Castration-Resistant Prostate Cancer Refractory to Second-Generation Androgen Receptor Axis-Targeted Agents: Opportunities and Challenges


Abstract
:1. Introduction
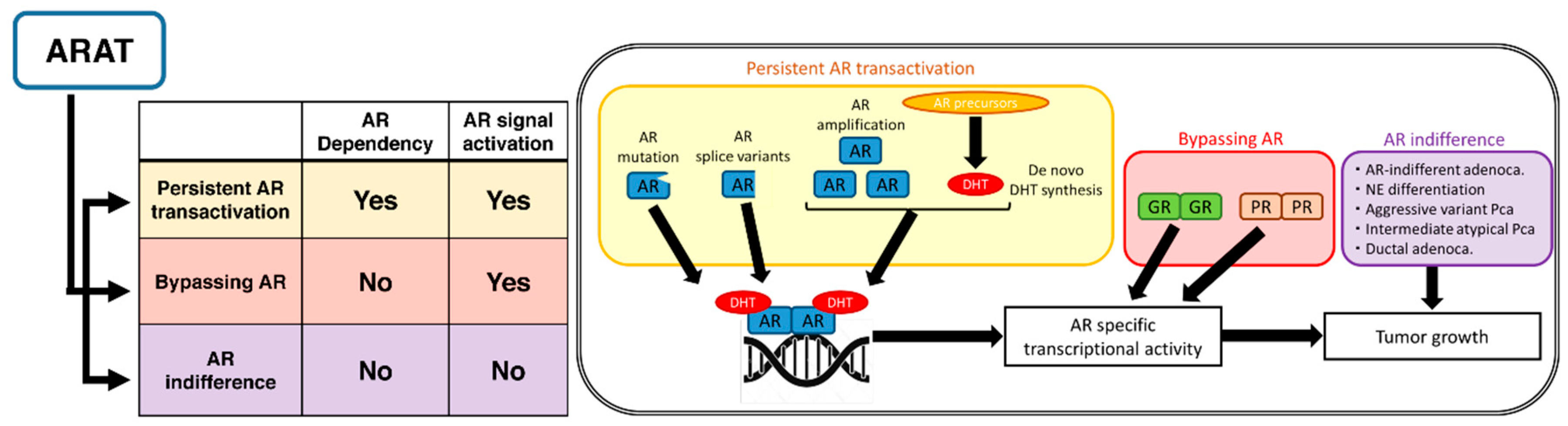
2. Mechanisms for Resistance to ARAT Agents Classified by AR Signaling Activation
2.1. Persistent AR Transactivation
2.1.1. Ligand-Dependent Activation of AR
De Novo Synthesis of DHT
AR Amplification/Overexpression
AR Mutations
2.1.2. Ligand-Independent Activation of AR
AR Splice Variants
AR Mutant
Crosstalk with Other Oncogenic Signaling Pathways
2.2. Bypassing AR
2.2.1. GR Overexpression
2.2.2. PR Upregulation
2.3. AR Indifference
3. Next-Generation ARAT Agents
4. Future Perspectives
4.1. Molecular Mechanisms for AR Overexpression
4.2. How Can We Know the Real Driving Mechanism in Individual Patients?
4.3. Novel Therapeutic Agents Multiply Questions for Clinical Use
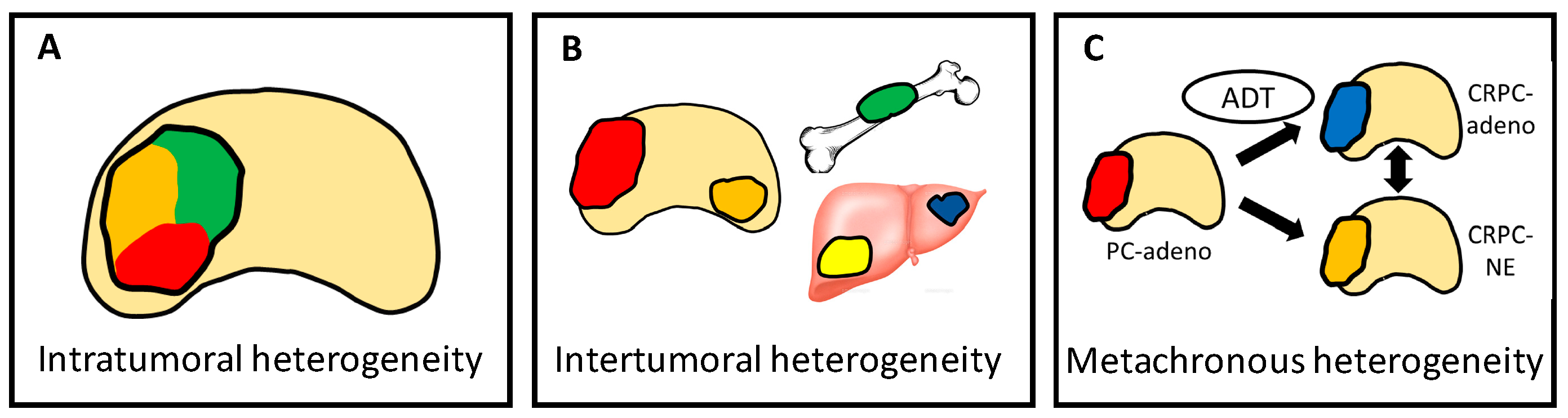
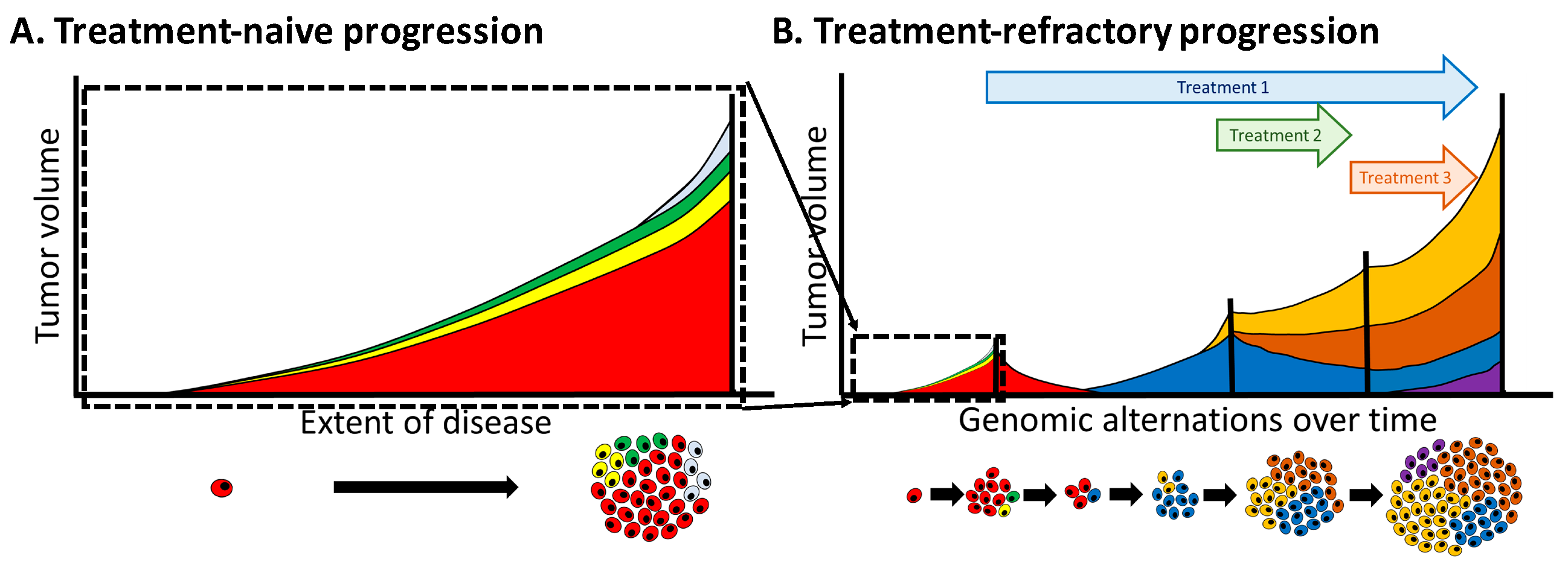
4.4. Heterogeneity Is Multi-Dimensional
4.5. Overcoming Heterogeneity that Drives Treatment Resistance
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Inoue, T.; Kamba, T.; Ogawa, O. Experimental evidence of persistent androgen-receptor-dependency in castration-resistant prostate cancer. Int. J. Mol. Sci. 2013, 14, 15615–15635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet (Lond. Engl.) 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.A.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantanos, T.; Evans, C.P.; Tombal, B.; Thompson, T.C.; Montironi, R.; Isaacs, W.B. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur. Urol. 2015, 67, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocrine-Rel. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Sadar, M.D. Androgen receptor targeted therapies in castration-resistant prostate cancer: Bench to clinic. Int. J. Urol. Off. J. Jpn. Urol. Assoc. 2016, 23, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachostergios, P.J.; Puca, L.; Beltran, H. Emerging Variants of Castration-Resistant Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: Induction of steroidogenesis and androgen receptor splice variants. Clin. Cancer Res. 2011, 17, 5913–5925. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N.; McPhaul, M.J.; Auchus, R.J. “Getting from here to there”—Mechanisms and limitations to the activation of the androgen receptor in castration-resistant prostate cancer. J. Investig. Med. 2010, 58, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Li, R.; Kuri, B.; Lotan, Y.; Roehrborn, C.G.; Liu, J.; Vessella, R.; Nelson, P.S.; Kapur, P.; Guo, X.; et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell 2013, 154, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Zhu, Y.; Yang, J.C.; Nadiminty, N.; Gaikwad, N.W.; Evans, C.P.; Gao, A.C. Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer Res. 2015, 75, 1413–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Armstrong, C.M.; Lou, W.; Lombard, A.; Evans, C.P.; Gao, A.C. Inhibition of AKR1C3 Activation Overcomes Resistance to Abiraterone in Advanced Prostate Cancer. Mol. Cancer Ther. 2017, 16, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Taplin, M.E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Loriot, Y.; Beraldi, E.; Zhang, F.; Wyatt, A.W.; Al Nakouzi, N.; Mo, F.; Zhou, T.; Kim, Y.; Monia, B.P.; et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin. Cancer Res. 2015, 21, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, A.W.; Azad, A.A.; Volik, S.V.; Annala, M.; Beja, K.; McConeghy, B.; Haegert, A.; Warner, E.W.; Mo, F.; Brahmbhatt, S.; et al. Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Romanel, A.; Gasi Tandefelt, D.; Conteduca, V.; Jayaram, A.; Casiraghi, N.; Wetterskog, D.; Salvi, S.; Amadori, D.; Zafeiriou, Z.; Rescigno, P.; et al. Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med. 2015, 7, 312re10. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Casadio, V.; Conteduca, V.; Burgio, S.L.; Menna, C.; Bianchi, E.; Rossi, L.; Carretta, E.; Masini, C.; Amadori, D.; et al. Circulating cell-free AR and CYP17A1 copy number variations may associate with outcome of metastatic castration-resistant prostate cancer patients treated with abiraterone. Br. J. Cancer 2015, 112, 1717–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, S.; Casadio, V.; Conteduca, V.; Lolli, C.; Gurioli, G.; Martignano, F.; Schepisi, G.; Testoni, S.; Scarpi, E.; Amadori, D.; et al. Circulating AR copy number and outcome to enzalutamide in docetaxel-treated metastatic castration-resistant prostate cancer. Oncotarget 2016, 7, 37839–37845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annala, M.; Vandekerkhove, G.; Khalaf, D.; Taavitsainen, S.; Beja, K.; Warner, E.W.; Sunderland, K.; Kollmannsberger, C.; Eigl, B.J.; Finch, D.; et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; Watson, P.A.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2013, 2, e00499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.J.; Sowalsky, A.G.; Gao, S.; Cai, C.; Voznesensky, O.; Schaefer, R.; Loda, M.; True, L.D.; Ye, H.; Troncoso, P.; et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res. 2015, 21, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Carreira, S.; Romanel, A.; Goodall, J.; Grist, E.; Ferraldeschi, R.; Miranda, S.; Prandi, D.; Lorente, D.; Frenel, J.S.; Pezaro, C.; et al. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 2014, 6, 254ra125. [Google Scholar] [CrossRef] [PubMed]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed]
- Ware, K.E.; Garcia-Blanco, M.A.; Armstrong, A.J.; Dehm, S.M. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocrine-Rel. Cancer 2014, 21, T87–T103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Xie, N.; Sun, S.; Plymate, S.; Mostaghel, E.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014, 33, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Alsagabi, M.; Fan, D.; Bova, G.S.; Tewfik, A.H.; Dehm, S.M. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011, 71, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hwang, T.H.; Oseth, L.A.; Hauge, A.; Vessella, R.L.; Schmechel, S.C.; Hirsch, B.; Beckman, K.B.; Silverstein, K.A.; Dehm, S.M. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene 2012, 31, 4759–4767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Laere, B.; van Dam, P.J.; Whitington, T.; Mayrhofer, M.; Diaz, E.H.; Van den Eynden, G.; Vandebroek, J.; Del-Favero, J.; Van Laere, S.; Dirix, L.; et al. Comprehensive Profiling of the Androgen Receptor in Liquid Biopsies from Castration-resistant Prostate Cancer Reveals Novel Intra-AR Structural Variation and Splice Variant Expression Patterns. Eur. Urol. 2017, 72, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Henzler, C.; Li, Y.; Yang, R.; McBride, T.; Ho, Y.; Sprenger, C.; Liu, G.; Coleman, I.; Lakely, B.; Li, R.; et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun. 2016, 7, 13668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.C.; Li, Y.; Dehm, S.M. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J. Biol. Chem. 2012, 287, 19736–19749. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhan, Y.; Qi, Y.; Cao, B.; Bai, S.; Xu, W.; Gambhir, S.S.; Lee, P.; Sartor, O.; Flemington, E.K.; et al. Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Res. 2015, 75, 3663–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Alvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O.; et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014, 5, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.C.; Selth, L.A.; Li, Y.; Nyquist, M.D.; Miao, L.; Bradner, J.E.; Raj, G.V.; Tilley, W.D.; Dehm, S.M. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucl. Acids Res. 2015, 43, 5880–5897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, M.; Ho, Y.; Hillman, D.W.; Van Etten, J.L.; Henzler, C.; Yang, R.; Sperger, J.M.; Li, Y.; Tseng, E.; Hon, T.; et al. Androgen Receptor Variant AR-V9 Is Coexpressed with AR-V7 in Prostate Cancer Metastases and Predicts Abiraterone Resistance. Clin. Cancer Res. 2017, 23, 4704–4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Isaacs, W.B.; Luo, J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate 2011, 71, 1656–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyquist, M.D.; Li, Y.; Hwang, T.H.; Manlove, L.S.; Vessella, R.L.; Silverstein, K.A.; Voytas, D.F.; Dehm, S.M. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17492–17497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Gao, S.; Valencia, K.; Owiredu, J.; Han, W.; de Waal, E.; Macoska, J.A.; Cai, C. A novel nonsense mutation in androgen receptor confers resistance to CYP17 inhibitor treatment in prostate cancer. Oncotarget 2017, 8, 6796–6808. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Smith, D.A.; Memarzadeh, S.; Lowell, C.A.; Cooper, J.A.; Witte, O.N. Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 6579–6584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Lamoureux, F.; Crafter, C.; Davies, B.R.; Beraldi, E.; Fazli, L.; Kim, S.; Thaper, D.; Gleave, M.E.; Zoubeidi, A. Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol. Cancer Ther. 2013, 12, 2342–2355. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Kim, S.; Cordonnier, T.; Crafter, C.; Davies, B.R.; Fazli, L.; Gleave, M.E.; Zoubeidi, A. Combination AZD5363 with Enzalutamide Significantly Delays Enzalutamide-resistant Prostate Cancer in Preclinical Models. Eur. Urol. 2015, 67, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Reid, A.H.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Banuelos, C.A.; Imamura, Y.; Leung, J.K.; Caley, D.P.; Wang, J.; Mawji, N.R.; Sadar, M.D. Cotargeting Androgen Receptor Splice Variants and mTOR Signaling Pathway for the Treatment of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.P.; Ramalingam, S.; Gediya, L.; Kwegyir-Afful, A.K.; Njar, V.C. Simultaneous targeting of androgen receptor (AR) and MAPK-interacting kinases (MNKs) by novel retinamides inhibits growth of human prostate cancer cell lines. Oncotarget 2015, 6, 3195–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Kach, J.; Long, T.M.; Selman, P.; Tonsing-Carter, E.Y.; Bacalao, M.A.; Lastra, R.R.; de Wet, L.; Comiskey, S.; Gillard, M.; VanOpstall, C.; et al. Selective Glucocorticoid Receptor Modulators (SGRMs) Delay Castrate-Resistant Prostate Cancer Growth. Mol. Cancer Ther. 2017, 16, 1680–1692. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, L.; Xie, N.; Xue, H.; Fazli, L.; Buttyan, R.; Wang, Y.; Gleave, M.; Dong, X. Expression and function of the progesterone receptor in human prostate stroma provide novel insights to cell proliferation control. J. Clin. Endocrinol. Metab. 2013, 98, 2887–2896. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Gupta, N.S.; Wang, W.; Toubaji, A.; Haffner, M.C.; Chaux, A.; Hicks, J.L.; Meeker, A.K.; Bieberich, C.J.; De Marzo, A.M.; et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod. Pathol. 2011, 24, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.C.; Dancer, J.Y.; Wang, Y.; Aparicio, A.; Navone, N.M.; Troncoso, P.; Czerniak, B.A. TMPRSS2-ERG gene fusion in small cell carcinoma of the prostate. Hum. Pathol. 2011, 42, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Zhang, S.; Yao, J.L.; Huang, J.; Lopez-Beltran, A.; Shen, S.; Osunkoya, A.O.; MacLennan, G.T.; Montironi, R.; Cheng, L. ERG-TMPRSS2 rearrangement is shared by concurrent prostatic adenocarcinoma and prostatic small cell carcinoma and absent in small cell carcinoma of the urinary bladder: Evidence supporting monoclonal origin. Modern Pathol. 2011, 24, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, S.; Wyatt, A.W.; Lin, D.; Lysakowski, S.; Zhang, F.; Kim, S.; Tse, C.; Wang, K.; Mo, F.; Haegert, A.; et al. The Placental Gene PEG10 Promotes Progression of Neuroendocrine Prostate Cancer. Cell Rep. 2015, 12, 922–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef] [PubMed]
- Dalal, K.; Che, M.; Que, N.S.; Sharma, A.; Yang, R.; Lallous, N.; Borgmann, H.; Ozistanbullu, D.; Tse, R.; Ban, F.; et al. Bypassing Drug Resistance Mechanisms of Prostate Cancer with Small Molecules that Target Androgen Receptor-Chromatin Interactions. Mol. Cancer Ther. 2017, 16, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Shore, N.D. Darolutamide (ODM-201) for the treatment of prostate cancer. Exp. Opin. Pharmacother. 2017, 18, 945–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toren, P.J.; Kim, S.; Pham, S.; Mangalji, A.; Adomat, H.; Guns, E.S.; Zoubeidi, A.; Moore, W.; Gleave, M.E. Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Mol. Cancer Ther. 2015, 14, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Cai, C.; Gao, S.; Simon, N.I.; Shen, H.C.; Balk, S.P. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin. Cancer Res. 2014, 20, 4075–4085. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, B.; Eisenberger, M.A.; Rettig, M.B.; Chu, F.; Pili, R.; Stephenson, J.J.; Vogelzang, N.J.; Koletsky, A.J.; Nordquist, L.T.; Edenfield, W.J.; et al. Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.T.; Haugk, K.; McKiernan, J.S.; Gulati, R.; Cheng, H.H.; Maes, J.L.; Dumpit, R.F.; Nelson, P.S.; Montgomery, B.; McCune, J.S.; et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS ONE 2018, 13, e0198389. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, D.Y.; Spisak, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szallasi, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e13. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447.e19. [Google Scholar] [CrossRef] [PubMed]
- Obinata, D.; Takayama, K.; Takahashi, S.; Inoue, S. Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer. Cancers 2017, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Sydes, M.R.; Spears, M.R.; Mason, M.D.; Clarke, N.W.; Dearnaley, D.P.; de Bono, J.S.; Attard, G.; Chowdhury, S.; Cross, W.; Gillessen, S.; et al. Adding abiraterone or docetaxel to long-term hormone therapy for prostate cancer: Directly randomised data from the STAMPEDE multi-arm, multi-stage platform protocol. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Kyriakopoulos, C.E.; Chen, Y.H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Gravis, G.; Boher, J.M.; Chen, Y.H.; Liu, G.; Fizazi, K.; Carducci, M.A.; Oudard, S.; Joly, F.; Jarrard, D.M.; Soulie, M.; et al. Burden of Metastatic Castrate Naive Prostate Cancer Patients, to Identify Men More Likely to Benefit from Early Docetaxel: Further Analyses of CHAARTED and GETUG-AFU15 Studies. Eur. Urol. 2018, 73, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Agents | Mechanism of Action | Clinical Trials |
|---|---|---|
| Apalutamide (ARN-509) | Second-generation AR antagonist | NCT01946204 (SPARTAN trial) |
| Darolutamide (ODM-201) | Second-generation AR antagonist | NCT02200614 |
| TRC253 | Second-generation AR antagonist | NCT02987829 |
| Seviteronel (VT-464) | Lyase-selective inhibitor of CYP17A1 | NCT02130700 NCT02445976 NCT02012920 |
| Galeterone (TOK-001) | Dual CYP17 inhibitor and AR antagonist | NCT02438007 |
| EPI-506 | N-terminal domain AR inhibitor | NCT02606123 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kita, Y.; Goto, T.; Akamatsu, S.; Yamasaki, T.; Inoue, T.; Ogawa, O.; Kobayashi, T. Castration-Resistant Prostate Cancer Refractory to Second-Generation Androgen Receptor Axis-Targeted Agents: Opportunities and Challenges. Cancers 2018, 10, 345. https://doi.org/10.3390/cancers10100345
Kita Y, Goto T, Akamatsu S, Yamasaki T, Inoue T, Ogawa O, Kobayashi T. Castration-Resistant Prostate Cancer Refractory to Second-Generation Androgen Receptor Axis-Targeted Agents: Opportunities and Challenges. Cancers. 2018; 10(10):345. https://doi.org/10.3390/cancers10100345
Chicago/Turabian StyleKita, Yuki, Takayuki Goto, Shusuke Akamatsu, Toshinari Yamasaki, Takahiro Inoue, Osamu Ogawa, and Takashi Kobayashi. 2018. "Castration-Resistant Prostate Cancer Refractory to Second-Generation Androgen Receptor Axis-Targeted Agents: Opportunities and Challenges" Cancers 10, no. 10: 345. https://doi.org/10.3390/cancers10100345