Abstract
Tight regulation of gene transcription is essential for normal development, tissue homeostasis, and disease-free survival. Enhancers are distal regulatory elements in the genome that provide specificity to gene expression programs and are frequently misregulated in cancer. Recent studies examined various enhancer-driven malignant dependencies and identified different approaches to specifically target these programs. In this review, we describe numerous features that make enhancers good transcriptional targets in cancer therapy and discuss different approaches to overcome enhancer perturbation. Interestingly, a number of approved therapeutic agents, such as cyclosporine, steroid hormones, and thiazolidinediones, actually function by affecting enhancer landscapes by directly targeting very specific transcription factor programs. More recently, a broader approach to targeting deregulated enhancer programs has been achieved via Bromodomain and Extraterminal (BET) inhibition or perturbation of transcription-related cyclin-dependent kinases (CDK). One challenge to enhancer-targeted therapy is proper patient stratification. We suggest that monitoring of enhancer RNA (eRNA) expression may serve as a unique biomarker of enhancer activity that can help to predict and monitor responsiveness to enhancer-targeted therapies. A more thorough investigation of cancer-specific enhancers and the underlying mechanisms of deregulation will pave the road for an effective utilization of enhancer modulators in a precision oncology approach to cancer treatment.
Keywords:
enhancers; BET inhibitors; CDK7 inhibitors; HDAC inhibitors; transcription factors; eRNAs; cancer 1. Introduction
Cancer is a disease of aberrant transcription which is dependent on mechanisms enabling deregulated gene expression [1,2,3]. Enhancers are short genomic elements or clusters of elements, which are bound by tissue- or cell type-specific transcription factors (TFs) that activate target gene transcription in a distal and autonomous manner [4]. Soon after their discovery, enhancers were reported to drive differential transcriptional regulation in a more diverse and versatile manner compared to transcriptional regulation occurring (primarily) at proximal promoter regions [5]. Accordingly, it is not surprising that misregulation of these transcriptional hubs was linked to various diseases, including cancer [6,7,8]. For example, a chromosomal rearrangement in acute myeloid leukemia (AML) was found to bring an enhancer into the proximity of the oncogenic MDS1 and EVI1 complex locus (MECOM), precipitating the malignancy [9]. The amplification of enhancers was also shown to play a role in the pathophysiology of prostate cancer and neuroblastoma [10,11]. Furthermore, hijacking of enhancers led to the activation of the oncogenic Growth Factor Independent 1 family in medulloblastoma [12]. Additionally, reprogramming of the enhancer landscape in pancreatic cancer was reported to play a significant role in promoting a more aggressive phenotype [13,14,15]. Moreover, enhancers were implicated in therapy resistance in leukemia [16]. Accordingly, the eminent implication of enhancers in diseases led to the development of the term “enhanceropathies” and enhancer biology has become a focal point of interest when investigating novel therapeutic targets in cancer [17]. In this report, we review recent studies supporting the rationale of targeting enhancers in cancer. Additionally, we summarize the reported use of enhancer modulators in different cancer types. Finally, we discuss the challenges facing the use of enhancer modulators in the clinical setting.
2. Targeting Transcription Factor-Related Programs in Cancer
Sequence-specific binding of transcription factors (TF) underlies the selective activation of enhancers in different systems [18]. TFs provide a high degree of specificity in gene regulation by binding to their cognate DNA sequences across the genome to activate (or repress) transcription via recruitment of various co-activators, such as chromatin remodeling proteins and histone modifying enzymes [19,20]. Certain TFs have been identified to be lineage-specific and drive the differentiation of certain cellular states through the activation of different enhancer repertoires [19,21]. Moreover, it was reported that certain TFs, including the majority of tissue-specific TFs, display a larger number of binding sites at distal enhancers compared to proximal promoters [22]. Accordingly, agents specifically targeting the function of such transcription factors will, in turn, perturb the activity of the select set of enhancers controlled by the given TF.
Notably, direct manipulation of individual enhancer activity has recently been achieved via gene editing approaches such as CRISPR-Cas9. For example, fetal hemoglobin was effectively induced by disrupting the binding site of the TF GATA binding protein 1 (GATA1) at the upstream enhancer of the fetal hemoglobin repressor BAF chromatin remodeling complex subunit BCL11A (BCL11A) [23]. Additionally, using catalytically dead Cas9-Krüppel-associated box domain (dCas9-KRAB) to silence various enhancer constituents of the oncogenic sphingosine kinase 1 (SPHK1) led to a decrease in its levels, which was associated with attenuated proliferation and migration in hepatocellular carcinoma [24,25]. Recruitment of a trans-enhancer by a customized dCas9 was more recently shown to activate target gene transcription in various cancer cell lines [26]. While these approaches illustrate examples of direct manipulation of enhancers in certain contexts, they have limited applications as therapeutic options due to various reasons. Firstly, the lack of effective, safe and specific delivery options of the Cas9 system remains a major challenge in applying these machineries in therapy. One of the novel approaches currently under study to deliver the Cas9 system includes multistage delivery nanoparticle (MDNP), which still requires further validation [27]. Additionally, the use of permanent gene editing in humans faces numerous ethical complications. Most importantly, these approaches can only affect a single target, which is rarely sufficient to combat highly plastic malignancies. Furthermore, the ramifications of permanent changes in the genome sequence are poorly understood and may lead to irreversible adverse events.
While genetic manipulation of enhancer function remains challenging, an established clinically relevant approach to modulate enhancer activity is the perturbation or activation of the sequence-specific transcription factor(s) that are required for enhancer function. A primary example of such targeting is the perturbation or activation of steroid hormone receptors in various cancers, such as breast cancer [28,29,30], prostate cancer [31,32,33], and lymphomas [34,35]. For example, 70% of breast cancers are estrogen receptor-positive (ER+) and are, at least initially, highly responsive to endocrine therapy [36]. Estrogen receptor-alpha (ERα) is a master transcription factor in breast cancer which can be activated by estradiol. Estrogen binding to the ligand-binding domain of ERα leads to conformational changes which promote dimerization, subsequent binding to specific targets in the genome called estrogen response elements (EREs), and recruitment of co-activator proteins [37]. When ERα localization was investigated throughout the genome, it was quickly recognized that it rarely binds to promoter regions, but rather shows a tendency to localize to enhancer regions [38]. Interestingly, ERα was recently reported to nucleate phase-separated condensates at highly active enhancers [39], thereby promoting transcription at these extremely active hubs [40]. Thus, while endocrine therapy has been a central approach for treating a large number of breast cancer patients for over four decades, it has only recently been appreciated that the main molecular mechanisms by which tamoxifen and similar steroid hormone receptor antagonists exert their effects is by the modulation of enhancer activity. This principle is also applicable to other steroid hormone receptors, where (positively or negatively) targeting the enhancer function of the androgen receptor (AR) [41,42,43] or glucocorticoid receptor [44] has been shown to be very effective in other malignancies.
Other therapeutic agents that are used for other indications, such as the insulin sensitizing thiazolidinediones, also modulate the enhancer landscape by acting as agonists for the nuclear receptor Peroxisome Proliferator-Activated Receptor-γ (PPARG) [45]. In this case, treatment with rosiglitazone leads to the selective activation of PPARG-occupied enhancers. While thiazolidinediones were reported to have an inhibitory proliferative effect in hepatocellular and esophageal cancers, enhancer modulation by glitazones in cancer is still not very well studied [46,47,48]. Interestingly, a retrospective study observed a significant negative correlation of administration of thiazolidinediones and colorectal cancer, suggesting these agonists may have a protective role in preventing cancer [49]. Accordingly, those drugs which were originally developed for other indications may have a potential unlocked utility in some cancers. However, further investigation is needed to confirm the efficacy of these agents in this context.
While targeting nuclear receptors is one of the best examples of how directly perturbing transcription factor activity can be achieved, other frequently utilized therapeutic agents have a similar mechanism of action. For example, calcineurin inhibitors, which attenuate the calcium-dependent translocation of the Nuclear Factor of Activated T cells (NFAT), were shown to have growth inhibitory effects in various types of cancer, such as hepatocellular carcinoma, melanoma, and retinoblastoma [50,51,52]. NFAT was shown to elicit its effects at enhancer regions in blood vessel maturation [53] and function together with STAT3 at enhancers downstream of KRAS signaling in pancreatic cancer [54]. Thus, the use of calcineurin inhibitors will directly impact the activity of NFAT-driven enhancer programs and can be a promising approach in cancer therapy, especially in cases such as breast and pancreatic cancer where the NFAT pathway has been shown to be activated [45,55]. Conversely, cyclosporine has been implicated in increased risk for squamous cell carcinoma due to an increase of Activating Transcription Factor 3 (ATF3), which suppresses p53-induced senescence [56]. Thus, the context-specific effects of this drug should be closely considered before harnessing its enhancer-perturbing activity. Another approach to modulate the effects of a transcription factor can be by targeting its stability. For example, the Hypoxia Inducible Factor Alpha subunit (HIF1A) is a transcription factor that is known for its role in mediating the hypoxic response and it was shown to correlate with poorer prognosis in various types of cancer [57,58]. Enhancers also underlie the activity of HIF1A in modulating target gene expression [15,59,60,61]. Interestingly, topoisomerase I inhibitors were observed to inhibit the translation of HIF1A and may therefore function in part by this mechanism in the case of cancers where this factor plays a significant role [62].
As these drugs target particular transcriptional programs driven by the activity of a highly specific group of enhancers, their effectiveness has shed light on the benefits of targeting deregulated enhancer programs in specific disease contexts. Accordingly, targeting deregulated enhancer activity is, in fact, already an established paradigm and mainstay in the clinical treatment of cancer.
3. Perturbing Enhancer Activity by Therapeutic Agents and Inhibitors
Expansion of our understanding of complex processes controlling gene regulation has uncovered numerous novel targets that can (potentially) be therapeutically modulated in cancer. However, such exponential growth in knowledge rendered the task of identifying and investing in a select few effective and relatively safe transcriptional targets immensely challenging. An ideal transcriptional target in cancer therapy would necessarily exhibit certain attributes which can lead to a perceptible change in the quality of life, prognosis, and the therapeutic management of patients. To achieve this, such a target must be amenable to inhibition, preferably by small molecules that have good bioavailability at the target site with an acceptable half-life (i.e., good pharmacokinetic and pharmacodynamics properties). Importantly, the ideal target should also have a specificity that spares non-transformed cells, thereby avoiding or minimizing any potential unwanted side effects caused by perturbations of normal cellular processes in healthy tissue (adverse or severe adverse events). Additionally, this target should be indispensable (non-redundant) to cancer cells, rendering them highly dependent on such a target. Finally, this dependence should ideally be shared by all or a high percentage of the malignant cell population in a given patient. As some enhancers or enhancer programs exhibit these characteristics, they provide promising transcriptional targets (Figure 1).
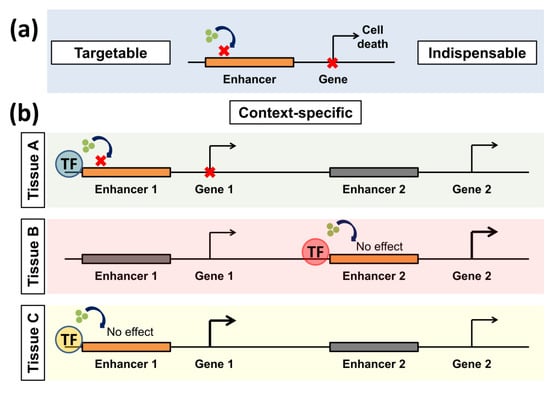
Figure 1.
Positive features rendering enhancers good transcriptional targets. (a) Enhancers can be pharmacologically manipulated using different small molecule inhibitors (indicated by green dots). They are also indispensable for cancer cells as they activate important oncogenes. (b) Enhancers are also context-specific. In this example, enhancers are activated by different transcription factors (TFs) in various tissues (A–C). The same inhibitor affects only a specific enhancer in a tissue if it is activated by a certain TF. Thus the illustrated inhibitor only affects Enhancer 1 in tissue A but not C. It also has no effect on the other active enhancers in tissue B. Gray enhancers are inactive, while orange ones are active. Bold arrows represent active transcription.
3.1. Enhancers Are Context-Specific and Indispensable for Cancer Cells
One of the major characteristics of a subgroup of enhancers is context-specificity. In general, enhancers direct lineage-specific transcriptional programs in a more predominant manner compared to promoters [63]. Around half of the enhancers identified in different tissues including brain, heart, ovaries, and placenta were tissue-specific [64]. Consistently, enhancers were the most distinctive identifying feature of tissue of origin upon analysis of hundreds of patient samples and human cell lines [65]. Interestingly, not only is there a distinct pattern of enhancer activation through tissues, but it was also observed that the interaction between active enhancers and their target genes are highly variable in various tissues as well [66]. Notably, interactions between enhancers and their target genes are variable in different systems and show even more tissue-specificity than differential activation of enhancers themselves, thus providing an additional layer of complexity.
In addition to having tissue-specificity, in order to be safely targetable, enhancers must exhibit specific activation in malignant cells compared to the tissue of origin. In this case, this activation should represent a new cancer cell-specific dependence that is not shared by healthy cells from the same tissue. Consistently, aberrant hypermethylation of enhancers in renal cancer cells led them to be more sensitive to the DNA methyltransferse (DNMT) inhibitor, decitabine, compared to healthy renal tissue [67]. More specifically, loss of the X-linked gene Lysine Demethylase 6A (KDM6A) resulted in a gender-specific aberrant activation of a set of enhancers leading to an aggressive phenotype of pancreatic cancer [68]. Importantly, inactivation of enhancers through Bromodomain and Extraterminal (BET) inhibitor treatment was effective in targeting this specific subtype of pancreatic cancer compared to other subtypes. This shows that enhancer specificity can extend to certain subtypes of cancer, adding a further layer of specificity and potentially increasing safety in targeting those elements.
3.2. Activity of Enhancers Can Be Pharmacologically Perturbed
Enhancers were shown to be specifically targetable by various small molecule inhibitors. Preferential dependence of enhancers on Bromodomain and Extraterminal (BET) proteins has been consistently reported in various cancer types such as lymphoma [69], ovarian cancer [70], breast cancer [71,72], pancreatic cancer [68,73], leukemia [74], multiple myeloma, and glioblastoma [74,75]. Other modulators with reported efficacy on enhancers include inhibitors of the transcriptional cyclin dependent kinases-7 (CDK7) and -9 (CDK9).
3.2.1. Epigenetic Modulators
Epigenetic regulation enables cells to control gene transcription in a manner complementary to sequence-specific transcription factor-based mechanisms. Such regulatory mechanisms include post-translational modification of histones, DNA methylation, nucleosome remodeling, and non-coding RNAs (ncRNAs) [76]. Histone marks do not act independently of one another, but rather cooperate to control gene transcription in what is referred to as “histone crosstalk” [77]. Eminent factors in the epigenetic machinery are so-called epigenetic “readers”, which recognize specific histone marks and recruit additional effectors [78]. An extensively studied example is the BET family of proteins, which each contain two bromodomains that can interact with acetylated lysine residues on target proteins via a hydrophobic pocket, thereby endowing BET proteins with the ability to recognize acetyl marks on chromatin [79]. JQ1 is a thienodiazepine that displaces the BET family member Bromodomain containing 4 (BRD4) from acetylated lysines by forming hydrogen bonds with a conserved asparagine residue that is situated in the hydrophobic pocket of BRD4 [80]. Many other BET inhibitors have also been developed, such as I-BET151, I-BET762, and OTX-015 [80,81,82]. In Diffuse Large B-Cell Lymphoma, BET inhibitors showed a marked effect on a subset of enhancers, termed super enhancers, that are highly enriched with BRD4 [69]. Super enhancers (SEs) were first identified as major drivers of gene expression that are highly enriched with transcription factor binding sites and include clusters of highly active distal regulatory elements [75,83]. SEs were observed to drive lineage-specific programs in various systems, such as epithelial differentiation, mesenchymal multipotency, and estrogen-dependent mammary gland malignancy, and showed sensitivity to BET inhibition [75,84,85,86]. Consistently, treating ovarian cancer cells with BET inhibitors diminished the activity of a super enhancer activating the chemoresistance-related aldehyde dehydrogenase and led to increased sensitivity to cisplatin treatment [70]. Additionally, treating various sensitive colorectal cancer cells with BET inhibitors attenuated the activity of enhancers gained in cancer compared to normal crypts [87]. While different super enhancer programs were identified in various subtypes of ependymomas, a general sensitivity to BET inhibition was reported in ependymoma cells [88]. The same pattern of activation of distinct BET-dependent super enhancers was also reported in chronic lymphocytic leukemia [89]. Enhancers driving the transcription of receptor tyrosine kinases that play a fundamental role in gastrointestinal stromal tumors have also shown dependence on BET family members [90].
Other important members of the epigenetic machinery include writers which act by selectively adding chemical moieties to a specific histone residue. Histone acetyltransferases (HATs), such as p300 and CREB-binding protein (CBP), transfer an acetyl group from acetyl-CoA to histone tails [91]. Inhibiting HATs in pancreatic cancer affected the activation of a certain subset of enhancers that are enriched by the Wnt-signaling transcription factor, Transcription Factor 7 Like 2 (TCF7L2) [86]. Furthermore, the Polycomb Repressive Complex-1 (PRC1) and -2 (PRC2) are extensively studied complexes which mediate monoubiquitination of H2A at lysine 119 (H2Aub1) and tri-methylation of histone 3 lysine 27 (H3K27me3), respectively [92]. As H2K27me3 is a histone mark which is highly associated with gene inactivation [93,94], targeting constituents of the PRC complex led to a specific de-depression of a specific set of enhancers in leukemia [95]. Consistently, targeting the catalytic subunit of the PRC2 complex, Enhancer of Zeste Homolog 2 (EZH2), led to the de-depression of enhancers controlling the pro-apoptotic B cell lymphoma-2 like 11 (BIM), thereby mediating apoptosis in breast cancer cells [96].
Other classes of important epigenetic factors include “erasers”, enzymes that remove histone marks [97]. This includes histone deacetylases (HDACs), which mediate the removal of lysine acetylation and consist of multiple classes that can also mediate de-acetylation of non-histone proteins [98]. HDAC inhibitors were found to affect the enhancer landscape in colorectal, pancreatic, and breast cancer [96,99,100]. While methylation was previously considered to be an irreversible modification, Lysine-Specific Demethylase 1 (LSD1, also called KDM1A) was identified in 2002 as a selective mediator of the de-methylation of histone 3 lysine 4 [101,102]. Mono-methylation and tri-methylation of histone 3 lysine 4 (H3K4me1 and H3K4me3) are known marks for gene activation at distal and proximal regulatory regions, respectively [76,103]. LSD1 inhibitors affect a specific subset of enhancers controlling differentiation in acute myeloid leukemia by disrupting their interaction with the SNAG-domain transcription repressor GFI1 [104]. LSD1 has also been shown to influence enhancer activity in a number of other systems including embryonic stem cell differentiation [105], androgen receptor function in prostate cancer [106,107], and ERα activity in breast cancer [108]. Altogether, epigenetic modulators provide a variety of targets which can be manipulated to modulate the cancer-specific enhancer landscape and affect transcriptional programs. While current research is largely focused on BET inhibitors and their role in affecting enhancers, many other epigenetic inhibitors may potentially also be used in the context of enhancer activity manipulation as further mechanisms and contexts are better defined.
3.2.2. Cyclin-Dependent Kinase Inhibitors
The recruitment of RNA Polymerase II (RNA Pol II) to the proximal promoter enables the initiation of transcription, which is signified by the phosphorylation of serine 5 within the heptapeptide repeats of the carboxy-terminal domain (CTD) and the subsequent capping of nascent RNA [109,110]. Pol II is frequently temporarily paused by the Negative Elongation Factor (NELF) and DRB-sensitivity Inducing Factor (DSIF) within the first 100 nucleotides after the transcription start site (TSS) [109,111,112]. Thereby, this promoter proximal pausing has been regarded as a crucial rate-limiting step for gene transcription in metazoans [113]. To proceed to productive elongation, the Positive Transcription Elongation Factor-b (P-TEFb) phosphorylates Pol II at serine 2 of the CTD as well as components of both the NELF and DSIF complexes [114,115] via its catalytic subunit Cyclin-Dependent Kinase-9 (CDK9) bound to the cognate cyclin T1 [116]. These phosphorylation events release Pol II from promoter proximal pausing and allow transcription elongation until polyadenylation sequences are transcribed, which leads to the cleavage and subsequent polyadenylation of mRNAs [117]. Notably, inhibition of CDK9 not only attenuates transcription elongation of the pre-mRNA, but also decreases eRNA production at enhancer regions [118]. Consistent with complementary functions in controlling enhancer function, the combination of CDK9 inhibition along with BET inhibitors has shown enhanced effects in both AML [119] and malignant rhabdoid tumor cells [120]. As a monotherapy, CDK9 inhibitors were observed to highly inhibit the expression of genes associated with super enhancers, such as MYC [121]. In chordoma, a highly aggressive tumor of the bone, inhibition of CDK9 and CDK7 has been reported to be highly effective [122].
As part of the TFIIH complex, CDK7 plays an important role in gene transcription via phosphorylation of the Pol II CTD at Ser5 [123,124]. It was reported that phosphorylation of the CTD by CDK7 leads to the dissociation of the CTD with DNA and the initiation of transcription [125]. CDK7 also plays a dual role in controlling cell cycle progression by phosphorylating and activating CDK1 and CDK2 [126]. Inhibition of CDK7 by the covalent inhibitor, THZ1, was found to be highly toxic to cancer cells, presumably by specific inactivation of super enhancers [127]. Indeed, super enhancers controlling the MYCN proto-oncogene were selectively inactivated by THZ1 in neuroblastoma [128]. Interestingly, several reports followed observing a selective perturbation of super enhancer programs by inhibition of CDK7 in small cell lung cancer [129], triple-negative breast cancer [130], ovarian cancer [131], esophageal carcinoma [132], melanoma [133], gliobastoma multiforme [134], and pancreatic cancer [135]. However, inhibition of CDK7 was reported to increase characteristics associated with metastasis in colorectal cancer cells [136]. THZ1 was found to attenuate the normal transition of the various stages of transcription starting from initiation into elongation [137]. Conversely, while THZ1 appears to preferentially affect super enhancer-associated genes, this effect does not appear to be due to altered RNA Pol II activity directly at the enhancers themselves [138]. Moreover, a more recent report suggests that the effects of THZ1 on super enhancer-associated genes may, in fact, be due to the off-target inhibition of CDK12 and CDK13, rather than CDK7 [139]. Thus, further studies to understand the exact mechanism of selective attenuation of super enhancer activation are necessary before the use of CDK7 (or CDK12/13) inhibitors can be precisely tested in the clinical setting on a mechanistic basis. Currently, two early-phase clinical studies are ongoing to investigate the use of CDK7 inhibitors in patients with advanced solid malignancies (NCT03363893, NCT03134638).
In addition to the previously mentioned regulators, the Mediator complex plays a crucial role in transcriptional regulation [140,141]. Mediator is a large multi-subunit complex that plays a crucial role in the assembly and activation of the pre-initiation complex (PIC) by forming a bridge between various sequence-specific transcription factors and components of the PIC [142]. In addition to its important role at gene promoters, Mediator is reported to connect initiating promoters with active distal enhancers through chromatin loop formation [143]. The first evidence of chromatin loop formation where a distal region affected the transcription of a target gene promoter was reported in 1984 by Dunn et al. [144] in bacteria. Approximately 20 years later, cohesin, which also plays a central role in sister-chromatid adhesion, was revealed to orchestrate the formation of DNA loops with the help of the insulator, CCTC-Binding Factor (CTCF), and the cohesin loader, Nipped-B-Like (NIPBL) [145,146,147]. Mediator was found to bind cohesin and NIPBL to bring active enhancers and promoters into close proximity [143]. As Mediator is composed of approximately 30 subunits, it can have different conformations [148]. One conformation includes the kinase module containing cyclin-dependent kinase 8 (CDK8), which does not appear to directly phosphorylate the RNA Pol II CTD, but was shown to have more preference toward affecting active enhancers [149]. In contrast to BET and CDK7 inhibition, CDK8 inhibitors have been reported to have an activating effect on super enhancers in leukemic cells [150]. Interestingly, given the proposed dose-dependent function of these enhancers, leukemic cells were still impaired in their growth following treatment with a CDK8 inhibitor. Additionally, CDK8 inhibitors were reported to be crucial in mediating the transcriptional effects of HIF1A [151] as well as beta-catenin in colorectal cancer [152]. Accordingly, CDK8 inhibition can lead to changes in the enhancer landscape by various mechanisms. A number of the described targets which modulate enhancer activity are illustrated and summarized in Figure 2.
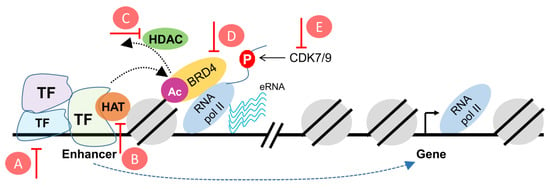
Figure 2.
Schematic representation of putative targets to reprogram the enhancer landscape in cancer. (A) modulators of transcription factors, (B) HAT inhibitors, (C) HDAC inhibitors, (D) BET inhibitors, (E) CDK7/9 inhibitors. HAT: Histone acetyltransferase; HDAC: Histone deacetylase; BET: Bromodomain and extraterminal; CDK: Cyclin-dependent kinases.
4. Challenges Facing the Utility of Enhancer Modulators in the Clinical Setting
While targeting transcriptional enhancers is still under investigation, compensatory resistance mechanisms upon inhibition of active enhancers have already been described. For example, the BET inhibitor JQ1 was reported to induce resistance mediated by transcriptional activation in bromodomain-independent pathways in castration-resistant prostate cancer [153]. Interestingly, this resistance uncovered an alternative dependency on CDK9-mediated activation of androgen receptor signaling. In pancreatic cancer, upregulation of the GLI Family Zinc Finger 2 (GLI2) was found to enable resistance to BET inhibition [154]. In leukemia, resistance to BET inhibition was partly caused by an increase in Wnt-signaling pathway activity [155,156]. Interestingly, while MAPK/ERK kinase inhibition (MEKi) sensitized colorectal cancer cells to BET inhibition, BET inhibitors sensitized MEKi-resistant cells in breast cancer [71,157]. Similarly, inhibitors of the Nuclear Factor Kappa-light-chain-enhancer of activated B cells (NFKB) pathway led to sensitization to BET inhibitors in uveal melanoma [158]. Coupling the inhibition of BET-dependent enhancers with targeting of other transcriptome regulating axes such as E2F-dependent promoters was suggested as an effective approach to target gene transcription in multiple myeloma [159]. Consequently, it is not unlikely that using enhancer modulators will reveal various challenges for their use in the clinical setting. These will include the development of resistance in addition to the difficulty in predicting responsiveness.
Notably, while the context-specific properties of enhancers can be leveraged in precise targeting of particular enhancer programs, it will also lead to inconsistent performances in various cancer entities. Accordingly, it is not surprising that enhancer modulators may show highly promising effects in one cancer type and fail in another. For example, inhibition of EZH2 was reported to be effective in lymphomas where aberrant histone methylation is caused by mutation in this particular regulator [160]. On the other hand, the depletion of EZH2 activity caused by histone 3.3 (H3.3) mutations in diffuse intrinsic pontine gliomas are major precipitants of the disease [161]. Additionally, malignant peripheral nerve sheath tumor (MPNST) is largely caused by mutations in the Ras signaling suppressor neurofibromin 1 (NF1) leading to activation of Ras signaling and usually accompanied by mutations in p53 and the PRC2 complex members, SUZ12 and EED, which lead to a loss of H3K27me3 [162,163,164]. Thus, treatment with EZH2 inhibitors in certain contexts (such as neurofibromatosis type 1) may actually exacerbate or precipitate tumorigenesis or tumor progression. This apparent discrepancy is very likely due to the importance of a balanced level of histone modifications to ensure homeostasis, as well as the signaling, genetic, and epigenetic context-dependency. Another example includes the use of HDAC inhibitors in various types of cancer. Multiple HDAC inhibitors have been approved by the United States Food and Drug Administration (FDA) for use in indications such as relapsed and refractory cutaneous T-cell lymphoma, peripheral T cell lymphoma, and multiple myeloma [165]. Conversely, clinical trials showed limited effects of HDAC inhibitors in other cancer types, such as head and neck [166], breast [167], ovarian [168], and pancreatic cancer [169]. However, better effects were observed in some cases when HDAC inhibitors were used in combination with other therapies such as BET inhibitors [73], chemoradiation [170], and VEGF inhibitors [171]. These observations underscore the complexity of the clinical use of enhancer modulators and the major challenges facing these agents. They also support the need for a deeper understanding of aberrant enhancer programming in different malignancies and how this relates to the effectiveness of enhancer-targeting therapies.
As described above, a major challenge in the clinical utilization of enhancer-targeting approaches to cancer therapy is the identification of potentially responsive patients for proper stratification. A breakthrough in this may be the use of so-called “eRNAs” (enhancer ribonucleic acids) as clinical pathologic indicators of active enhancer programs in tumor cells. Although distal intergenic enhancer regions were not previously thought to be transcribed, it has since been shown that transcription occurs at these regions in contradiction to the general trends of energy conservation inside the cell [172]. The functions and mechanisms of the resulting eRNA products are still not fully elucidated [173]. However, eRNAs appear to provide excellent markers of enhancer activity that likely outperform information on the occupancy of transcription activators or histone marks [42]. Functionally, eRNAs were reported to augment gene transcription as their knockdown led to decreased target gene transcription [174,175]. Furthermore, chromatin loop formation and eRNA production were reported to precede transcription of the mRNA [176] and, more recently, eRNAs were reported to promote the formation of phase-separated nuclear interchromatin granules associated with actively transcribed genes [39]. Irrespective of their function, we suggest that these products may provide particularly useful clinical markers for predicting and monitoring therapeutic responsiveness and resistance (Figure 3).
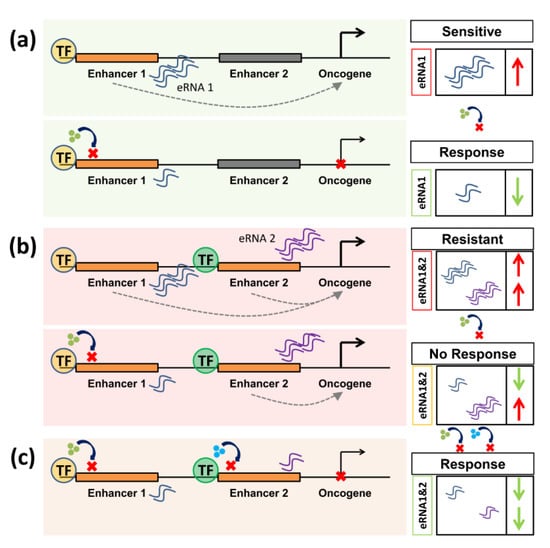
Figure 3.
Enhancer RNAs (eRNAs) are putative biomarkers for responsiveness and resistance in perturbation of enhancer activity. (a) In a responsive context, inhibiting an enhancer leads to a decrease in the activity of oncogenic target genes. In this case, high levels of eRNA can predict responsiveness to a specific inhibitor by providing a direct readout of enhancer activity. (b) In case of resistance, compensating mechanisms such as the activation of a different enhancer program can occur. Thereby, high levels of different eRNAs can predict resistance to a certain therapy. (c) To re-sensitize cells, compensatory mechanisms should also be targeted to ensure therapeutic success.
This is of particular interest as eRNAs were reported to be highly enriched at tissue-specific enhancers [177]. Indeed, the eRNA CCAT1 was proposed as a therapeutic biomarker that can predict responsiveness to BET inhibition [178]. Interestingly, an enhancer identified in prostate cancer, which is associated with the gene encoding prostate-specific antigen (PSA) and the resulting eRNA, was shown to play a central role in controlling gene transcription in prostate cancer cells [179]. Additionally, Kaczkowski et al. [180] identified 90 eRNAs that are generally upregulated in cancer cells upon screening over 200 cell lines and approximately 300 primary samples. Identification of eRNAs has been made feasible due to the development of techniques such as global run-on sequencing (GRO-seq) [181], transient transcriptome sequencing (TT-seq) [182], precision nuclear run-on sequencing (PRO-seq) [183], and, more recently, chromatin run-on and sequencing (ChRO-seq) [184]. Notably, length-extension ChRO-seq enables the detection of nascent RNA from tissue samples that were stored for longer periods of up to 30 years. Thus, current technologies allow us to more easily identify eRNAs from patient samples irrespective of sample quality, further enabling a potential utilization of eRNAs as enhancer biomarkers. Importantly, manipulation of these shortly-lived transcripts by cell-permeable synthetic antisense oligonucleotides (ASOs) [185] can also affect target gene transcription and may be an effective mechanism of perturbing enhancer activity [186].
Notably, other technical challenges face the elucidation of the mechanisms of enhancer functions and their targeting. Enhancers are usually identified using highly complex bioinformatic analyses that are not always accessible to clinicians and scientists alike. Identification of important enhancers has also been accompanied by the emergence of different subclasses of enhancers. Since the recent identification of super enhancers in 2013 [6,75], approximately 300 scientific papers discussing this subclass have been published. Another class of enhancers called “stretch enhancers” are sometimes used interchangeably with super enhancers [187,188]. This comparison is, however, somewhat inaccurate as studies indicate that stretch enhancers only meet the requirement of spanning long stretches of DNA but, unlike super enhancers, are not necessarily rich with transcription factors or cell-specific [189]. An additional subclass includes “shadow enhancers”, which are a group of “secondary” enhancers that are superfluous and redundant to an active enhancer, thereby ensuring the precision of gene transcriptional regulation [190]. Such a concept, which was first identified in Drosophila, has also been reported in mammals [191]. This led to the sometimes imprecise use of the term “shadow enhancers” to describe typical enhancers, which are not necessarily supportive of other enhancers and may play decisive roles in tissue- and cancer-specific gene regulation in their own right. A clearer definition of these new classifications will significantly help in a better and more precise understanding of enhancer activity and its modulation.
Another crucial hurdle facing the investigation of the role of enhancers in transcriptional activation is the complexity of defining the target genes of each enhancer. In the cell, targets of enhancers are not necessarily in close linear (genomic) proximity and can be separated by many unaffected genes [192]. As previously reported, interactions between enhancers and their target genes are variable in different systems and can show more tissue specificity than differential activation of enhancers themselves [66]. Chromatin conformation capture assays to detect interactions between cis-regulatory elements were first established in 2002 and have been followed by many techniques that extended our knowledge about the interactions between enhancers and their target promoters [193,194,195,196]. As these techniques are difficult to perform in patient samples and are generally not very cost-effective, identification of target genes in a concise manner in patient samples remains challenging.
5. Conclusions and Future Directions
Aberrant transcriptional regulation is one of the characteristics of malignancy which can be most efficiently and specifically manipulated through enhancer elements. A greater breadth of knowledge about activated enhancers or super enhancers, interconnected with dependencies and biomarkers, may play a significant role in the optimization of therapy for patients suffering from cancer and other diseases. In conclusion, enhancers exhibit many attributes of an ideal transcriptional target and are highly promising to be leveraged in cancer therapy and management. This is due to the fact that they are targetable, specific, and indispensable. They also frequently produce products (eRNA) that may potentially be utilized as predicative biomarkers and/or for monitoring therapeutic effectiveness. However, the targeting of compensatory mechanisms in response to their modulation should be considered as well. Given the clear significance of targeting enhancers in cancer, more studies are needed to further expand the currently available agents modulating the activity of these extremely important transcription targets.
As newer technologies to modulate targets become available in the future, our abilities to manipulate gene expression programs in cancer would be exponentially expanded. Our group and others [14,15] uncovered a role of the transcription factor deltaNp63 as a major driver of a more aggressive subtype of pancreatic cancer (squamous subtype) by activating a squamous enhancer/super enhancer program. The fact that deltaNp63 overexpression was sufficient to activate the squamous program suggests that this transcription factor may be an Achilles heel in this particular subtype [14]. DeltaNp63 was reported to be highly expressed in cancers and molecular subtypes of cancer that are squamous or basal in their nature, such as breast, head and neck, lung, and esophageal carcinoma [197,198,199,200]. We suggest that these findings may serve as a basis and rationale for the next step in precision oncology and the design of future basket trials where the effect of targeting deltaNp63 and its downstream activated enhancers can be investigated in malignancies that are divergent in origin but similar in the enhancer programs that drive their identity and aggressiveness.
Author Contributions
F.H.H. and S.A.J. conceived the ideas for this work and wrote this manuscript. Portions of this manuscript were included in the doctoral dissertation of F.H.H.
Funding
This research received no external funding.
Acknowledgments
We would like to acknowledge Zeynab Najafova, Xin Wang, and Ana Patricia Kutschat for numerous helpful conversations and Volker Ellenrieder and Elisabeth Hessmann for providing a central framework for understanding the complexities of clinical translation of basic research findings.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Suzuki, A.; Makinoshima, H.; Wakaguri, H.; Esumi, H.; Sugano, S.; Kohno, T.; Tsuchihara, K.; Suzuki, Y. Aberrant transcriptional regulations in cancers: Genome, transcriptome and epigenome analysis of lung adenocarcinoma cell lines. Nucleic Acids Res. 2014, 42, 13557–13572. [Google Scholar] [CrossRef]
- Lu, B.; Klingbeil, O.; Tarumoto, Y.; Somerville, T.D.D.; Huang, Y.H.; Wei, Y.; Wai, D.C.; Low, J.K.K.; Milazzo, J.P.; Wu, X.S.; et al. A Transcription Factor Addiction in Leukemia Imposed by the MLL Promoter Sequence. Cancer Cell 2018, 34, 970–981.e8. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Toohey, M.G.; Morley, K.L.; Peterson, D.O. Multiple hormone-inducible enhancers as mediators of differential transcription. Mol. Cell. Biol. 1986, 6, 4526–4538. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Vahedi, G.; Kanno, Y.; Furumoto, Y.; Jiang, K.; Parker, S.C.; Erdos, M.R.; Davis, S.R.; Roychoudhuri, R.; Restifo, N.P.; Gadina, M.; et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 2015, 520, 558–562. [Google Scholar] [CrossRef]
- Sun, W.; Yao, S.; Tang, J.; Liu, S.; Chen, J.; Deng, D.; Zeng, C. Integrative analysis of super enhancer SNPs for type 2 diabetes. PLoS ONE 2018, 13, e0192105. [Google Scholar] [CrossRef]
- Groschel, S.; Sanders, M.A.; Hoogenboezem, R.; de Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; van der Velden, V.H.J.; Havermans, M.; Avellino, R.; van Lom, K.; et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef]
- Takeda, D.Y.; Spisak, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szallasi, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e3. [Google Scholar] [CrossRef]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E.; et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 2018, 50, 515–523. [Google Scholar] [CrossRef]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stutz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20. [Google Scholar] [CrossRef]
- Somerville, T.D.D.; Xu, Y.; Miyabayashi, K.; Tiriac, H.; Cleary, C.R.; Maia-Silva, D.; Milazzo, J.P.; Tuveson, D.A.; Vakoc, C.R. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep. 2018, 25, 1741–1755.e7. [Google Scholar] [CrossRef]
- Hamdan, F.H.; Johnsen, S.A. DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc. Natl. Acad. Sci. USA 2018, 115, E12343–E12352. [Google Scholar] [CrossRef]
- Knoechel, B.; Roderick, J.E.; Williamson, K.E.; Zhu, J.; Lohr, J.G.; Cotton, M.J.; Gillespie, S.M.; Fernandez, D.; Ku, M.; Wang, H.; et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 364–370. [Google Scholar] [CrossRef]
- Smith, E.; Shilatifard, A. Enhancer biology and enhanceropathies. Nat. Struct. Mol. Biol. 2014, 21, 210–219. [Google Scholar] [CrossRef]
- Palstra, R.J.; Grosveld, F. Transcription factor binding at enhancers: Shaping a genomic regulatory landscape in flux. Front. Genet. 2012, 3, 195. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef]
- Najafova, Z.; Tirado-Magallanes, R.; Subramaniam, M.; Hossan, T.; Schmidt, G.; Nagarajan, S.; Baumgart, S.J.; Mishra, V.K.; Bedi, U.; Hesse, E.; et al. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017, 45, 127–141. [Google Scholar] [CrossRef]
- Lan, X.; Witt, H.; Katsumura, K.; Ye, Z.; Wang, Q.; Bresnick, E.H.; Farnham, P.J.; Jin, V.X. Integration of Hi-C and ChIP-seq data reveals distinct types of chromatin linkages. Nucleic Acids Res. 2012, 40, 7690–7704. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nature Med. 2019. [Google Scholar] [CrossRef]
- Tsang, F.H.; Law, C.T.; Tang, T.C.; Cheng, C.L.; Chin, D.W.; Tam, W.V.; Wei, L.; Wong, C.C.; Ng, I.O.; Wong, C.M. Aberrant Super-Enhancer Landscape in Human Hepatocellular Carcinoma. Hepatology 2019. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Xu, X.; Gao, J.; Dai, W.; Wang, D.; Wu, J.; Wang, J. Gene activation by a CRISPR-assisted trans enhancer. eLife 2019, 8, e45973. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, K.; Wang, C.; Zhang, Z.; Zheng, C.; Zhao, Y.; Zheng, Y.; Liu, C.; An, Y.; Shi, L.; et al. Multistage Delivery Nanoparticle Facilitates Efficient CRISPR/dCas9 Activation and Tumor Growth Suppression In Vivo. Adv. Sci. (Weinh) 2018, 6, 1801423. [Google Scholar] [CrossRef]
- Ward, H.W. Anti-oestrogen therapy for breast cancer: A trial of tamoxifen at two dose levels. Br. Med. J. 1973, 1, 13–14. [Google Scholar] [CrossRef]
- Moseson, D.L.; Sasaki, G.H.; Kraybill, W.G.; Leung, B.S.; Davenport, C.E.; Fletcher, W.S. The use of antiestrogens tamoxifen and nafoxidine in the treatment of human breast cancer in correlation with estrogen receptor values. A phase II study. Cancer 1978, 41, 797–802. [Google Scholar] [CrossRef]
- Manni, A.; Trujillo, J.E.; Marshall, J.S.; Brodkey, J.; Pearson, O.H. Antihormone treatment of stage IV breast cancer. Cancer 1979, 43, 444–450. [Google Scholar] [CrossRef]
- Labrie, F.; Dupont, A.; Belanger, A.; Lefebvre, F.A.; Cusan, L.; Raynaud, J.P.; Husson, J.M.; Fazekas, A.T. New hormonal therapy in prostate cancer: Combined use of a pure antiandrogen and an LHRH agonist. Horm. Res. 1983, 18, 18–27. [Google Scholar] [CrossRef]
- Moguilewsky, M.; Fiet, J.; Tournemine, C.; Raynaud, J.P. Pharmacology of an antiandrogen, anandron, used as an adjuvant therapy in the treatment of prostate cancer. J. Steroid Biochem. 1986, 24, 139–146. [Google Scholar] [CrossRef]
- Soloway, M.S. Newer methods of hormonal therapy for prostate cancer. Urology 1984, 24, 30–38. [Google Scholar]
- Yamamoto, K.R.; Stampfer, M.R.; Tomkins, G.M. Receptors from glucocorticoid-sensitive lymphoma cells and two clases of insensitive clones: Physical and DNA-binding properties. Proc. Natl. Acad. Sci. USA 1974, 71, 3901–3905. [Google Scholar] [CrossRef]
- Gehring, U.; Mohit, B.; Tomkins, G.M. Glucocorticoid action on hybrid clones derived from cultured myeloma and lymphoma cell lines. Proc. Natl. Acad. Sci. USA 1972, 69, 3124–3127. [Google Scholar] [CrossRef]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S.M. Treatment of estrogen receptor-positive breast cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006, 38, 1289–1297. [Google Scholar] [CrossRef]
- Nair, S.J.; Yang, L.; Meluzzi, D.; Oh, S.; Yang, F.; Friedman, M.J.; Wang, S.; Suter, T.; Alshareedah, I.; Gamliel, A.; et al. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat. Struct. Mol. Biol. 2019, 26, 193–203. [Google Scholar] [CrossRef]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef]
- Makkonen, H.; Kauhanen, M.; Paakinaho, V.; Jaaskelainen, T.; Palvimo, J.J. Long-range activation of FKBP51 transcription by the androgen receptor via distal intronic enhancers. Nucleic Acids Res. 2009, 37, 4135–4148. [Google Scholar] [CrossRef]
- Wang, D.; Garcia-Bassets, I.; Benner, C.; Li, W.; Su, X.; Zhou, Y.; Qiu, J.; Liu, W.; Kaikkonen, M.U.; Ohgi, K.A.; et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Pihlajamaa, P.; Sahu, B.; Lyly, L.; Aittomaki, V.; Hautaniemi, S.; Janne, O.A. Tissue-specific pioneer factors associate with androgen receptor cistromes and transcription programs. EMBO J. 2014, 33, 312–326. [Google Scholar] [CrossRef] [PubMed]
- McDowell, I.C.; Barrera, A.; D’Ippolito, A.M.; Vockley, C.M.; Hong, L.K.; Leichter, S.M.; Bartelt, L.C.; Majoros, W.H.; Song, L.; Safi, A.; et al. Glucocorticoid receptor recruits to enhancers and drives activation by motif-directed binding. Genome Res. 2018, 28, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Quang, C.T.; Leboucher, S.; Passaro, D.; Fuhrmann, L.; Nourieh, M.; Vincent-Salomon, A.; Ghysdael, J. The calcineurin/NFAT pathway is activated in diagnostic breast cancer cases and is essential to survival and metastasis of mammary cancer cells. Cell Death Dis. 2015, 6, e1658. [Google Scholar] [CrossRef]
- Takashima, T.; Fujiwara, Y.; Higuchi, K.; Arakawa, T.; Yano, Y.; Hasuma, T.; Otani, S. PPAR-gamma ligands inhibit growth of human esophageal adenocarcinoma cells through induction of apoptosis, cell cycle arrest and reduction of ornithine decarboxylase activity. Int. J. Oncol. 2001, 19, 465–471. [Google Scholar] [PubMed]
- Fujii, D.; Yoshida, K.; Tanabe, K.; Hihara, J.; Toge, T. The ligands of peroxisome proliferator-activated receptor (PPAR) gamma inhibit growth of human esophageal carcinoma cells through induction of apoptosis and cell cycle arrest. Anticancer Res. 2004, 24, 1409–1416. [Google Scholar] [PubMed]
- Motomura, W.; Takahashi, N.; Nagamine, M.; Sawamukai, M.; Tanno, S.; Kohgo, Y.; Okumura, T. Growth arrest by troglitazone is mediated by p27Kip1 accumulation, which results from dual inhibition of proteasome activity and Skp2 expression in human hepatocellular carcinoma cells. Int. J. Cancer 2004, 108, 41–46. [Google Scholar] [CrossRef]
- Liao, K.F.; Lin, C.L.; Lai, S.W. Association between colorectal cancer and thiazolidinediones administration in a case-control study. BioMedicine 2019, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: Regulation and function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef]
- Wang, S.; Kang, X.; Cao, S.; Cheng, H.; Wang, D.; Geng, J. Calcineurin/NFATc1 pathway contributes to cell proliferation in hepatocellular carcinoma. Dig. Dis. Sci. 2012, 57, 3184–3188. [Google Scholar] [CrossRef] [PubMed]
- Perotti, V.; Baldassari, P.; Bersani, I.; Molla, A.; Vegetti, C.; Tassi, E.; Dal Col, J.; Dolcetti, R.; Anichini, A.; Mortarini, R. NFATc2 is a potential therapeutic target in human melanoma. J. Invest. Dermatol. 2012, 132, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Norrmen, C.; Ivanov, K.I.; Cheng, J.; Zangger, N.; Delorenzi, M.; Jaquet, M.; Miura, N.; Puolakkainen, P.; Horsley, V.; Hu, J.; et al. FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J. Cell Biol. 2009, 185, 439–457. [Google Scholar] [CrossRef]
- Baumgart, S.; Chen, N.M.; Siveke, J.T.; Konig, A.; Zhang, J.S.; Singh, S.K.; Wolf, E.; Bartkuhn, M.; Esposito, I.; Hessmann, E.; et al. Inflammation-induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer Discov. 2014, 4, 688–701. [Google Scholar] [CrossRef]
- Buchholz, M.; Schatz, A.; Wagner, M.; Michl, P.; Linhart, T.; Adler, G.; Gress, T.M.; Ellenrieder, V. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006, 25, 3714–3724. [Google Scholar] [CrossRef]
- Wu, X.; Nguyen, B.C.; Dziunycz, P.; Chang, S.; Brooks, Y.; Lefort, K.; Hofbauer, G.F.; Dotto, G.P. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature 2010, 465, 368–372. [Google Scholar] [CrossRef]
- Baba, Y.; Nosho, K.; Shima, K.; Irahara, N.; Chan, A.T.; Meyerhardt, J.A.; Chung, D.C.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am. J. Pathol. 2010, 176, 2292–2301. [Google Scholar] [CrossRef]
- Wu, Y.; Yun, D.; Zhao, Y.; Wang, Y.; Sun, R.; Yan, Q.; Zhang, S.; Lu, M.; Zhang, Z.; Lu, D.; et al. Down regulation of RNA binding motif, single-stranded interacting protein 3, along with up regulation of nuclear HIF1A correlates with poor prognosis in patients with gastric cancer. Oncotarget 2017, 8, 1262–1277. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.T.; Chua, M.S.; Whitlock, J.P., Jr.; Shin, Y.C.; Song, W.H.; Kim, Y.; Eom, C.Y.; An, W.G. Rodent-specific hypoxia response elements enhance PAI-1 expression through HIF-1 or HIF-2 in mouse hepatoma cells. Int. J. Oncol. 2010, 37, 1627–1638. [Google Scholar]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef]
- Tausendschon, M.; Rehli, M.; Dehne, N.; Schmidl, C.; Doring, C.; Hansmann, M.L.; Brune, B. Genome-wide identification of hypoxia-inducible factor-1 and -2 binding sites in hypoxic human macrophages alternatively activated by IL-10. Biochim. Biophys. Acta 2015, 1849, 10–22. [Google Scholar] [CrossRef]
- Rapisarda, A.; Uranchimeg, B.; Sordet, O.; Pommier, Y.; Shoemaker, R.H.; Melillo, G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: Mechanism and therapeutic implications. Cancer Res. 2004, 64, 1475–1482. [Google Scholar] [CrossRef]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef]
- Xiong, L.; Kang, R.; Ding, R.; Kang, W.; Zhang, Y.; Liu, W.; Huang, Q.; Meng, J.; Guo, Z. Genome-wide Identification and Characterization of Enhancers Across 10 Human Tissues. Int. J. Biol. Sci. 2018, 14, 1321–1332. [Google Scholar] [CrossRef]
- Cao, Q.; Anyansi, C.; Hu, X.; Xu, L.; Xiong, L.; Tang, W.; Mok, M.T.S.; Cheng, C.; Fan, X.; Gerstein, M.; et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. 2017, 49, 1428–1436. [Google Scholar] [CrossRef]
- He, B.; Chen, C.; Teng, L.; Tan, K. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2191–E2199. [Google Scholar] [CrossRef]
- Hu, C.Y.; Mohtat, D.; Yu, Y.; Ko, Y.A.; Shenoy, N.; Bhattacharya, S.; Izquierdo, M.C.; Park, A.S.; Giricz, O.; Vallumsetla, N.; et al. Kidney cancer is characterized by aberrant methylation of tissue-specific enhancers that are prognostic for overall survival. Clin. Cancer Res. 2014, 20, 4349–4360. [Google Scholar] [CrossRef]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018, 33, 512–526.e8. [Google Scholar] [CrossRef]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef]
- Zawistowski, J.S.; Bevill, S.M.; Goulet, D.R.; Stuhlmiller, T.J.; Beltran, A.S.; Olivares-Quintero, J.F.; Singh, D.; Sciaky, N.; Parker, J.S.; Rashid, N.U.; et al. Enhancer Remodeling during Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex. Cancer Discov. 2017, 7, 302–321. [Google Scholar] [CrossRef]
- Nagarajan, S.; Hossan, T.; Alawi, M.; Najafova, Z.; Indenbirken, D.; Bedi, U.; Taipaleenmaki, H.; Ben-Batalla, I.; Scheller, M.; Loges, S.; et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014, 8, 460–469. [Google Scholar] [CrossRef]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nature Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.S.; et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Lee, J.S.; Smith, E.; Shilatifard, A. The language of histone crosstalk. Cell 2010, 142, 682–685. [Google Scholar] [CrossRef]
- Musselman, C.A.; Lalonde, M.E.; Cote, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef] [PubMed]
- Mirguet, O.; Lamotte, Y.; Donche, F.; Toum, J.; Gellibert, F.; Bouillot, A.; Gosmini, R.; Nguyen, V.L.; Delannee, D.; Seal, J.; et al. From ApoA1 upregulation to BET family bromodomain inhibition: Discovery of I-BET151. Bioorg. Med. Chem. Lett. 2012, 22, 2963–2967. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef]
- Bojcsuk, D.; Nagy, G.; Balint, B.L. Inducible super-enhancers are organized based on canonical signal-specific transcription factor binding elements. Nucleic Acids Res. 2017, 45, 3693–3706. [Google Scholar] [CrossRef]
- Gerard, D.; Schmidt, F.; Ginolhac, A.; Schmitz, M.; Halder, R.; Ebert, P.; Schulz, M.H.; Sauter, T.; Sinkkonen, L. Temporal enhancer profiling of parallel lineages identifies AHR and GLIS1 as regulators of mesenchymal multipotency. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Saiakhova, A.; Corradin, O.; Luppino, J.M.; Lovrenert, K.; Bartels, C.F.; Morrow, J.J.; Mack, S.C.; Dhillon, G.; Beard, L.; et al. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat. Commun. 2017, 8, 14400. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018, 553, 101–105. [Google Scholar] [CrossRef]
- Ott, C.J.; Federation, A.J.; Schwartz, L.S.; Kasar, S.; Klitgaard, J.L.; Lenci, R.; Li, Q.; Lawlor, M.; Fernandes, S.M.; Souza, A.; et al. Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia. Cancer Cell 2018, 34, 982–995.e7. [Google Scholar] [CrossRef]
- Hemming, M.L.; Lawlor, M.A.; Andersen, J.L.; Hagan, T.; Chipashvili, O.; Scott, T.G.; Raut, C.P.; Sicinska, E.; Armstrong, S.A.; Demetri, G.D.; et al. Enhancer Domains in Gastrointestinal Stromal Tumor Regulate KIT Expression and Are Targetable by BET Bromodomain Inhibition. Cancer Res. 2019, 79, 994–1009. [Google Scholar] [CrossRef]
- Xu, Y.M.; Du, J.Y.; Lau, A.T. Posttranslational modifications of human histone H3: An update. Proteomics 2014, 14, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Willson, T.A.; Wakefield, M.J.; Trounson, E.; Hilton, D.J.; Blewitt, M.E.; Oshlack, A.; Majewski, I.J. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011, 39, 7415–7427. [Google Scholar] [CrossRef]
- Arthur, R.K.; Ma, L.; Slattery, M.; Spokony, R.F.; Ostapenko, A.; Negre, N.; White, K.P. Evolution of H3K27me3-marked chromatin is linked to gene expression evolution and to patterns of gene duplication and diversification. Genome Res. 2014, 24, 1115–1124. [Google Scholar] [CrossRef]
- Xu, B.; On, D.M.; Ma, A.; Parton, T.; Konze, K.D.; Pattenden, S.G.; Allison, D.F.; Cai, L.; Rockowitz, S.; Liu, S.; et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 2015, 125, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.P.; Ling, K. EZH2 and histone deacetylase inhibitors induce apoptosis in triple negative breast cancer cells by differentially increasing H3 Lys(27) acetylation in the BIM gene promoter and enhancers. Oncol. Lett. 2017, 14, 5735–5742. [Google Scholar] [CrossRef] [PubMed]
- Torres, I.O.; Fujimori, D.G. Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr. Opin. Struct. Biol. 2015, 35, 68–75. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Sanchez, G.J.; Richmond, P.A.; Bunker, E.N.; Karman, S.S.; Azofeifa, J.; Garnett, A.T.; Xu, Q.; Wheeler, G.E.; Toomey, C.M.; Zhang, Q.; et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018, 46, 1756–1776. [Google Scholar] [CrossRef]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef]
- Sawan, C.; Herceg, Z. Histone modifications and cancer. Adv. Genet. 2010, 70, 57–85. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.M.R.; Wiseman, D.H.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 2018, 22, 3641–3659. [Google Scholar] [CrossRef]
- Whyte, W.A.; Bilodeau, S.; Orlando, D.A.; Hoke, H.A.; Frampton, G.M.; Foster, C.T.; Cowley, S.M.; Young, R.A. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 2012, 482, 221–225. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Metzger, E.; Imhof, A.; Patel, D.; Kahl, P.; Hoffmeyer, K.; Friedrichs, N.; Muller, J.M.; Greschik, H.; Kirfel, J.; Ji, S.; et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature 2010, 464, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Kwon, Y.S.; Nunez, E.; Cardamone, M.D.; Hutt, K.R.; Ohgi, K.A.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G.; et al. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc. Natl. Acad. Sci. USA 2008, 105, 19199–19204. [Google Scholar] [CrossRef] [PubMed]
- Buratowski, S. Progression through the RNA polymerase II CTD cycle. Mol. Cell 2009, 36, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef]
- Scheidegger, A.; Nechaev, S. RNA polymerase II pausing as a context-dependent reader of the genome. Biochem. Cell Biol. 2016, 94, 82–92. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shibata, H.; Handa, H. Transcription elongation factors DSIF and NELF: Promoter-proximal pausing and beyond. Biochim. Biophys. Acta 2013, 1829, 98–104. [Google Scholar] [CrossRef]
- Kwak, H.; Lis, J.T. Control of transcriptional elongation. Ann. Rev. Genet. 2013, 47, 483–508. [Google Scholar] [CrossRef]
- Nechaev, S.; Adelman, K. Pol II waiting in the starting gates: Regulating the transition from transcription initiation into productive elongation. Biochim. Biophys. Acta 2011, 1809, 34–45. [Google Scholar] [CrossRef]
- Marshall, N.F.; Price, D.H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef]
- Liu, X.; Kraus, W.L.; Bai, X. Ready, pause, go: Regulation of RNA polymerase II pausing and release by cellular signaling pathways. Trends Biochem. Sci. 2015, 40, 516–525. [Google Scholar] [CrossRef]
- Washburn, R.S.; Gottesman, M.E. Regulation of transcription elongation and termination. Biomolecules 2015, 5, 1063–1078. [Google Scholar] [CrossRef]
- Hah, N.; Murakami, S.; Nagari, A.; Danko, C.G.; Kraus, W.L. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013, 23, 1210–1223. [Google Scholar] [CrossRef]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef]
- Moreno, N.; Holsten, T.; Mertins, J.; Zhogbi, A.; Johann, P.; Kool, M.; Meisterernst, M.; Kerl, K. Combined BRD4 and CDK9 inhibition as a new therapeutic approach in malignant rhabdoid tumors. Oncotarget 2017, 8, 84986–84995. [Google Scholar] [CrossRef]
- Boffo, S.; Damato, A.; Alfano, L.; Giordano, A. CDK9 inhibitors in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 36. [Google Scholar] [CrossRef]
- Sharifnia, T.; Wawer, M.J.; Chen, T.; Huang, Q.Y.; Weir, B.A.; Sizemore, A.; Lawlor, M.A.; Goodale, A.; Cowley, G.S.; Vazquez, F.; et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nature Med. 2019, 25, 292–300. [Google Scholar] [CrossRef]
- Fisher, R.P.; Morgan, D.O. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 1994, 78, 713–724. [Google Scholar] [CrossRef]
- Schwartz, B.E.; Larochelle, S.; Suter, B.; Lis, J.T. Cdk7 is required for full activation of Drosophila heat shock genes and RNA polymerase II phosphorylation in vivo. Mol. Cell. Biol. 2003, 23, 6876–6886. [Google Scholar] [CrossRef]
- Lolli, G. Binding to DNA of the RNA-polymerase II C-terminal domain allows discrimination between Cdk7 and Cdk9 phosphorylation. Nucleic Acids Res. 2009, 37, 1260–1268. [Google Scholar] [CrossRef]
- Larochelle, S.; Merrick, K.A.; Terret, M.E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Zhang, Z.; Peng, H.; Wang, X.; Yin, X.; Ma, P.; Jing, Y.; Cai, M.C.; Liu, J.; Zhang, M.; Zhang, S.; et al. Preclinical Efficacy and Molecular Mechanism of Targeting CDK7-Dependent Transcriptional Addiction in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 1739–1750. [Google Scholar] [CrossRef]
- Jiang, Y.Y.; Lin, D.C.; Mayakonda, A.; Hazawa, M.; Ding, L.W.; Chien, W.W.; Xu, L.; Chen, Y.; Xiao, J.F.; Senapedis, W.; et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 2017, 66, 1358–1368. [Google Scholar] [CrossRef]
- Eliades, P.; Abraham, B.J.; Ji, Z.; Miller, D.M.; Christensen, C.L.; Kwiatkowski, N.; Kumar, R.; Njauw, C.N.; Taylor, M.; Miao, B.; et al. High MITF Expression Is Associated with Super-Enhancers and Suppressed by CDK7 Inhibition in Melanoma. J. Invest. Dermatol. 2018, 138, 1582–1590. [Google Scholar] [CrossRef]
- Meng, W.; Wang, J.; Wang, B.; Liu, F.; Li, M.; Zhao, Y.; Zhang, C.; Li, Q.; Chen, J.; Zhang, L.; et al. CDK7 inhibition is a novel therapeutic strategy against GBM both in vitro and in vivo. Cancer Manag. Res. 2018, 10, 5747–5758. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Geng, J.; Zhang, L.; Wang, Y.; Niu, N.; Fang, Y.; Liu, F.; Shi, J.; Zhang, Z.G.; Sun, Y.W.; et al. THZ1 reveals CDK7-dependent transcriptional addictions in pancreatic cancer. Oncogene 2019. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, L.; Jiang, G.; Chen, Z.; Li, J.; An, P.; Chen, L.; Du, J.; Wang, H. Targeting CDK7 increases the stability of Snail to promote the dissemination of colorectal cancer. Cell Death Differ. 2018. [Google Scholar] [CrossRef]
- Nilson, K.A.; Guo, J.; Turek, M.E.; Brogie, J.E.; Delaney, E.; Luse, D.S.; Price, D.H. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol. Cell 2015, 59, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Sampathi, S.; Acharya, P.; Zhao, Y.; Wang, J.; Stengel, K.R.; Liu, Q.; Savona, M.R.; Hiebert, S.W. The CDK7 inhibitor THZ1 alters RNA polymerase dynamics at the 5′ and 3′ ends of genes. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Olson, C.M.; Liang, Y.; Leggett, A.; Park, W.D.; Li, L.; Mills, C.E.; Elsarrag, S.Z.; Ficarro, S.B.; Zhang, T.; Duster, R.; et al. Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem. Biol. 2019. [Google Scholar] [CrossRef]
- Kornberg, R.D. Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci. 2005, 30, 235–239. [Google Scholar] [CrossRef]
- Poss, Z.C.; Ebmeier, C.C.; Taatjes, D.J. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 575–608. [Google Scholar] [CrossRef]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Dunn, T.M.; Hahn, S.; Ogden, S.; Schleif, R.F. An operator at -280 base pairs that is required for repression of araBAD operon promoter: Addition of DNA helical turns between the operator and promoter cyclically hinders repression. Proc. Natl. Acad. Sci. USA 1984, 81, 5017–5020. [Google Scholar] [CrossRef]
- Wendt, K.S.; Yoshida, K.; Itoh, T.; Bando, M.; Koch, B.; Schirghuber, E.; Tsutsumi, S.; Nagae, G.; Ishihara, K.; Mishiro, T.; et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008, 451, 796–801. [Google Scholar] [CrossRef]
- Bell, A.C.; West, A.G.; Felsenfeld, G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 1999, 98, 387–396. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Z.; Bando, M.; Itoh, T.; Deardorff, M.A.; Clark, D.; Kaur, M.; Tandy, S.; Kondoh, T.; Rappaport, E.; et al. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009, 7, e1000119. [Google Scholar] [CrossRef]
- Lewis, B.A.; Reinberg, D. The mediator coactivator complex: Functional and physical roles in transcriptional regulation. J. Cell Sci. 2003, 116, 3667–3675. [Google Scholar] [CrossRef]
- Petrenko, N.; Jin, Y.; Wong, K.H.; Struhl, K. Mediator Undergoes a Compositional Change during Transcriptional Activation. Mol. Cell 2016, 64, 443–454. [Google Scholar] [CrossRef]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef]
- Pawar, A.; Gollavilli, P.N.; Wang, S.; Asangani, I.A. Resistance to BET Inhibitor Leads to Alternative Therapeutic Vulnerabilities in Castration-Resistant Prostate Cancer. Cell Rep. 2018, 22, 2236–2245. [Google Scholar] [CrossRef]
- Kumar, K.; Raza, S.S.; Knab, L.M.; Chow, C.R.; Kwok, B.; Bentrem, D.J.; Popovic, R.; Ebine, K.; Licht, J.D.; Munshi, H.G. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci. Rep. 2015, 5, 9489. [Google Scholar] [CrossRef]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Neitzel, L.R.; Loganathan, S.N.; Tang, N.; Qin, L.; Crispi, E.E.; Guo, Y.; Knapp, S.; Beauchamp, R.D.; et al. The MAPK Pathway Regulates Intrinsic Resistance to BET Inhibitors in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2027–2037. [Google Scholar] [CrossRef]
- Ambrosini, G.; Do, C.; Tycko, B.; Realubit, R.B.; Karan, C.; Musi, E.; Carvajal, R.D.; Chua, V.; Aplin, A.E.; Schwartz, G.K. Inhibition of NF-kappaB-dependent signaling enhances sensitivity and overcomes resistance to BET inhibition in uveal melanoma. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Fulciniti, M.; Lin, C.Y.; Samur, M.K.; Lopez, M.A.; Singh, I.; Lawlor, M.A.; Szalat, R.E.; Ott, C.J.; Avet-Loiseau, H.; Anderson, K.C.; et al. Non-overlapping Control of Transcriptome by Promoter- and Super-Enhancer-Associated Dependencies in Multiple Myeloma. Cell Rep. 2018, 25, 3693–3705.e6. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nature Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Basu, T.N.; Gutmann, D.H.; Fletcher, J.A.; Glover, T.W.; Collins, F.S.; Downward, J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 1992, 356, 713–715. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Haigentz, M., Jr.; Kim, M.; Sarta, C.; Lin, J.; Keresztes, R.S.; Culliney, B.; Gaba, A.G.; Smith, R.V.; Shapiro, G.I.; Chirieac, L.R.; et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 2012, 48, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Wong, C.; et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A California Cancer Consortium study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef] [PubMed]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef]
- Teknos, T.N.; Grecula, J.; Agrawal, A.; Old, M.O.; Ozer, E.; Carrau, R.; Kang, S.; Rocco, J.; Blakaj, D.; Diavolitsis, V.; et al. A phase 1 trial of Vorinostat in combination with concurrent chemoradiation therapy in the treatment of advanced staged head and neck squamous cell carcinoma. Invest. New Drugs 2018. [Google Scholar] [CrossRef]
- Pili, R.; Liu, G.; Chintala, S.; Verheul, H.; Rehman, S.; Attwood, K.; Lodge, M.A.; Wahl, R.; Martin, J.I.; Miles, K.M.; et al. Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: A multicentre, single-arm phase I/II clinical trial. Br. J. Cancer 2017, 116, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef]
- Schaukowitch, K.; Joo, J.Y.; Liu, X.; Watts, J.K.; Martinez, C.; Kim, T.K. Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 2014, 56, 29–42. [Google Scholar] [CrossRef]
- Liang, J.; Zhou, H.; Gerdt, C.; Tan, M.; Colson, T.; Kaye, K.M.; Kieff, E.; Zhao, B. Epstein–Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 14121. [Google Scholar] [CrossRef]
- Pnueli, L.; Rudnizky, S.; Yosefzon, Y.; Melamed, P. RNA transcribed from a distal enhancer is required for activating the chromatin at the promoter of the gonadotropin alpha-subunit gene. Proc. Natl. Acad. Sci. USA 2015, 112, 4369–4374. [Google Scholar] [CrossRef]
- Kim, Y.W.; Lee, S.; Yun, J.; Kim, A. Chromatin looping and eRNA transcription precede the transcriptional activation of gene in the β-globin locus. Biosci. Rep. 2015, 35, e00179. [Google Scholar] [CrossRef]
- Cheng, J.H.; Pan, D.Z.; Tsai, Z.T.; Tsai, H.K. Genome-wide analysis of enhancer RNA in gene regulation across 12 mouse tissues. Sci. Rep. 2015, 5, 12648. [Google Scholar] [CrossRef]
- McCleland, M.L.; Mesh, K.; Lorenzana, E.; Chopra, V.S.; Segal, E.; Watanabe, C.; Haley, B.; Mayba, O.; Yaylaoglu, M.; Gnad, F.; et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Investig. 2016, 126, 639–652. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, L.; Ren, S.; Wang, L.; Blackburn, P.R.; McNulty, M.S.; Gao, X.; Qiao, M.; Vessella, R.L.; Kohli, M.; et al. Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep. 2016, 15, 599–610. [Google Scholar] [CrossRef]
- Kaczkowski, B.; Tanaka, Y.; Kawaji, H.; Sandelin, A.; Andersson, R.; Itoh, M.; Lassmann, T.; Hayashizaki, Y.; Carninci, P.; Forrest, A.R. Transcriptome Analysis of Recurrently Deregulated Genes across Multiple Cancers Identifies New Pan-Cancer Biomarkers. Cancer Res. 2016, 76, 216–226. [Google Scholar] [CrossRef]
- Core, L.J.; Waterfall, J.J.; Lis, J.T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008, 322, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Schwalb, B.; Michel, M.; Zacher, B.; Fruhauf, K.; Demel, C.; Tresch, A.; Gagneur, J.; Cramer, P. TT-seq maps the human transient transcriptome. Science 2016, 352, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Mahat, D.B.; Kwak, H.; Booth, G.T.; Jonkers, I.H.; Danko, C.G.; Patel, R.K.; Waters, C.T.; Munson, K.; Core, L.J.; Lis, J.T. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc. 2016, 11, 1455–1476. [Google Scholar] [CrossRef]
- Chu, T.; Rice, E.J.; Booth, G.T.; Salamanca, H.H.; Wang, Z.; Core, L.J.; Longo, S.L.; Corona, R.J.; Chin, L.S.; Lis, J.T.; et al. Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme. Nat. Genet. 2018, 50, 1553–1564. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef]
- Parker, S.C.J.; Stitzel, M.L.; Taylor, D.L.; Orozco, J.M.; Erdos, M.R.; Akiyama, J.A.; van Bueren, K.L.; Chines, P.S.; Narisu, N.; Program, N.C.S.; et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. USA 2013, 110, 17921–17926. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.X.; Erdos, M.R.; Parker, S.C.J.; Collins, F.S. Motif signatures in stretch enhancers are enriched for disease-associated genetic variants. Epigenetics Chromatin 2015, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mathelier, A.; Zhang, X. Super-enhancers are transcriptionally more active and cell type-specific than stretch enhancers. Epigenetics 2018, 13, 910–922. [Google Scholar] [CrossRef]
- Hong, J.W.; Hendrix, D.A.; Levine, M.S. Shadow enhancers as a source of evolutionary novelty. Science 2008, 321, 1314. [Google Scholar] [CrossRef]
- Cao, K.; Collings, C.K.; Marshall, S.A.; Morgan, M.A.; Rendleman, E.J.; Wang, L.; Sze, C.C.; Sun, T.; Bartom, E.T.; Shilatifard, A. SET1A/COMPASS and shadow enhancers in the regulation of homeotic gene expression. Genes Dev. 2017, 31, 787–801. [Google Scholar] [CrossRef]
- Benabdallah, N.S.; Bickmore, W.A. Regulatory Domains and Their Mechanisms. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef]
- Bretz, A.C.; Gittler, M.P.; Charles, J.P.; Gremke, N.; Eckhardt, I.; Mernberger, M.; Mandic, R.; Thomale, J.; Nist, A.; Wanzel, M.; et al. ΔNp63 activates the Fanconi anemia DNA repair pathway and limits the efficacy of cisplatin treatment in squamous cell carcinoma. Nucleic Acids Res. 2016, 44, 3204–3218. [Google Scholar] [CrossRef]
- Kumakura, Y.; Rokudai, S.; Iijima, M.; Altan, B.; Yoshida, T.; Bao, H.; Yokobori, T.; Sakai, M.; Sohda, M.; Miyazaki, T.; et al. Elevated expression of DeltaNp63 in advanced esophageal squamous cell carcinoma. Cancer Sci. 2017, 108, 2149–2155. [Google Scholar] [CrossRef]
- Coates, P.J.; Nenutil, R.; Holcakova, J.; Nekulova, M.; Podhorec, J.; Svoboda, M.; Vojtesek, B. p63 isoforms in triple-negative breast cancer: DeltaNp63 associates with the basal phenotype whereas TAp63 associates with androgen receptor, lack of BRCA mutation, PTEN and improved survival. Virchows Arch. 2018, 472, 351–359. [Google Scholar] [CrossRef]
- Orzol, P.; Nekulova, M.; Holcakova, J.; Muller, P.; Votesek, B.; Coates, P.J. DeltaNp63 regulates cell proliferation, differentiation, adhesion, and migration in the BL2 subtype of basal-like breast cancer. Tumour Biol. 2016, 37, 10133–10140. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).