Loss of Cyclin-Dependent Kinase Inhibitor Alters Oncolytic Adenovirus Replication and Promotes More Efficient Virus Production


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
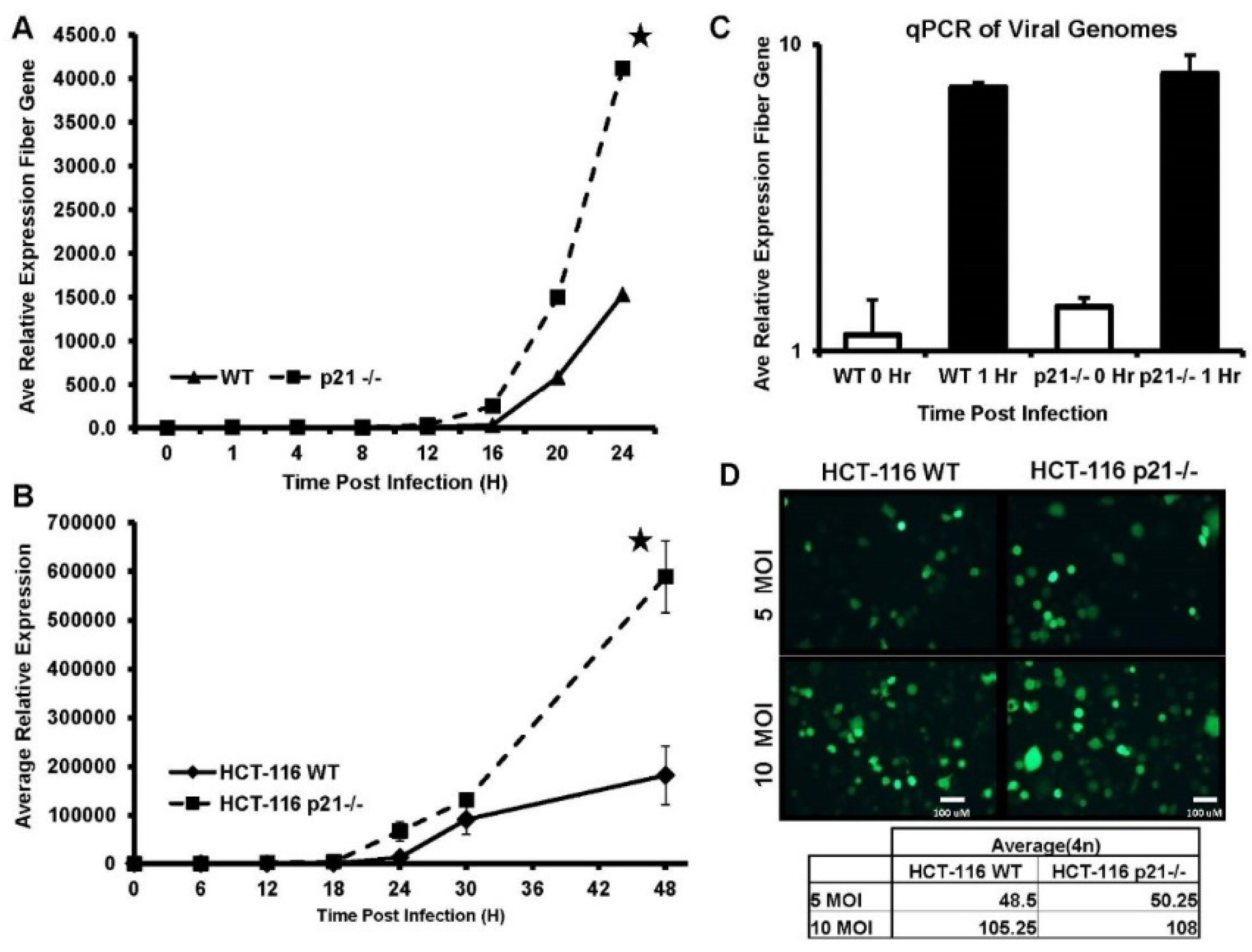
2.1. DNA Replication Is Significantly Increased in the Absence of p21
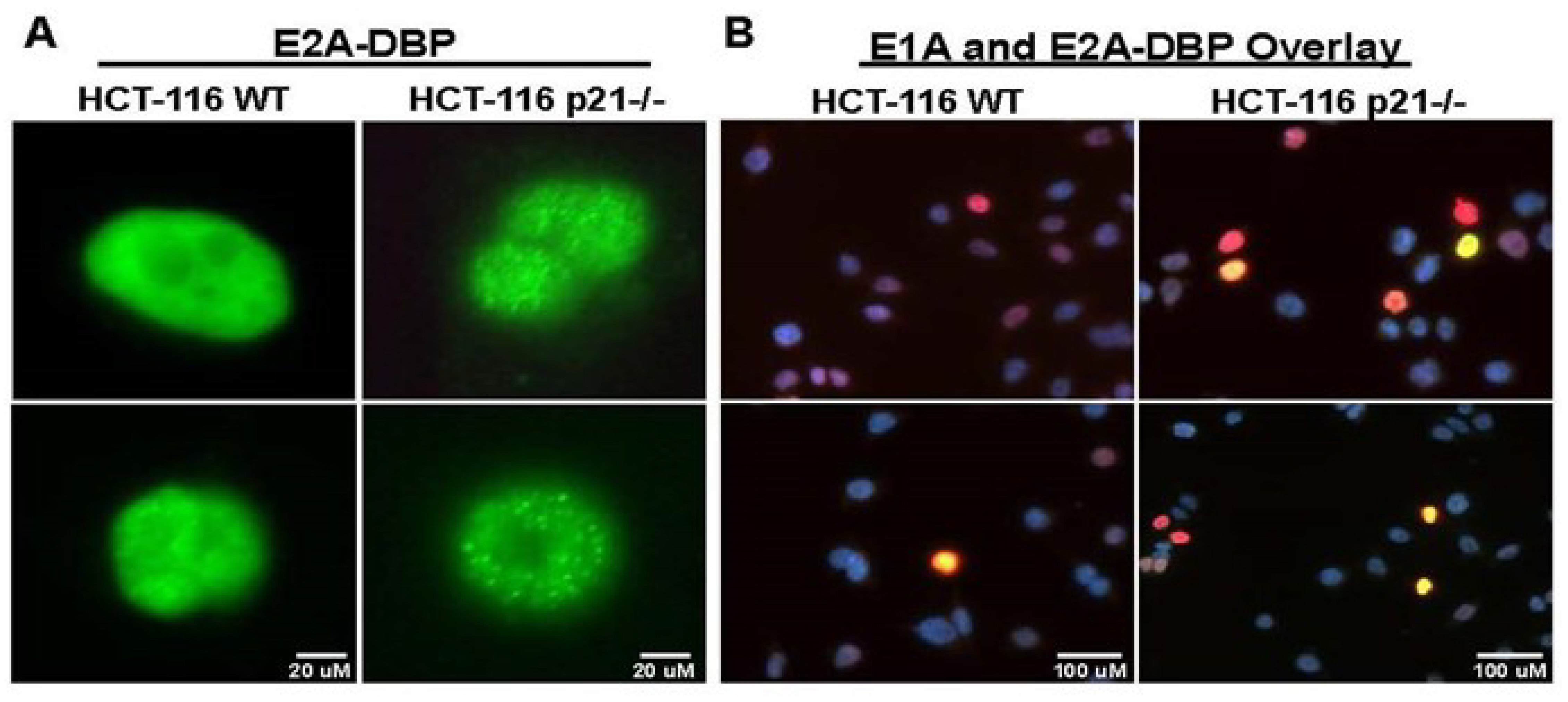
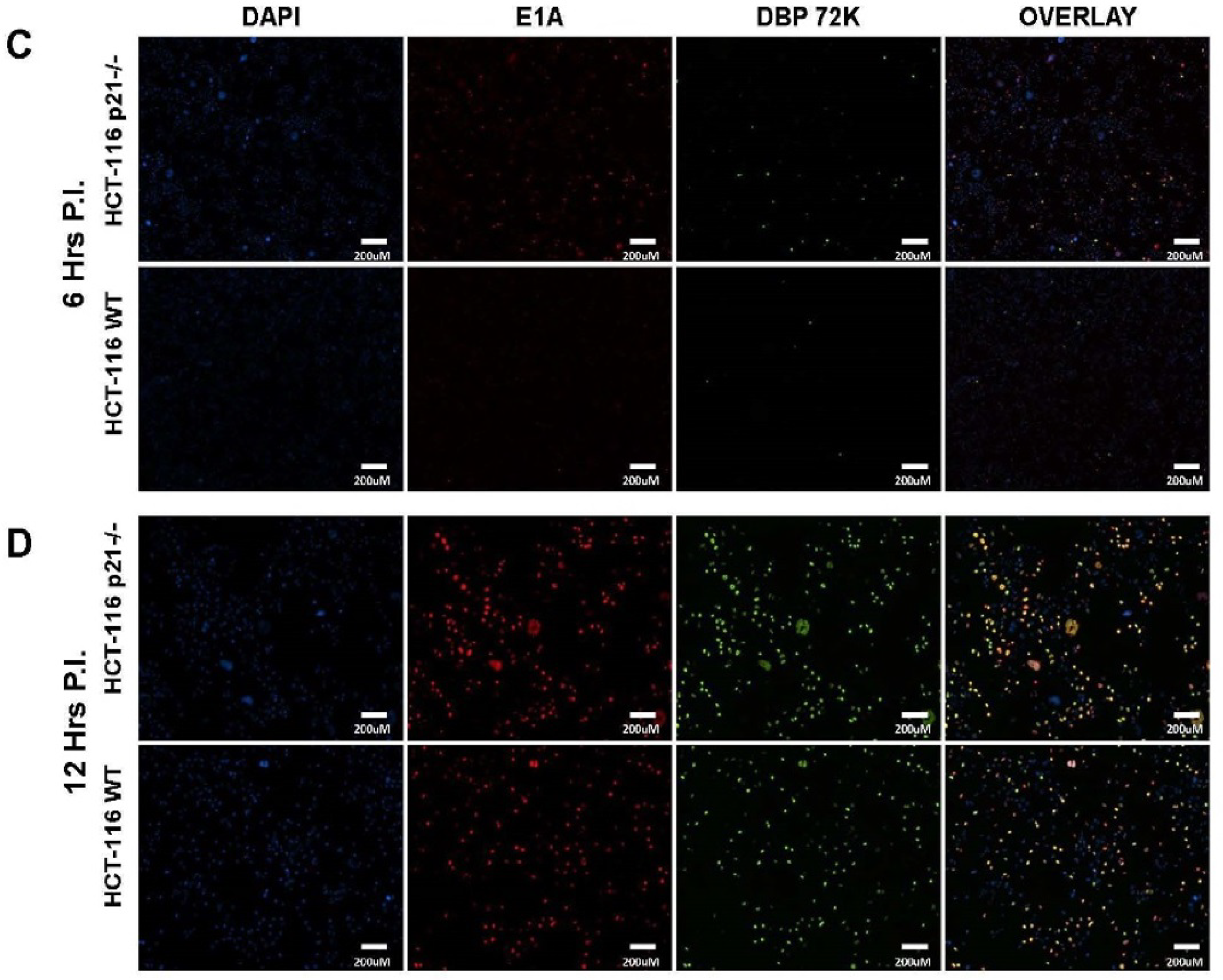
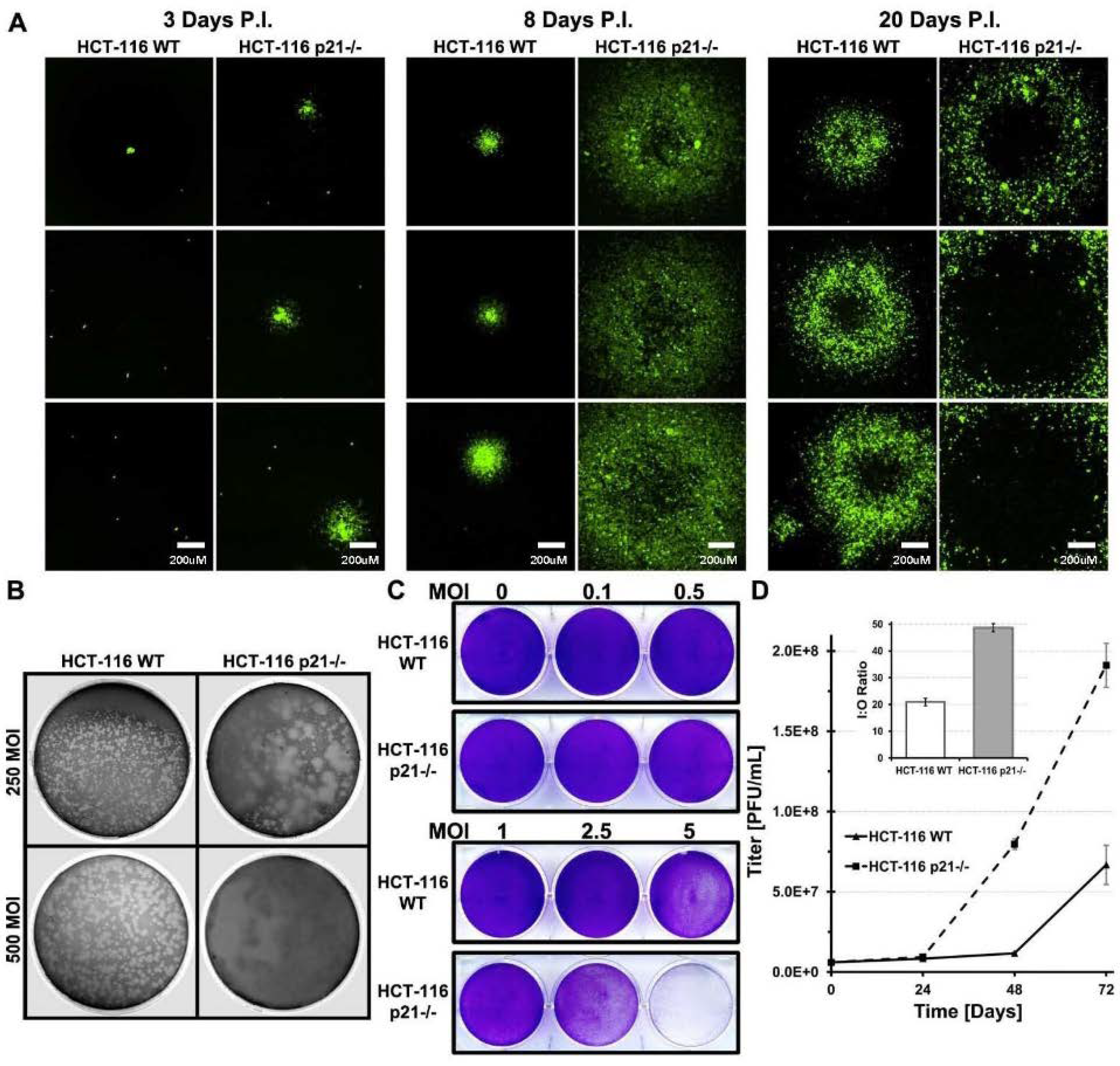
2.2. Oncolytic Viral DNA Replication Foci Form Earlier in p21−/− Cells
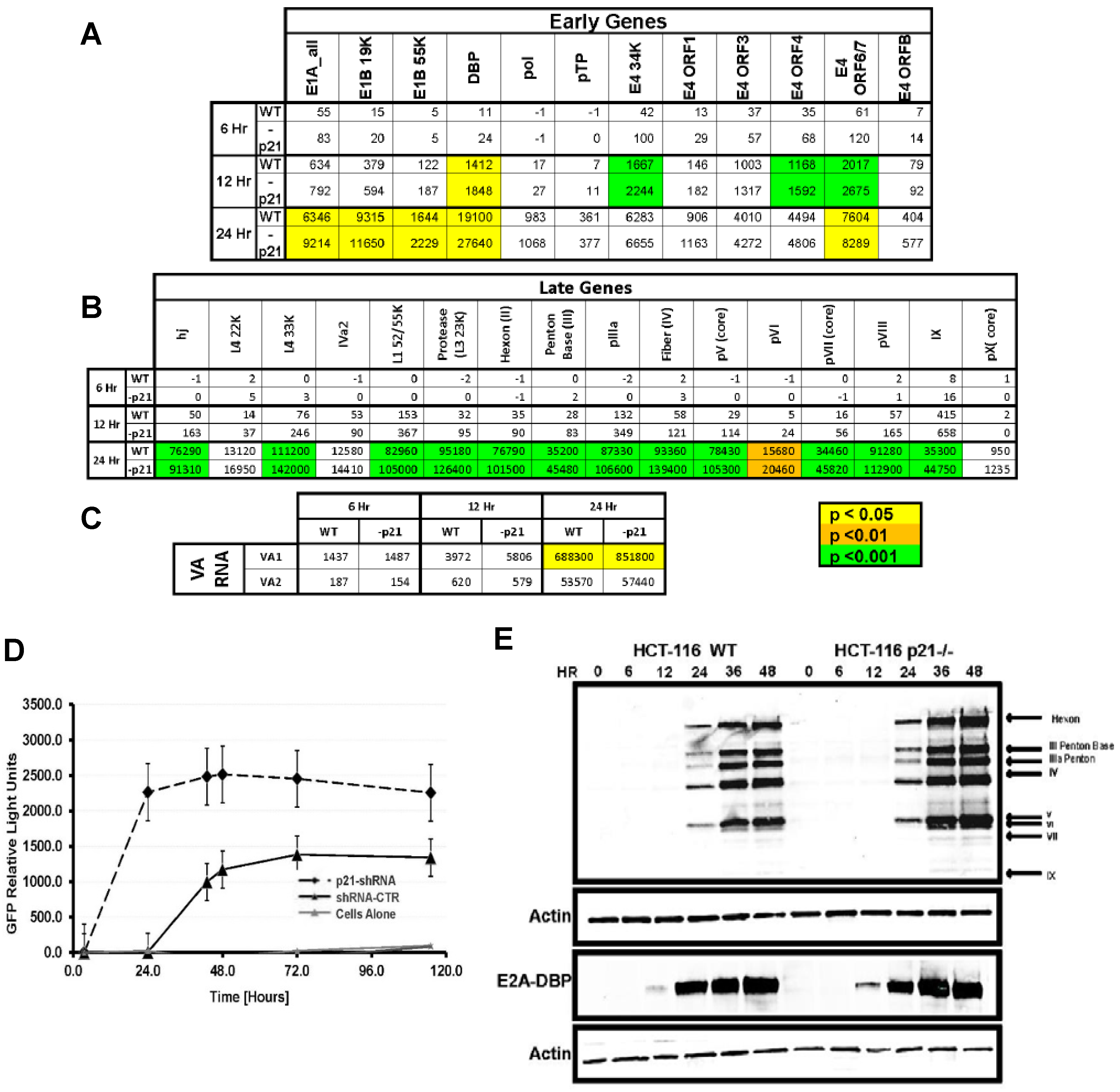
2.3. Viral Transcription and Late Gene Translation Is Higher in p21−/− Cells
2.4. HCT-116 p21 WT Infected Cells Produce Smaller Plaques Which Develop Slower
2.5. Lower Amount of Oncolytic Virus in p21 Intact Cells Is Independent of Infection Time
3. Materials and Methods
3.1. Cell Culture and Antibodies
3.2. Indirect Immunofluorescence
3.3. Western Blot Analysis
3.4. Viral Stock Preparation and Real-Time PCR
| Fiber Sense | CCCATTGGGGGATACAAAGGGAGGA |
| Fiber Antisense | GCAGATGAAGCGCGCGCAAGACCGT |
| E4 Sense | GTAATTCACCACCTCCCGGTA |
| E4 Antisense | GGCTCTCCACTGTCATTGTTC |
| Actin Sense | GTACCACTGGCATCGTGATGGACT |
| Actin Antisense | CCGCTCATTGCCAATGGTGAT |
3.5. Plaque Assay
3.6. Output to Input Assay
3.7. Nanostring nCounter Assay
3.8. Crystal Violet Staining
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chu, R.L.; Post, D.E.; Khuri, F.R.; Van Meir, E.G. Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clin. Cancer Res. 2004, 10, 5299–5312. [Google Scholar] [CrossRef] [PubMed]
- Hermiston, T.W.; Kuhn, I. Armed therapeutic viruses: Strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 2002, 9, 1022–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rein, D.T.; Breidenbach, M.; Curiel, D.T. Current developments in adenovirus-based cancer gene therapy. Future Oncol. 2006, 2, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, W.N.; Bilsland, A.; Hardie, M.; Evans, T.R. Drug insight: Cancer cell immortality-telomerase as a target for novel cancer gene therapies. Nat. Clin. Pract. Oncol. 2004, 1, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Rancourt, C.; Piche, A.; Gomez-Navarro, J.; Wang, M.; Alvarez, R.D.; Siegal, G.P.; Fuller, G.M.; Jones, S.A.; Curiel, D.T. Interleukin-6 modulated conditionally replicative adenovirus as an antitumor/cytotoxic agent for cancer therapy. Clin. Cancer Res. 1999, 5, 43–50. [Google Scholar] [PubMed]
- Li, Y.; McCadden, J.; Ferrer, F.; Kruszewski, M.; Carducci, M.; Simons, J.; Rodriguez, R. Prostate-specific expression of the diphtheria toxin a chain (dt-a): Studies of inducibility and specificity of expression of prostate-specific antigen promoter-driven dt-a adenoviral-mediated gene transfer. Cancer Res. 2002, 62, 2576–2582. [Google Scholar] [PubMed]
- Hoti, N.; Chowdhury, W.; Hsieh, J.T.; Sachs, M.D.; Lupold, S.E.; Rodriguez, R. Valproic acid, a histone deacetylase inhibitor, is an antagonist for oncolytic adenoviral gene therapy. Mol. Ther. 2006, 14, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Shiyanov, P.; Bagchi, S.; Adami, G.; Kokontis, J.; Hay, N.; Arroyo, M.; Morozov, A.; Raychaudhuri, P. P21 disrupts the interaction between CDK2 and the e2f-p130 complex. Mol. Cell. Biol. 1996, 16, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Hurwitz, J.; Massague, J. Cell-cycle inhibition by independent CDK and PCNA binding domains in p21cip1. Nature 1995, 375, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Hoti, N.; Chowdhury, W.H.; Mustafa, S.; Ribas, J.; Castanares, M.; Johnson, T.; Liu, M.; Lupold, S.E.; Rodriguez, R. Armoring crads with p21/Waf-1 shrnas: The next generation of oncolytic adenoviruses. Cancer Gene Ther. 2010, 17, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Shiina, M.; Lacher, M.D.; Christian, C.; Korn, W.M. Rna interference-mediated knockdown of p21(waf1) enhances anti-tumor cell activity of oncolytic adenoviruses. Cancer Gene Ther. 2009, 16, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, D.; Ghosh, M.K.; Mal, A.; Harter, M.L. Inactivation of p21 by E1A leads to the induction of apoptosis in DNA-damaged cells. J. Virol. 2001, 75, 9844–9856. [Google Scholar] [CrossRef] [PubMed]
- Keblusek, P.; Dorsman, J.C.; Teunisse, A.F.; Teunissen, H.; van der Eb, A.J.; Zantema, A. The adenoviral E1A oncoproteins interfere with the growth-inhibiting effect of the CDK-inhibitor p21(CIP1/WAF1). J. Gen. Virol. 1999, 80, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, R.O.; Pintel, D.J. Replication of minute virus of mice in murine cells is facilitated by virally induced depletion of p21. J. Virol. 2012, 86, 8328–8332. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Knutson, E.; Kurosky, A.; Albrecht, T. Degradation of p21cip1 in cells productively infected with human cytomegalovirus. J. Virol. 2001, 75, 3613–3625. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and PCAN-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997, 11, 2090–2100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Scadden, D.T.; Crumpacker, C.S. Primitive hematopoietic cells resist HIV-1 infection via p21. J. Clin. Investig. 2007, 117, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Schuur, E.R.; Lim, H.Y.; Henderson, G.A.; Simons, J.W.; Henderson, D.R. Prostate attenuated replication competent adenovirus (ARCA) CN706: A selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997, 57, 2559–2563. [Google Scholar] [PubMed]
- Reich, N.C.; Sarnow, P.; Duprey, E.; Levine, A.J. Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 1983, 128, 480–484. [Google Scholar] [CrossRef]
- Pombo, A.; Ferreira, J.; Bridge, E.; Carmo-Fonseca, M. Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO J. 1994, 13, 5075–5085. [Google Scholar] [PubMed]
- O’Shea, C.C.; Johnson, L.; Bagus, B.; Choi, S.; Nicholas, C.; Shen, A.; Boyle, L.; Pandey, K.; Soria, C.; Kunich, J.; et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 2004, 6, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Yakimovich, A.; Gumpert, H.; Burckhardt, C.J.; Lutschg, V.A.; Jurgeit, A.; Sbalzarini, I.F.; Greber, U.F. Cell-free transmission of human adenovirus by passive mass transfer in cell culture simulated in a computer model. J. Virol. 2012, 86, 10123–10137. [Google Scholar] [CrossRef] [PubMed]
- DeWeese, T.L.; van der Poel, H.; Li, S.; Mikhak, B.; Drew, R.; Goemann, M.; Hamper, U.; DeJong, R.; Detorie, N.; Rodriguez, R.; et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001, 61, 7464–7472. [Google Scholar] [PubMed]
- Hoti, N.; Li, Y.; Chen, C.L.; Chowdhury, W.H.; Johns, D.C.; Xia, Q.; Kabul, A.; Hsieh, J.T.; Berg, M.; Ketner, G.; et al. Androgen receptor attenuation of AD5 replication: Implications for the development of conditionally replication competent adenoviruses. Mol. Ther. 2007, 15, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.P.; Balajee, A.S.; Bohr, V.A. The C-terminal domain of p21 inhibits nucleotide excision repair in vitro and in vivo. Mol. Biol. Cell 1999, 10, 2119–2129. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Höti, N.; Johnson, T.J.; Chowdhury, W.H.; Rodriguez, R. Loss of Cyclin-Dependent Kinase Inhibitor Alters Oncolytic Adenovirus Replication and Promotes More Efficient Virus Production. Cancers 2018, 10, 202. https://doi.org/10.3390/cancers10060202
Höti N, Johnson TJ, Chowdhury WH, Rodriguez R. Loss of Cyclin-Dependent Kinase Inhibitor Alters Oncolytic Adenovirus Replication and Promotes More Efficient Virus Production. Cancers. 2018; 10(6):202. https://doi.org/10.3390/cancers10060202
Chicago/Turabian StyleHöti, Naseruddin, Tamara Jane Johnson, Wasim H. Chowdhury, and Ronald Rodriguez. 2018. "Loss of Cyclin-Dependent Kinase Inhibitor Alters Oncolytic Adenovirus Replication and Promotes More Efficient Virus Production" Cancers 10, no. 6: 202. https://doi.org/10.3390/cancers10060202