1. Introduction
Recombinant immunotoxins (RITs) are fusions of a targeting moiety and a toxin [
1]. While some immunotoxins use a natural ligand to target a specific receptor, others use an Fv or Fab fragment of a receptor-targeting antibody.
Pseudomonas aeruginosa (PE)-based immunotoxins usually contain a 38 kDa fragment of the
Pseudomonas aeruginosa exotoxin A (PE38), consisting of domains II and III of the native toxin; domain I is replaced by the Fv.
Once the Fv portion of the immunotoxin binds to its target receptor, the immunotoxin is internalized by endocytosis. In the endocytic compartment, the immunotoxin is cleaved by the pro-protein convertase (PC) furin, leading to the formation of two immunotoxin fragments; the Fv containing fragment and a second fragment consisting of most of domain II and the catalytic domain III. These two fragments are held together by a disulfide bond. The disulfide bond is reduced and the catalytic fragment is trafficked by late endosomes to the trans-golgi network (TGN), and there binds to the KDEL receptor, which traffics it to the endoplasmic reticulum (ER). From there, the toxin fragment is translocated into the cytosol. Once in the cytosol, domain III catalyzes the adenosine diphosphate ribosylation of a diphthamide modification on elongation factor 2 (eEF2), thereby inhibiting protein translation and ultimately causing apoptosis [
2,
3,
4].
Immunotoxins have been investigated in several clinical trials for treating cancer (reviewed in [
2,
3,
4,
5]). Some of the main challenges identified in these trials is non-specific animal toxicity and high immunogenicity producing anti-drug antibodies (ADA) that hamper immunotoxin efficacy [
5]. Clinical trials with immuno-suppressed leukemia patients, whose immune status diminished ADA formation, showed higher immunotoxin efficacy [
5,
6].
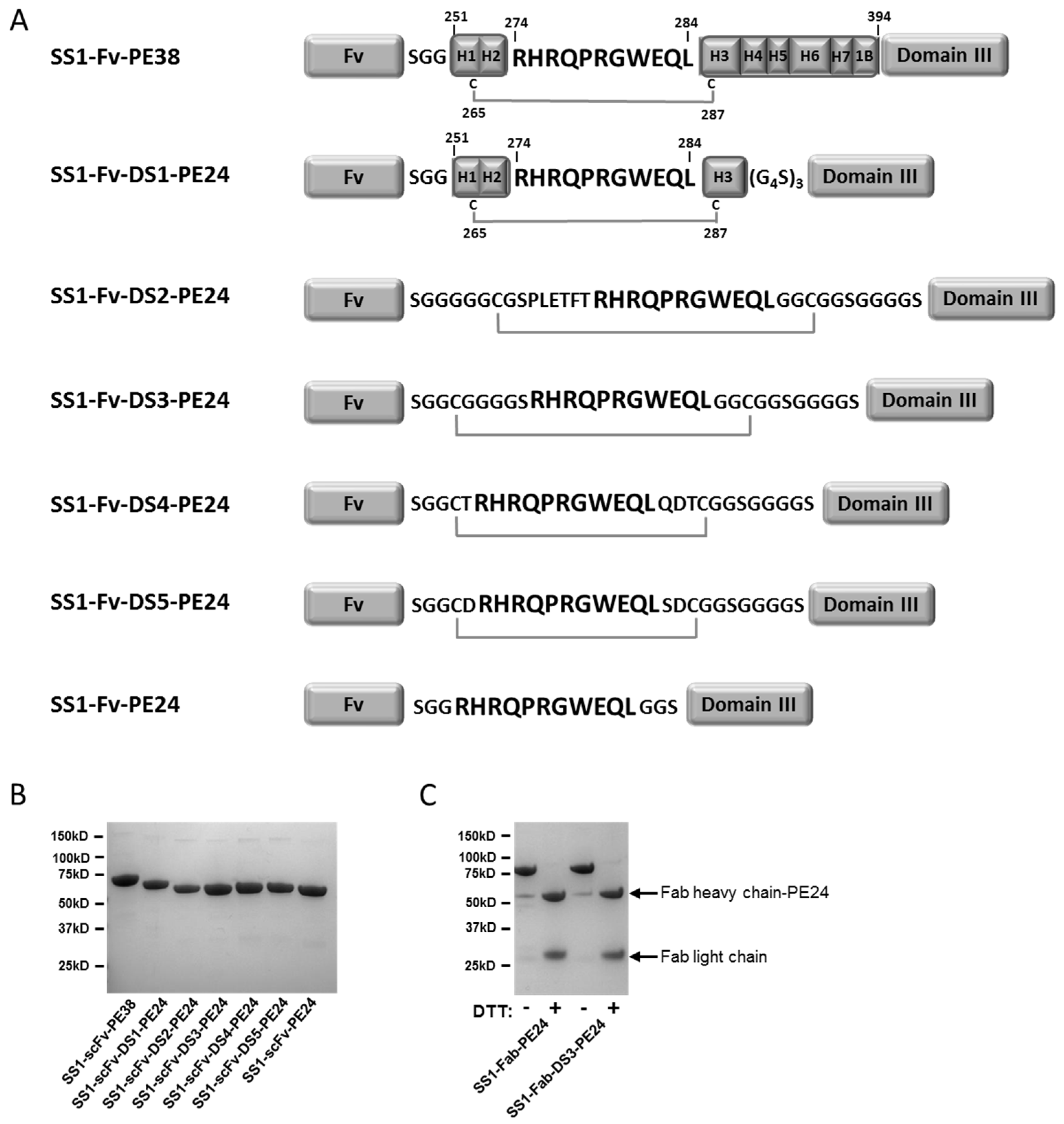
To try and prevent processing of PE38 toxins by antigen presenting cells, thereby reducing the immunogenicity of PE-based immunotoxins, a lysosomal protease resistant version of PE38 (designated “PE24”) was previously developed [
7]. While PE38 consists of two domains (domains II and III) of the full length exotoxin A, the PE24 toxin consists of only the catalytic domain III connected to the targeting Fv by the furin cleavage site (FCS) normally found in domain II (see
Figure 1). By removing domain II, processing by lysosomal enzymes is reduced, and immunogenic epitopes found within domain II are also removed. When combined with point mutations designed to disrupt B and T cell epitopes in the remaining domain III, a de-immunized PE24 molecule was produced, which was shown to be significantly less immunogenic in mouse models [
8,
9]. In addition to low immunogenicity, PE24 toxins show improved cytotoxic activity in multiple cell lines compared to PE38 toxins in vitro, and much lower non-specific toxicity in mice and rats, allowing higher doses to be given [
10,
11]. PE24 toxins therefore exhibit improved characteristics for clinical trials. However, in re-engineering of PE38 into PE24, the native disulfide bond around the FCS in domain II of PE38 was removed, leaving only a non-constrained FCS sequence between toxin domain III and the Fv targeting moiety.
Furin is a ubiquitous transmembrane serine endoprotease [
12]. As a member of the PC family, furin cleaves multiple pro-proteins along the secretory pathway [
12]. Transmembrane furin cycles between the TGN, the cell surface and endosomal compartments [
12,
13]. Production of a soluble, secreted form of furin has also been described in epithelial cells in vitro by posttranslational processing. Furin cleavage is necessary for activation of several toxins, including pseudomonas, diphtheria and anthrax toxins [
12]. While PE-based toxins are activated by furin cleavage after internalization in the early endosome, anthrax toxin is activated by furin cleavage at the cell surface, showing that furin is present and active at the plasma membrane [
14]. PE38 immunotoxins contain a naturally occurring disulfide bond around the FCS found in domain II. If these immunotoxins are cleaved by cell surface furin, the disulfide bond will prevent disassociation of the toxin and Fv until it is reduced within the cell following immunotoxin internalization. In contrast, the unprotected FCS found in PE24 based immunotoxins, lacking a protective disulfide bond, could be cleaved by furin in the tumor extra-cellular environment or at the cell surface before internalization, thereby deactivating PE24-based immunotoxins.
In this paper, we describe the design and preparation of RITs in which the furin cleavage site is stabilized by addition of different peptide bridges that form a disulfide bond across the furin site so that the two portions of the immunotoxin remain together until the disulfide bond is reduced. We have then characterized the properties of the most promising of these RITs.
2. Results
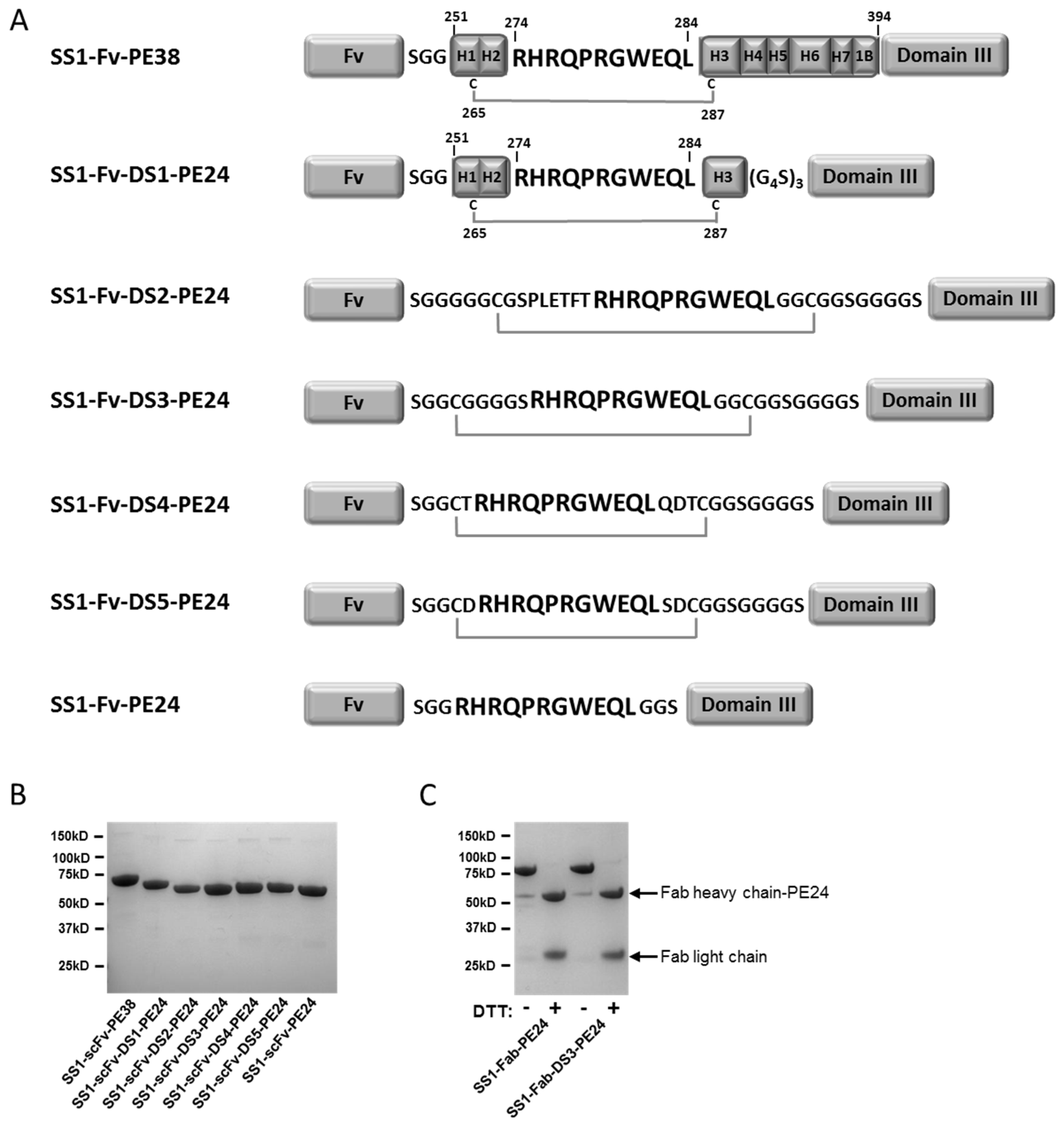
2.1. Design and Production of the DS-PE24 Immunotoxins
To bridge the furin cleavage site (FCS, “RHRQPRGWEQL”) with a disulfide bond, cysteine containing flanking peptides were designed and introduced into the anti-mesothelin SS1-PE24 immunotoxin (
Figure 1A). The first DS-PE24 construct is SS1-Fv-DS1-PE24. It retains helixes 1–3 of domain II (residues 251–301), which include the naturally occurring disulfide bond and the FCS, surrounded by Gly-Ser linkers. As removal of helixes 4–7 uncovers the hydrophobic core of domain II, we anticipated solubility problems with this construct. Therefore, four hydrophobic residues in the uncovered core were mutated into polar residues in order to increase solubility (L256Q, L259Q, V290T and L293Q).
SS1-Fv-DS2-PE24 contains helix 2 and the FCS, surrounded by long Gly-Ser linkers with two cysteines located the same number of residues away from the furin site as in the native domain II. SS1-Fv-DS3-PE24 is the simplest of the DS-PE24 constructs. The 11 amino acid FCS is bridged by two cysteines, six glycines and one serine.
The final two constructs, SS1-Fv-DS4-PE24 and SS1-Fv-DS5-PE24, contain sequences surrounding the FCS that were designed computationally using Rosetta Protein Design methods [
14,
15] to constrain the FCS in the native conformation found in domain II.
Because RITs combining a scFv and PE24 have a low molecular weight of around 50 kDa, and are subject to renal filtration resulting in a short in vivo half-life, the scFv targeting moiety was replaced with a Fab to increase size, reduce filtration by the kidney and increase half-life. Therefore, the parental SS1-PE24 immunotoxin and the SS1-DS3-PE24 construct were also produced as SS1-Fab immunotoxins (SS1-Fab-PE24 and SS1-Fab-DS3-PE24). All of the RITs were produced by standard protein refolding from
Escherichia coli (
E. coli) inclusion bodies (see Materials and methods) and were obtained as highly purified monomeric proteins of the correct molecular weight (
Figure 1B,C). Starting with 100 mg of total protein, all the RITs could be produced in large amounts by our standard refolding procedure (
Table 1).
2.2. The Disulfide Bond around the Furin Cleavage Site Is Formed
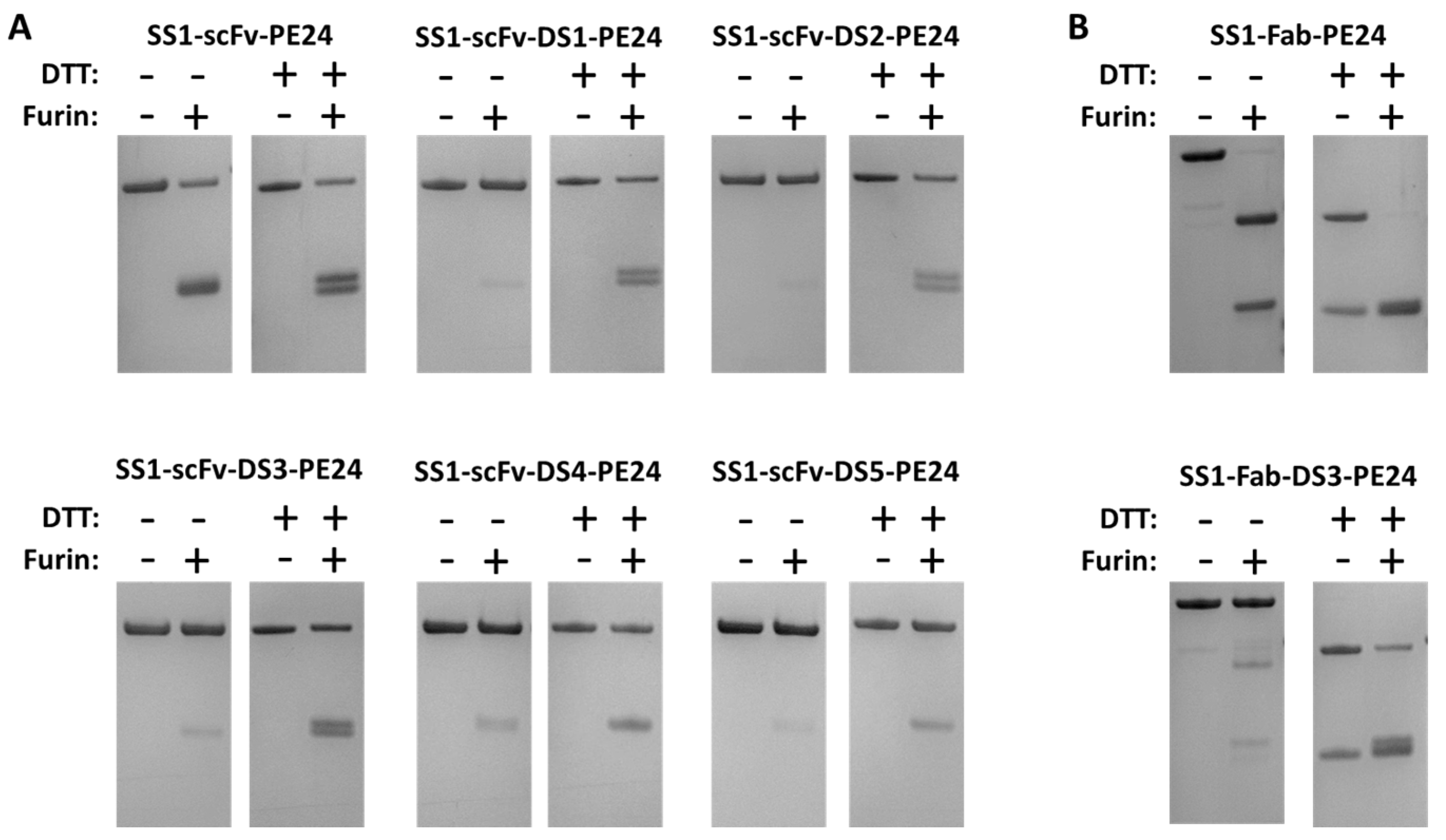
To show that a disulfide bond had formed around the FCS in our DS-PE24 immunotoxins, we treated the DS-PE24 proteins with soluble recombinant furin. We expected that even after furin cleavage, the DS-PE24 immunotoxins would still migrate as a single band on a non-reducing gel, because the two fragments would still be connected by the disulfide bond. We also expected that they would migrate as two bands in a reducing gel, because the disulfide bond would then be reduced.
As shown in
Figure 2, the parental SS1-scFv-PE24, which does not contain a disulfide bond between the toxin and the Fv, shows a lower molecular weight band after furin cleavage under non-reducing conditions, indicating separation of immunotoxin into an Fv and a toxin fragment with similar molecular weights (25.5 and 26.7 kDa, respectively). These two fragments are better resolved, when the proteins are run under reducing conditions, presumably because the disulfide bonds in the scFv are reduced changing the shape of the protein. Using soluble furin, we were unable to reach 100% immunotoxin cleavage, even under optimized conditions.
In contrast, SS1-scFv-DS1-PE24 and all the other DS-PE24 proteins migrated as a single band on a non-reducing gel when treated with furin. Only when analyzed on a non-reducing gel were low molecular weight bands after furin treatment observed, which correspond to the scFv and PE24. In all cases, some un-cleaved DS-PE24 protein remained, because the soluble furin could not digest all the added protein. The extent of cleavage varied among the five proteins. ScFv immunotoxins containing the DS1, DS2, or DS3 sequences were cleaved more completely than SS1-scFv-DS4-PE24 or SS1-scFv-DS5-PE24.
We also examined the cleavage of the two Fab immunotoxins with furin. As shown in
Figure 2B, SS1-Fab-PE24 was almost completely cleaved by furin as indicated by the disappearance of the high molecular weight band in the gel. The products represent the Fab and PE24. In contrast, furin treatment of SS1-Fab-DS3-PE24 had very little effect on the high molecular weight band when the protein was analyzed on a non-reducing gel, but the protein dissociated into lower molecular bands on a reducing gel after furin treatment. Under reducing conditions, the V
L-κ chain is also visible as part of the lower double band.
Overall, the formation of a FCS protective disulfide bond was observed in all of the DS-PE24 mutants to some degree. We could not fully quantify the percentage of molecules containing the disulfide bond due to lack of full cleavage by furin.
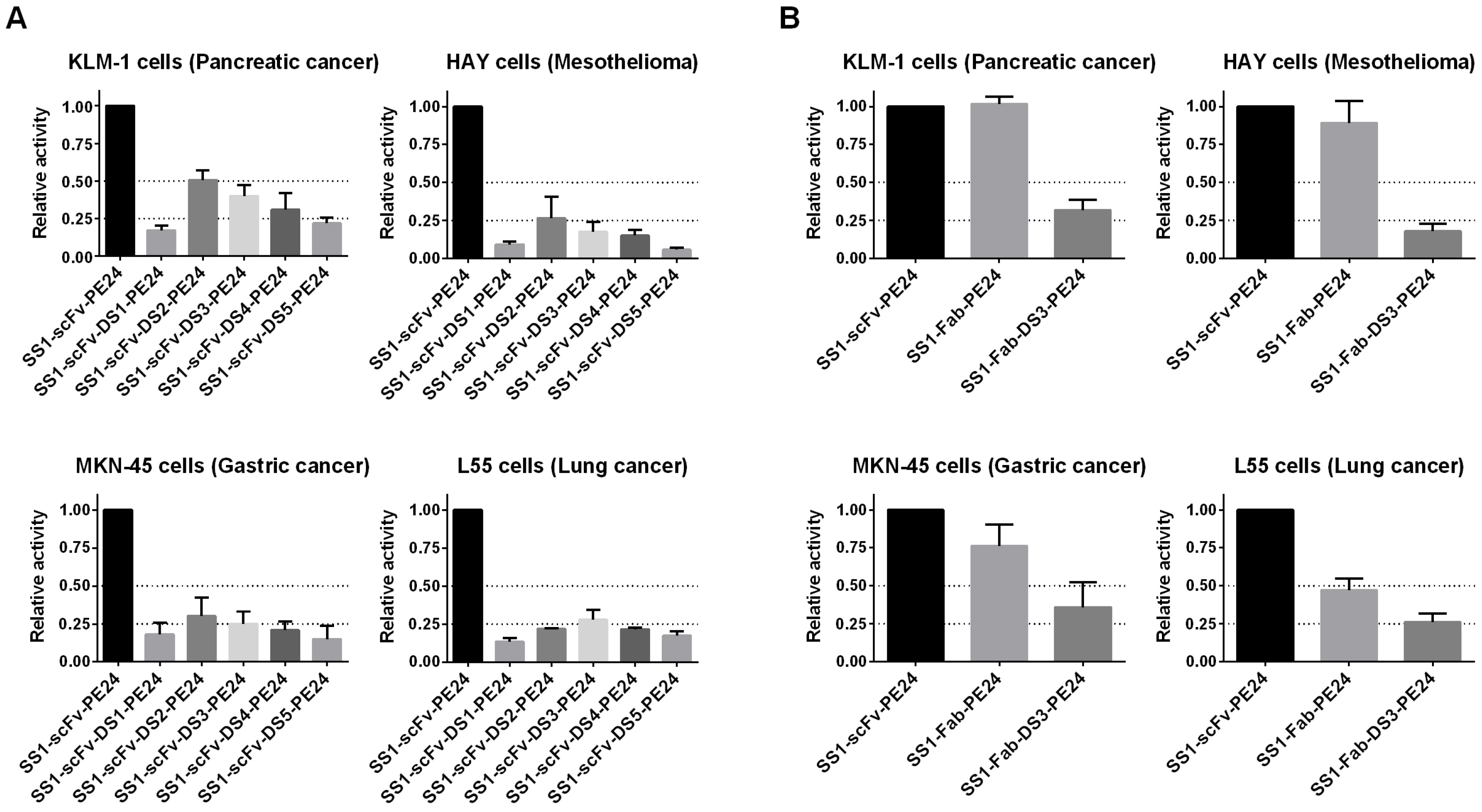
2.3. Cytotoxicity Assays
The cytotoxic activity of the different DS-PE24 immunotoxins was compared in a WST-8 cell growth inhibition assay on four different cell lines; the pancreatic cancer KLM-1 cell line, the mesothelioma HAY cell line, the gastric cancer MKN45 cell line and the lung cancer L55 cell line. All of these cancers express mesothelin, the target of the SS1 targeting moiety found in the tested immunotoxins. KLM-1 cells express high levels of mesothelin (60 × 10
3 sites/cell) [
15], as do HAY cells (unquantified) [
16] and MKN-45 cells (9.9 × 10
3 sites/cell) [
17]. On the other hand, L-55 cells express much lower amounts of mesothelin (unquantified) [
18,
19].
As shown in
Figure 3A and
Table 2, the most active DS-PE24 constructs were SS1-scFv-DS2-PE24 and SS1-scFv-DS3-PE24. However, they were about 2–6-fold less active than the parental SS1-scFv-PE24, depending on the cell line studied. SS1-scFv-DS1-PE24, which contains a large portion of domain II, exhibited much lower cytotoxic activity, with a reduction of 6-11 fold in cytotoxic activity. Of the two computationally designed linkers, SS1-scFv-DS4-PE24 exhibited cytotoxic activity that was only slightly lower than SS1-scFv-DS2-PE24 and SS1-scFv-DS3-PE24, while SS1-scFv-DS5-PE24 showed the lowest activity of the constructs with a 5–18-fold reduction.
Figure 3B and
Table 2 show data with the Fab containing immunotoxins. We found that SS1-Fab-PE24 and SS1-Fab-DS3-PE24 showed cytotoxic activities similar to the scFv immunotoxins. Since immunogenicity is a problem with PE immunotoxins, using a linker peptide with the least possible immunogenic sequences would be advantageous. The DS3 linker combines maximal cytotoxic activity with a linker consisting of glycines and serines that are not usually immunogenic. We therefore focused on DS-PE24 immunotoxins containing the DS3 linker in subsequent assays.
2.4. Stability Studies
The DS-PE24 immunotoxins were constructed to protect the FCS from extra-cellular cleavage by furin or other proteases in the circulation. To determine if the addition of the disulfide bond affects stability, both the parental PE24 and DS3-PE24 immunotoxins were tested in a serum stability assay (
Table 3). Both scFv immunotoxins (SS1-scFv-PE24 and SS1-scFv-DS3-PE24) and Fab immunotoxins (SS1-Fab-PE24 and SS1-Fab-DS3-PE24) were incubated in 95% human serum at 37 °C for 1, 6 and 24 h, and then tested for cytotoxic activity on the human pancreatic cell line KLM1. All molecules except SS1-Fab-DS3-PE24 showed no significant changes up to 6 h and a significant 1.6–2-fold decrease after 24 h (uncorrected Fisher’s LSD,
p < 0.05). SS1-Fab-DS3-PE24 showed no significant changes over the 24 h period. While the two DS-PE24 constructs, SS1-scFv-DS3-PE24 and SS1-Fab-DS3-PE24, seemed slightly more stable than the PE24 immunotoxins; these differences were not statistically significant (uncorrected Fisher’s LSD). We conclude that the addition of the disulfide bond around the FCS does not affect overall immunotoxin stability.
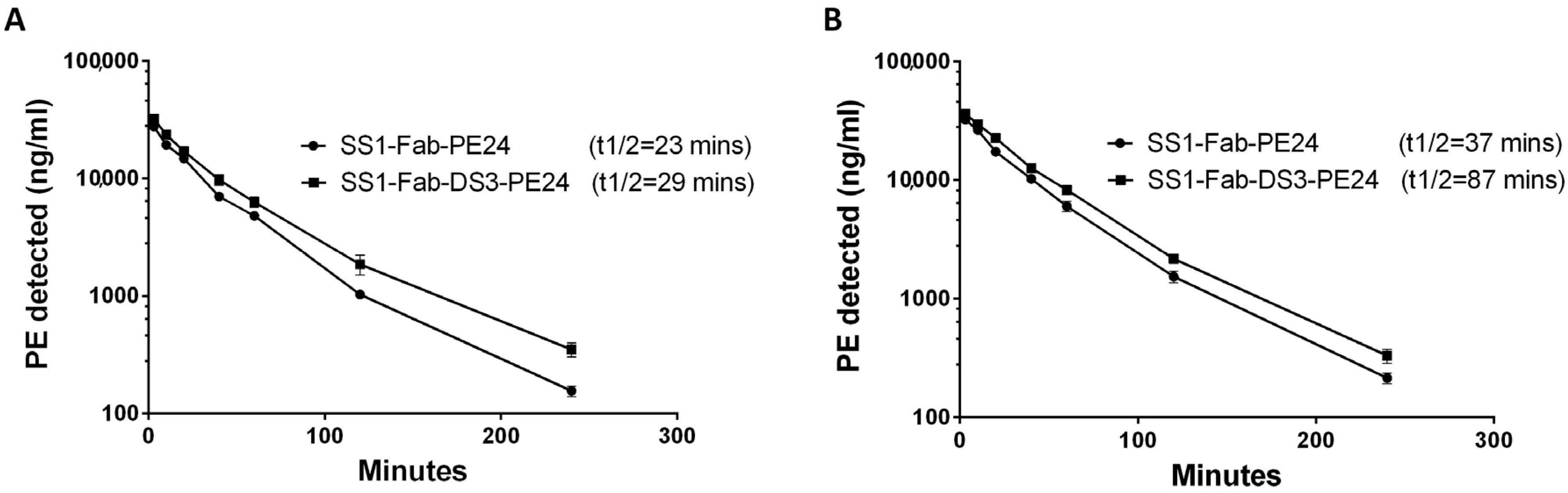
2.5. Half-Life Studies in Mice
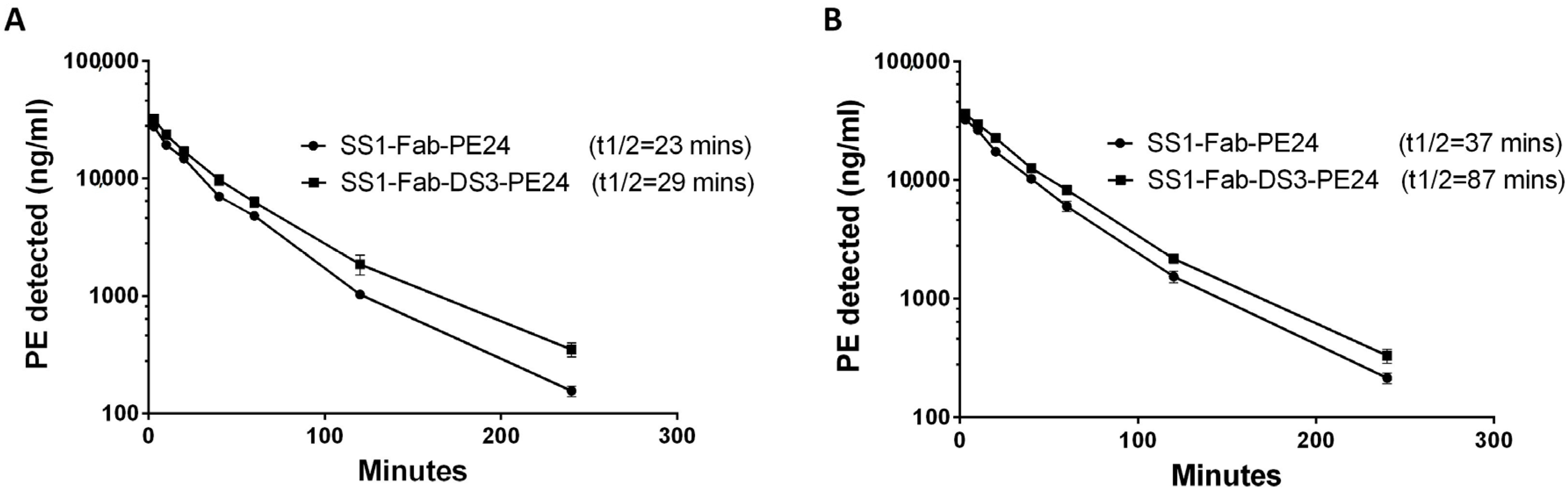
The Fab constructs (SS1-Fab-PE24 and SS1-Fab-DS3-PE24) were chosen for mouse studies because they were very active in vitro and are expected to have a longer half-life and more activity in mice. To measure the half-life, mice were injected with 30 μg of the SS1-Fab-PE24 or SS1-Fab-DS3-PE24 (three mice in each group). This dose is similar to the dose used to treat tumors in mice. Blood samples were obtained at different times, and the serum levels of immunotoxin were detected using ELISA (
Figure 4 and
Table 4). The half-life of the two immunotoxins was compared using a “two-phase decay” model that models the clearance of the drug into two phases, an initial fast phase and a slower second phase.
As shown in
Figure 4 and
Table 5, addition of the DS linker increased the slow phase half-life of SS1-Fab-DS3-PE24 from 23 to 29.8 min (a 29% increase) in the first experiment and from 37.6 to 87.9 min (a 133% increase) in the second experiment. In both experiments, the addition of the disulfide bond around the FCS caused a decrease in clearance in both the fast and slow clearance phases, as well as an increase in the area under the curve (AUC) (
Table 5). The reason for the difference in half-lives between the two mouse experiments is probably due to the different amounts of blood drawn for the blood level ELISAs (40 µL vs. 10 µL), because the larger volumes removed substantial amounts of immunotoxin from the blood stream.
2.6. Anti-Tumor Experiments
The anti-tumor activity of SS1-Fab-DS3-PE24 was compared with the parental SS1-Fab-PE24 immunotoxin with respect to the growth of pancreatic cancer KLM-1 tumors in mice. Mice were implanted with 5 million KLM-1 tumor cells in the right flank on day 0 and treatment began on day 8 when the tumors reached about 150 mm
3 in size. Mice were treated every other day (×3) with either 50 μg of either RIT or saline and tumor size monitored over several weeks. As shown in
Figure 5, both immunotoxins show a 9-day tumor growth delay (from 25 days to 34 days) to reach a tumor volume of 600 mm
3, but showed no significant difference from each other. None of the treated mice showed significant weight loss, showing the given doses to be well tolerated. Noticeably, the SS1-Fab-DS3-PE24 immunotoxin, while being less active in vitro, shows wild-type anti-tumor activity in vivo.
4. Materials and Methods
4.1. Construction, Expression and Protein Purification
Double-stranded gene fragments (gBlocks, Integrated DNA Technologies, Coralville, IA, USA) corresponding to the desired sequences were ordered and ligated into our standard laboratory immunotoxin production vector as described previously [
31] using the Gibson Assembly Master Mix (New England Biolabs, Ipswich, MA, USA) according to manufacturer’s instructions. All RITs were expressed and purified from
E. coli inclusion bodies as previously described [
31]. Briefly, inclusion bodies from
E. coli were prepared and dissolved in GTE buffer (6 M guanidine-HCl, 100 mM Tris-HCl, 2 mM EDTA). The denatured proteins were then refolded in 100 mL Tris-HCl, 1 mM EDTA, 0.5 M arginine, 0.5 M NDSB-201, pH 10 for 32 h, and then dialyzed against 30 mM Tris-HCl, 0.1 M urea for 40 h. The refolded proteins were purified using ion-exchange and size exclusion chromatography.
4.2. Computational Design of DS-PE24 Linkers
Rosetta Protein Design methods [
32,
33] were used to design linkers that constrain the FCS in the native conformation found in domain II. For this, the furin cleavage site (FCS) was extended by appending 2–5 amino acids on each side and the backbone conformations of these appended residues were sampled in torsional space and analyzed for placement of disulfide bonds with ideal geometry. Around 20,000 different sequences were designed using Rosetta sequence design methods [
33], and two of the lowest energy sequences were selected for experimental characterization (DS4 and DS5).
4.3. Cell Growth Inhibition Assay
The cytotoxic activity of the RITs was evaluated using the WST-8 cell counting kit (Dojindo Molecular Technologies, Rockville, MD, USA). Cells were incubated with varying concentrations of immunotoxin for 72 h, after which the WST-8 reagent was added. Ninety-six well plates were read at OD 450. Readings were normalized to the phosphate buffered saline (PBS) only positive control and the IC50 (concentration inhibiting growth by 50%) for each construct was calculated using a variable four-parameter slope, non-linear regression fit with Graphpad Prism vs. 6.01. One hundred percent cell killing was achieved using 2.3 μg/mL of staurosporine (Sigma, St. Louis, MO, USA) as a control. Each assay contained four replicates of each concentration and the assays were repeated three to four times. The KLM1 pancreatic cancer cell line was provided by Dr. U. Rudloff (NCI, Bethesda, MD, USA); the L55 lung adenocarcinoma cell line was provided by Dr. S. Albelda (University of Pennsylvania, Philadelphia, PA, USA); the HAY mesothelioma cell line was provided by the Stehlin Foundation for Cancer Research (Houston, TX, USA); the gastric cancer MKN45 cell line was provided by Dr. Takao Yamori (Pharmaceuticals and Medical Device Agency, Tokyo, Japan).
4.4. In Vitro Furin Cleavage
Immunotoxins (2 µg) were diluted into furin cleavage buffer (100 mM Tris-HCl pH 7.5, 1 mM CaCl2) and incubated with 1.5 μg of recombinant human furin (Peprotech, Rocky Hill, NJ, USA) in a total volume of 20 µL for 16 h at 37 °C. Control samples were incubated without furin. All of the samples (±furin) were then split into two and analyzed under reducing (sample reducing reagent added, Life Technologies) and non-reducing conditions on 4%–12% Bis-Tris gradient polyacrylamide gels according to the manufacturer’s instructions (Novex, Life Technologies, Waltham, MA, USA).
4.5. Immunotoxin Stability in Human Serum
The parental PE24 containing constructs (SS1-scFv-PE24 and SS1-Fab-PE24) and the DS3-PE24 mutants (SS1-scFv-DS3-PE24 and SS1-Fab-DS3-PE24) were diluted to 15 µg/mL into non-heat inactivated 100% human AB serum (Gemini Bio-Products, West Sacramento, CA, USA). The immunotoxins were then incubated at 37 °C for 0, 1, 6, 24 and 48 h. After incubation, the samples were diluted 1:5 in PBS and used directly in the cell growth inhibition WST-8 assay described above. The assay was repeated three times.
4.6. In Vivo Half-Life
Balb/c mice (6–8 weeks, averaging 20 g) were injected intravenously with 30 µg of either SS1-Fab-PE24 or SS1-Fab-DS3-PE24 (
n = 3). Blood samples were drawn at 3, 10, 20, 40, 60, 120, and 240 min. The concentration of immunotoxins in the sera was measured in ELISA using a standard curve for each immunotoxin as described previously [
34]. Briefly, microtiter plates were coated with 50 μL of MSLN-rFc (a fusion protein consisting of rabbit IgG Fc fused to the mesothelin protein) (1 μg/mL) in PBS at 4 °C overnight. Diluted standards or serum samples were applied, followed by incubation with HRP-conjugated anti-PE antibody, IP12 [
35]. After washing, plates were developed using tetramethylbenzidine substrate kit (Thermo Scientific, Rockford, IL, USA). Serum half-life was calculated using a “two-phase decay” model in Graphpad Prism vs. 6.01. The animal protocol (LMB-005) was approved by the National Cancer Institute Animal Care and Use Committee and the animals handled according to institute guidelines.
4.7. Anti-Tumor Activity
Athymic nude mice were inoculated subcutaneously with 5 × 106 KLM1 cells in Matrigel on day 0. On day 8, when the average tumor volume was 150 mm3, mice were randomized into three groups and treated intravenously in the tail vein with either 200 μL vehicle (0.2% HSA in PBS) (n = 7), 1.5 mg/kg SS1-Fab-DS3-PE24 in 0.2% HSA (n = 10) or 1.5 mg/kg SS1-Fab-PE24 in 0.2% HSA (n = 10). Mice were treated qodx3 on days 8, 10 and 12. Weights and general health were monitored during treatment. Tumors were measured every 2–3 d until volumes reached 600 mm3. The animal protocol (LMB-014) was approved by the National Cancer Institute Animal Care and Use Committee and the animals were handled according to institute guidelines.
4.8. Graphs and Statistics
Graphs were plotted and analyzed using Graphpad Prism vs. 6.01 (GraphPad Software, Inc., La Jolla, CA, USA).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}