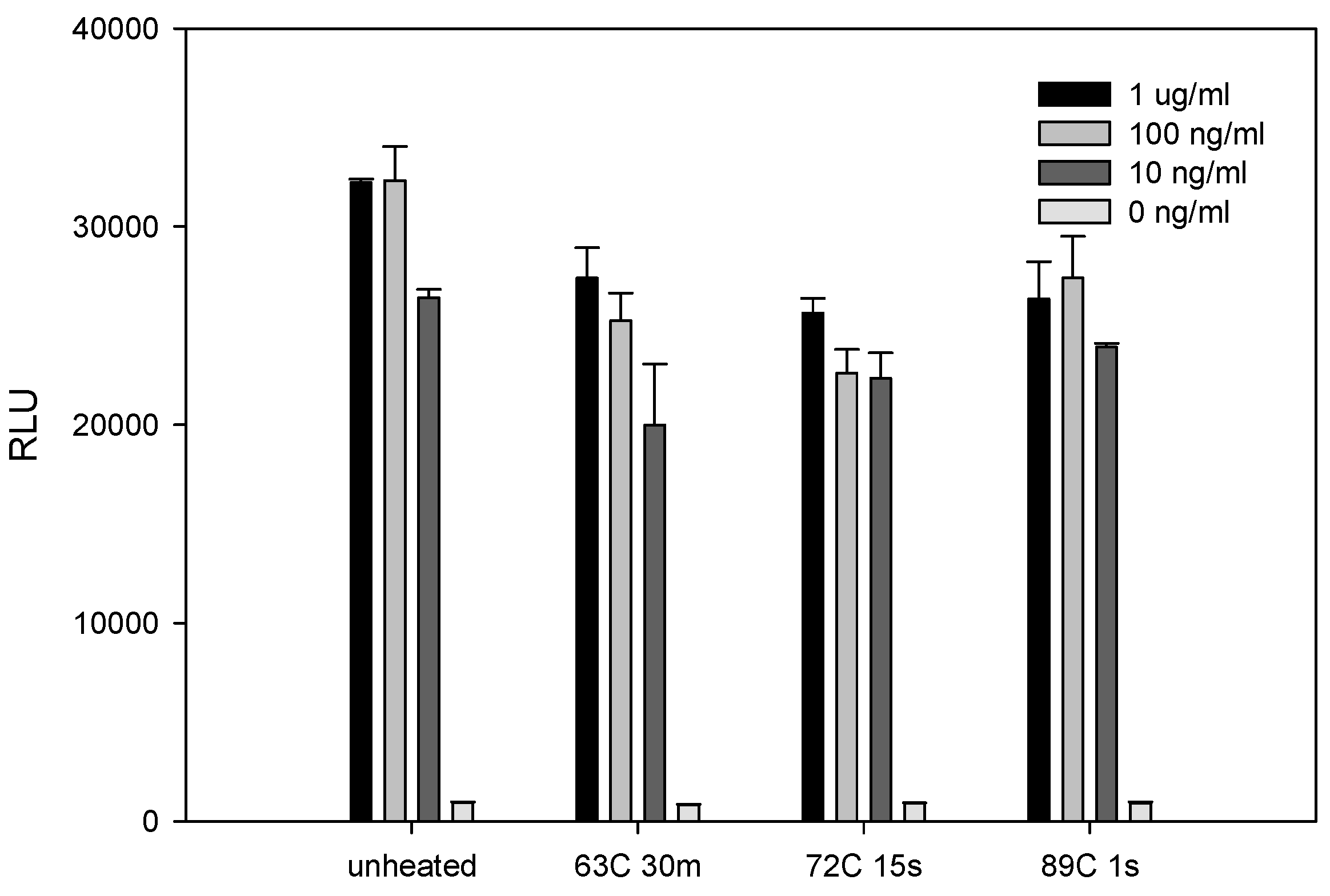
2.3. The Effect of Various Pasteurization Time-Temperature Conditions on SEE Activity
2.4. Matrix Effect on Heat Inactivation of SEE
2.5. Immunomagnetic Beads Remove Food Matrix Interference
2.6. Neutralizing Effect of SEE Antibody
3. Discussion
4. Materials and Methods
4.1. Toxin and Cell Lines
4.2. Ethics Statement
4.3. Splenocyte Isolation
4.4. SEE Magnetic Bead Preparation
4.5. Sample Binding and Disassociation of SEE from Beads
4.6. Ex Vivo Assay for SEE Detection
4.7. In Vitro Assay for SEE Detection
4.8. Neutralizing Effect of SEE Antibody
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| SEs | Staphylococcal enterotoxins |
| SEE | Staphylococcal enterotoxin type E |
| SEA | Staphylococcal enterotoxin type A |
| MHC II | Major histocompatibility complex class II |
| TCR | T-Cell receptor |
| NFAT | Nuclear factor of activated T-Cell |
| NFAT-RE | Nuclear factor of activated T-Cell response element |
| APC | Antigen presenting cell |
| TNF | Tumor necrosis factor |
| IFN | Interferon |
| MS | Mass spectrometry |
| ELISA | Enzyme-linked immunosorbent assay |
| PBS | Phosphate buffered saline |
| BSA | Bovine serum albumin |
| TBS | Tris buffered saline |
| BrdU | 5-bromo-2-deoxyuridine |
References
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef]
- Ahanotu, E.; Alvelo-Ceron, D.; Ravita, T.; Gaunt, E. Staphylococcal enterotoxin B as a biological weapon: Recognition, management, and surveillance of staphylococcal enterotoxin. Appl. Biosaf. 2006, 11, 120–126. [Google Scholar] [CrossRef]
- Hu, D.L.; Omoe, K.; Sashinami, H.; Shinagawa, K.; Nakane, A. Immunization with a nontoxic mutant of staphylococcal enterotoxin A, SEAD227A, protects against enterotoxin-induced emesis in house musk shrews. J. Infect. Dis. 2009, 199, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.; Cao, Y.; Xiao, F.; Zhang, J.; Li, H.; Hu, F. Staphylococcus aureus enterotoxin C2 mutants: Biological activity assay in vitro. J. Ind. Microbiol. Biotechnol. 2008, 35, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.O.; Grossman, D.; Kappler, J.W.; Marrack, P.; Rich, R.R.; Betley, M.J. Lack of complete correlation between emetic and T-cell-stimulatory activities of staphylococcal enterotoxins. Infect. Immun. 1993, 61, 3175–3183. [Google Scholar] [PubMed]
- Asao, T.; Kumeda, Y.; Kawai, T.; Shibata, T.; Oda, H.; Haruki, K.; Nakazawa, H.; Kozaki, S. An extensive outbreak of staphylococcal food poisoning due to low-fat milk in Japan: Estimation of enterotoxin A in the incriminated milk and powdered skim milk. Epidemiol. Infect. 2003, 130, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, R.; Do, P.M.; Hernlem, B.J. Auto-presentation of Staphylococcal enterotoxin A by mouse CD4+ T cells. Open J. Immunol. 2011, 1, 8–14. [Google Scholar] [CrossRef]
- Ikeda, T.; Tamate, N.; Yamaguchi, K.; Makino, S. Mass outbreak of food poisoning disease caused by small amounts of staphylococcal enterotoxins A and H. Appl. Environ. Microbiol. 2005, 71, 2793–2795. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, R.; Hernlem, B. TNF as biomarker for rapid quantification of active Staphylococcus enterotoxin A in food. Sensors 2012, 12, 5978–5985. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, R.; Hernlem, B.J. Quantitative analysis of staphylococcus enterotoxin A by differential expression of IFN-gamma in splenocyte and CD4(+) T-cells. Sensors 2014, 14, 8869–8876. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, R.; Do, P.M. In vitro cell-based assay for activity analysis of staphylococcal enterotoxin A in food. FEMS Immunol. Med. Microbiol. 2009, 56, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Mashouf, R.Y.; Hosseini, S.M.; Mousavi, S.M.; Arabestani, M.R. Prevalence of enterotoxin genes and antibacterial susceptibility pattern of Staphylococcus aureus strains isolated from animal originated foods in West of Iran. Oman Med. J. 2015, 30, 283–290. [Google Scholar] [CrossRef] [PubMed]
- McLauchlin, J.; Narayanan, G.L.; Mithani, V.; O’Neill, G. The detection of enterotoxins and toxic shock syndrome toxin genes in Staphylococcus aureus by polymerase chain reaction. J. Food. Prot. 2000, 63, 479–488. [Google Scholar] [PubMed]
- Ostyn, A.; De Buyser, M.L.; Guillier, F.; Groult, J.; Felix, B.; Salah, S.; Delmas, G.; Hennekinne, J.A. First evidence of a food poisoning outbreak due to staphylococcal enterotoxin type E, France, 2009. Euro Surveill 2010, 15, 1–4. [Google Scholar]
- Wieneke, A.A.; Roberts, D.; Gilbert, R.J. Staphylococcal food poisoning in the United Kingdom, 1969–1990. Epidemiol. Infect. 1993, 110, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Bergdoll, M.S.; Borja, C.R.; Robbins, R.N.; Weiss, K.F. Identification of enterotoxin E. Infect. Immun. 1971, 4, 593–595. [Google Scholar] [PubMed]
- Bennett, R.W. Staphylococcal enterotoxin and its rapid identification in foods by enzyme-linked immunosorbent assay-based methodology. J. Food Prot. 2005, 68, 1264–1270. [Google Scholar] [PubMed]
- Bergdoll, M.S. Monkey feeding test for staphylococcal enterotoxin. Methods Enzymol. 1988, 165, 324–333. [Google Scholar] [PubMed]
- Dupuis, A.; Hennekinne, J.A.; Garin, J.; Brun, V. Protein Standard Absolute Quantification (PSAQ) for improved investigation of staphylococcal food poisoning outbreaks. Proteomics 2008, 8, 4633–4636. [Google Scholar] [CrossRef] [PubMed]
- Park, C.E.; Akhtar, M.; Rayman, M.K. Nonspecific reactions of a commercial enzyme-linked immunosorbent assay kit (TECRA) for detection of staphylococcal enterotoxins in foods. Appl. Environ. Microbiol. 1992, 58, 2509–2512. [Google Scholar] [PubMed]
- Rasooly, R.; Hernlem, B.J. CD154 as a potential early molecular biomarker for rapid quantification analysis of active Staphylococcus enterotoxin A. FEMS Immunol. Med. Microbiol. 2011, 64, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Crist, S.A.; Sprague, D.L.; Ratliff, T.L. Nuclear factor of activated T cells (NFAT) mediates CD154 expression in megakaryocytes. Blood 2008, 111, 3553–3561. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.D.; Newton, M.E.; Weiss, A. CD28 and T cell antigen receptor signal transduction coordinately regulate interleukin 2 gene expression in response to superantigen stimulation. J. Exp. Med. 1992, 175, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.; Croteau, G.; Sekaly, R.P.; Kappler, J.; Marrack, P. HLA-DR alleles differ in their ability to present staphylococcal enterotoxins to T cells. J. Exp. Med. 1990, 172, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Hase, C.C.; Finkelstein, R.A. Bacterial extracellular zinc-containing metalloproteases. Microbiol. Rev. 1993, 57, 823–837. [Google Scholar] [PubMed]
- Bayles, K.W.; Iandolo, J.J. Genetic and molecular analyses of the gene encoding staphylococcal enterotoxin D. J. Bacteriol. 1989, 171, 4799–4806. [Google Scholar] [PubMed]
- Iandolo, J.J. Genetic analysis of extracellular toxins of Staphylococcus aureus. Annu. Rev. Microbiol. 1989, 43, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, R.; Hernlem, B.; Friedman, M. Low levels of aflatoxin B1, ricin, and milk enhance recombinant protein production in mammalian cells. PLoS ONE 2013, 8, e71682. [Google Scholar] [CrossRef] [PubMed]
- Barry, W.; Hudgins, L.; Donta, S.T.; Pesanti, E.L. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA 1992, 267, 3315–3316. [Google Scholar] [CrossRef] [PubMed]
- Kaul, R.; McGeer, A.; Norrby-Teglund, A.; Kotb, M.; Schwartz, B.; O’Rourke, K.; Talbot, J.; Low, D.E. Intravenous immunoglobulin therapy for streptococcal toxic shock syndrome—A comparative observational study. The Canadian Streptococcal Study Group. Clin. Infect. Dis. 1999, 28, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Bavari, S.; Hunt, R.E.; Ulrich, R.G. Divergence of human and nonhuman primate lymphocyte responses to bacterial superantigens. Clin. Immunol. Immunopathol. 1995, 76, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Hufnagle, W.O.; Tremaine, M.T.; Betley, M.J. The carboxyl-terminal region of staphylococcal enterotoxin type A is required for a fully active molecule. Infect. Immun. 1991, 59, 2126–2134. [Google Scholar] [PubMed]
- Kaminuma, O.; Kitamura, F.; Kitamura, N.; Hiroi, T.; Miyoshi, H.; Miyawaki, A.; Miyatake, S. Differential contribution of NFATc2 and NFATc1 to TNF-alpha gene expression in T cells. J. Immunol. 2008, 180, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.D.; Urban, R.G.; Strominger, J.L.; Robinson, H. Zinc regulates the function of two superantigens. Proc. Natl. Acad. Sci. USA 1992, 89, 5507–5511. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Dauwalder, O.; Brun, V.; Badiou, C.; Ferry, T.; Etienne, J.; Vandenesch, F.; Lina, G. Staphylococcus aureus superantigens elicit redundant and extensive human Vbeta patterns. Infect. Immun. 2009, 77, 2043–2050. [Google Scholar] [CrossRef] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasooly, R.; Do, P.; Hernlem, B. Sensitive, Rapid, Quantitative and in Vitro Method for the Detection of Biologically Active Staphylococcal Enterotoxin Type E. Toxins 2016, 8, 150. https://doi.org/10.3390/toxins8050150
Rasooly R, Do P, Hernlem B. Sensitive, Rapid, Quantitative and in Vitro Method for the Detection of Biologically Active Staphylococcal Enterotoxin Type E. Toxins. 2016; 8(5):150. https://doi.org/10.3390/toxins8050150
Chicago/Turabian StyleRasooly, Reuven, Paula Do, and Bradley Hernlem. 2016. "Sensitive, Rapid, Quantitative and in Vitro Method for the Detection of Biologically Active Staphylococcal Enterotoxin Type E" Toxins 8, no. 5: 150. https://doi.org/10.3390/toxins8050150