Membrane Type-1 Matrix Metalloproteinase Expression in Acute Myeloid Leukemia and Its Upregulation by Tumor Necrosis Factor-α

Abstract
:1. Introduction
2. Experimental Section
2.1. Patients and Cells
2.2. Gel-Based and Real-Time RT-PCR Analysis
2.3. Western Blot
2.4. Trans-Matrigel Invasion Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt # | Age/Sex | Diagnosis (WHO Classification) | Karyotype | Hb g/L | Plt 109/L | WBC 109/L | % Blasts in PB | % Blasts in BM | MT1-MMP |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 23/M | AML with inv(16) | 46,XY,inv(16)(p13.1q22)[16] | 135 | 47 | 179 | 70 | 63 | + |
| 2 | 22/M | AML without maturation | 51~54,XY,+Y,+4,+8,+10,+13,+20,+21,+22[CP20] | 77 | 19 | 166 | 87 | 94 | + |
| 3 | 75/M | Acute myelomonocytic leukemia | 46,XY,del(20)(q11.2)[6]/47,idem,+8[6]/~45,idem,+8,−10[cp2] | 100 | 28 | 80.9 | 65 | 62 | - |
| 4 | 44/F | Acute myelomonocytic leukemia | 46,XX[20] | 78 | 98 | 152.4 | 67 | 62 | + |
| 5 | 58/M | AML without maturation | 46,XY[20] | 144 | 33 | 59.8 | 73 | 83 | + |
| 6 | 23/M | AML with t(15:17) | 46,XY,t(15;17)(q22;q11)[6]/48,XY,der(9;15)t(9;15;17)(q22;q21),ider (17)(q10)t(15;17)(q22;q11),+ider (17)(q10)t(15;17)(q22;q21),+21[13] | 96 | 11 | 5.2 | 38 | 67 | + |
| 7 | 66/F | Acute myelomonocytic leukemia | 46,XX[19] | 99 | 55 | 15.1 | 50 | 72 | - |
| 8 | 37/F | Acute monoblastic leukemia | 48,XX,+8,+8[14]/96,idemx2[2]/46,XX[1] | 121 | 190 | 91.8 | 84 | 81 | + |
| 9 | 17/M | Acute myelomonocytic leukemia | 46,XY[20] | 52 | 27 | 101.2 | 44 | 51 | + |
| 10 | 78/M | AML without maturation | 46,XY[20] | 104 | 58 | 101.4 | 81 | 88 | + |
| 11 | 36/M | AML with t(15:17) | 46,XY,t(17;19;22)(q21;p13;q13).nuc ish 15q22(PMLx2),17q21(RARAx2)(PML con RARAx1)[225] | 143 | 18 | 94 | 89 | 92 | + |
| 12 | 52/M | Acute myelomonocytic leukemia | 46,XY[20] | 89 | 121 | 22.7 | 18 | 23 | + |
| 13 | 61/F | AML with t(15:17) | 15q22(PMLx2),17q21(RARAx2)(PML con RARAx1)[231] | 83 | 49 | 33.6 | 27.9 | 83 | + |
| 14 | 28/M | AML with t(15:17) | 46,XY,t(15;17)(q22;q21.1)[2]/46,XY,t(15;17)(q22;q21.1),add(20)(p?11.2)[18] | 91 | 21 | 21.6 | 84 | 77 | + |
| 15 | 49/F | AML without maturation | 46,XX[20] | 126 | 17 | 145.8 | 94 | 90 | + |
| 16 | 38/M | Acute myelomonocytic leukemia | 46,XY[20] | 64 | 87 | 44.1 | 49 | 59 | + |
| 17 | 46/M | Acute monocytic leukemia | 46,XY[20] | 92 | 16 | 66.7 | 58 | 89 | + |
| 18 | 77/M | AML with t(8;21) | 45,XY,−Y,t(8;21)(q22;q22)[10] | 67 | 69 | 6.3 | 62 | 51 | + |
| 19 | 62/M | AML with 11q23 abnormality | 46,XY,del(11)(q23) | 123 | 60 | 71.2 | 80 | 82 | + |
| 20 | 57/M | AML with multilineage dysplasia | 43,X,−Y[20],−3[20],−5[20],−[20]+8[3],der(3)t(3;13)(q21;q34)[20]add(17)(p11.2)[20],+mar[4][cp20] | 104 | 31 | 15.2 | 52 | 60 | + |
| 21 | 48/F | Acute monocytic leukemia | 46,XX[20] | 105 | 120 | 7.1 | 54 | 79 | + |
| 22 | 50/M | Acute monocytic leukemia | 46,XY[25] | 140 | 78 | 109.9 | 67 | 91 | + |
| 23 | 67/M | Biphenotypic acute leukemia | 45~47,XY,−2[7],−7[6], add(11)(q23)[5],−12 [4], +1~7mar[6] [cp6]/46, XY[1] nuc ish 11q23 (MLLstx2) (91.0%) | 98 | 72 | 2.5 | 13 | 49 | + |
| 24 | 72/M | AML NOC | ND | 84 | 39 | 207 | 87 | 64 | + |
| 25 | 72/M | AML NOC | 43,XY,−3,−7,der(8)t(8;15)(p23;q15), −9,−9,−13,−13,−15,−16, add(17)(p11.2),der(20)t(3;20) (q13.2;q13.1),dic(21,?)(q21;?),der (22)t(3;22)(p23;q13),−16,add(17) (p11.2),der(20)t(3;20) (q13.2;q13.1),dic(21;?)(q21;?), der(22)t(3;2)(p23;q13),+mar1, +mar2,+mar3,+mar4,+mar5, +mar6[3] | 87 | 8 | 10 | 25 | 22 | + |
| 26 | 75/F | AML with maturation | 44,XX,del(5)(q15q31),−7,−18[18]/45,sl,+9[3] | 119 | 46 | 5.8 | 36 | 33 | + |
| 27 | 47/M | AML without maturation | 45,XY,add(2)(p11.2),del(5)(q13q15),del(9)(q22),add(11)(p15),−17[19] | 92 | 21 | 19.3 | 83 | 62 | + |
| 28 | 61/F | Relapsed Acute myelomonocytic leukemia | 46,XX[20] | 85 | 37 | 112.6 | 87 | 73 | + |
| 29 | 54/F | AML with inv(16) | 46,XX,inv(16)(p13q22)[18] | 119 | 46 | 21 | 36 | 40 | + |
| 30 | 37/F | AML with maturation | 46,XX[20] | 83 | 42 | 188.5 | 91 | 77 | + |
| 31 | 60/F | AML with t(8;21) | 45,X,−X,t(8;21)(q22;q22)[18]/46, XX[2] | 88 | 44 | 8.8 | 65 | 66 | + |
| 32 | 74/F | AML without maturation | 47,XX,+13[11]/46,sl,−X[4] | 114 | 28 | 29.1 | 95 | 93 | + |
| 33 | 45/M | Relapsed AML with t(15;17) | 46,XY,t(15;17)(q22;q21)[10]/46,XY,idem,del(9)(q13q22)[6]/46,XY[2] | 116 | 18 | 10.0 | 60 | 79 | + |
| 34 | 69/M | AML with multilineage dysplasia | 45,XY,−7[20] | 123 | 14 | 93.9 | 36 | 35 | + |
| 35 | 66/F | Relapsed AML | 46,X,−X,+mar[15]/46,XX[4] | 106 | 46 | 3.3 | 21 | 87 | + |
| 36 | 66/M | AML with multilineage dysplasia | 43~44,XY,−5[20],del(7)(q31)[20],+8[20],−12[20],−13[20],+add(14) (q32)[20],−17[20],18[5],der(8) t(12;18) (q13;p11.3)[20],−20[20], +mar[13],+mar1x2[5],1dmin[15][cp20] | 99 | 19 | 18 | 30 | 46 | + |
| 37 | 68/M | AML with inv(16) | 46,XY,inv(16)(p13.1q22)[19] | 94 | 20 | 121.2 | 70 | 66 | + |
| 38 | 73/M | Relapsed AML | 46,XY[19] | 108 | 56 | 14.8 | 86 | 88 | + |
| 39 | 64/F | AML with maturation | 47,XX,+4[5]/46,XX[9] | 87 | 214 | 46.8 | 72 | 84 | + |
| 40 | 34/F | AML with maturation | 46,XX,del(9)(q13q22)[20] | 79 | 29 | 10.2 | 69 | 76 | + |
| 41 | 55/F | AML with multilineage dysplasia | 46,XX[18] | 105 | 68 | 39.6 | 23 | 60 | + |
| 42 | 66/M | AML with multilineage dysplasia | 47,XY,+8[18] | 99 | 11 | 79.6 | 88 | 89 | + |
| 43 | 50/F | Acute myelomonocytic leukemia | 45,XX,inv(3)(q21q26),−7[18] | 94 | 301 | 18.2 | 18 | 26 | + |
| Mean ± SD (n = 43) | 100.3 ± 20.8 | 56.6 ± 58.0 | 61.7 ± 58.2 | 60.1 ± 24.7 | 67.8 ± 20.3 |
2.5. SiRNA Electroporation
2.6. Flow Cytometric Analysis
2.7. Zymography
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Statistical Analysis
3. Results
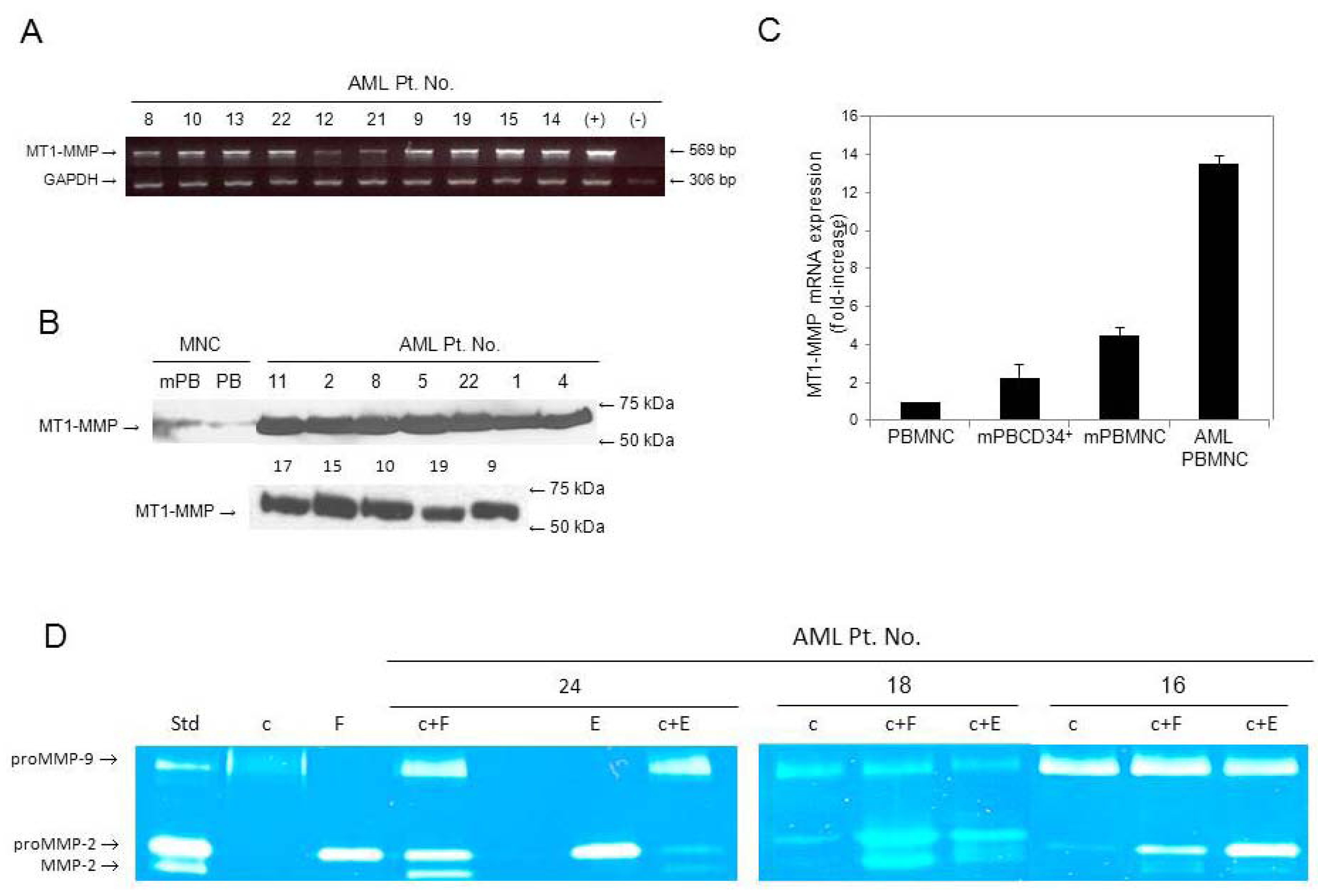
3.1. AML Cells Highly Express MT1-MMP in Contrast to Normal Cells
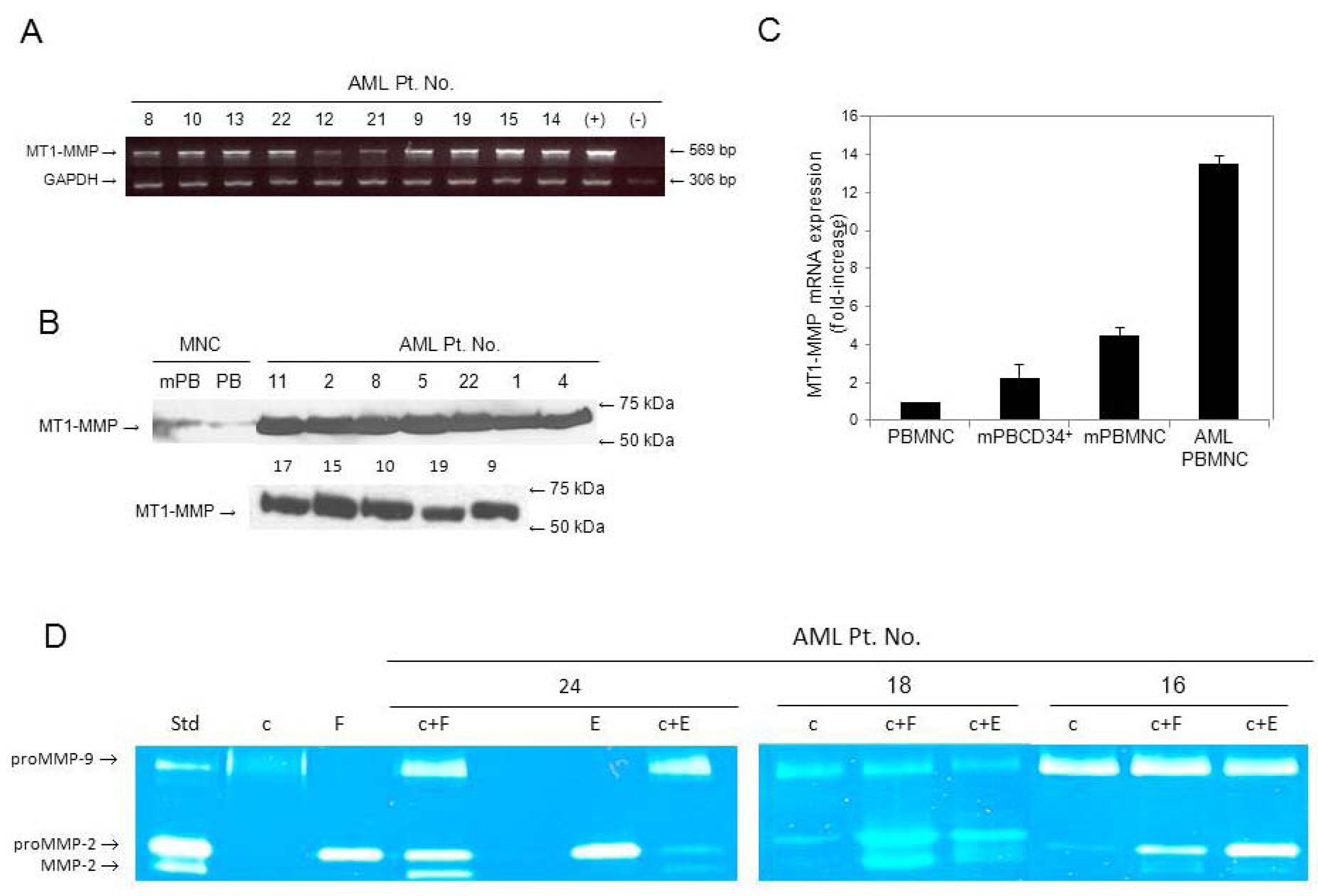
3.2. Secretion of proMMPs by AML Cells and Active MMP-2 in Co-Cultures with Stromal Cells
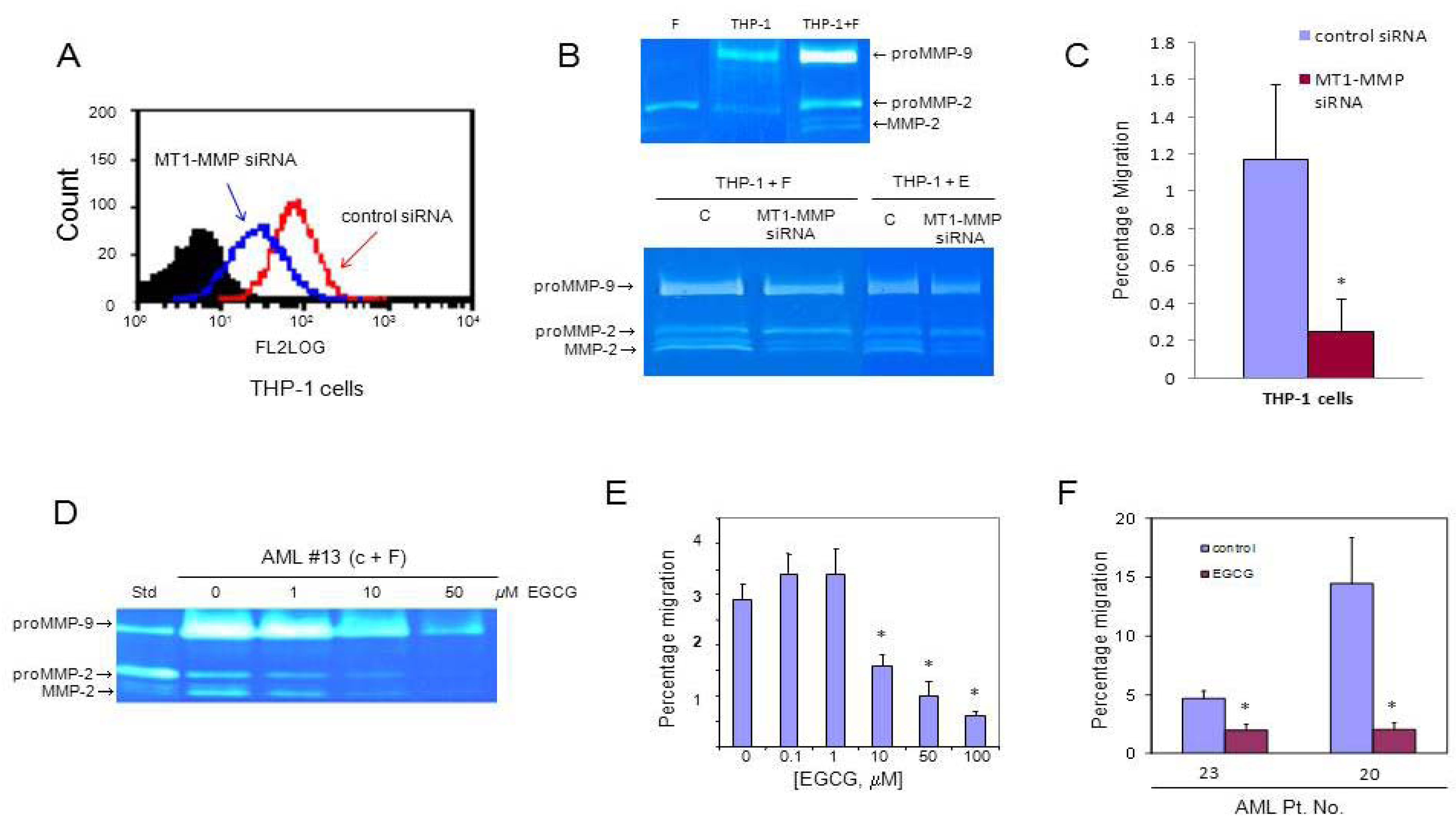
3.3. MT1-MMP Inhibition Reduces Cell Migration Across Reconstituted Basement Membrane
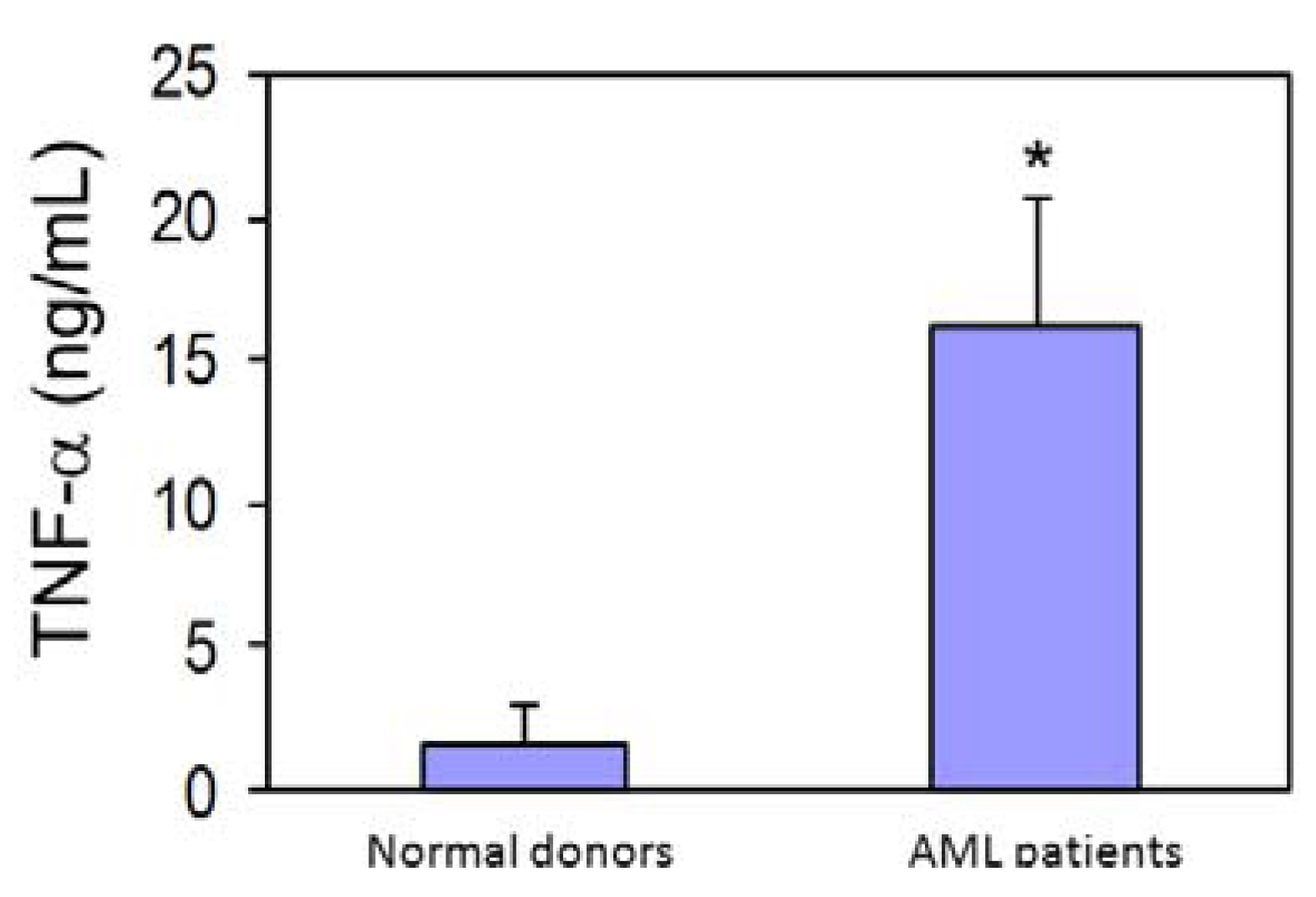
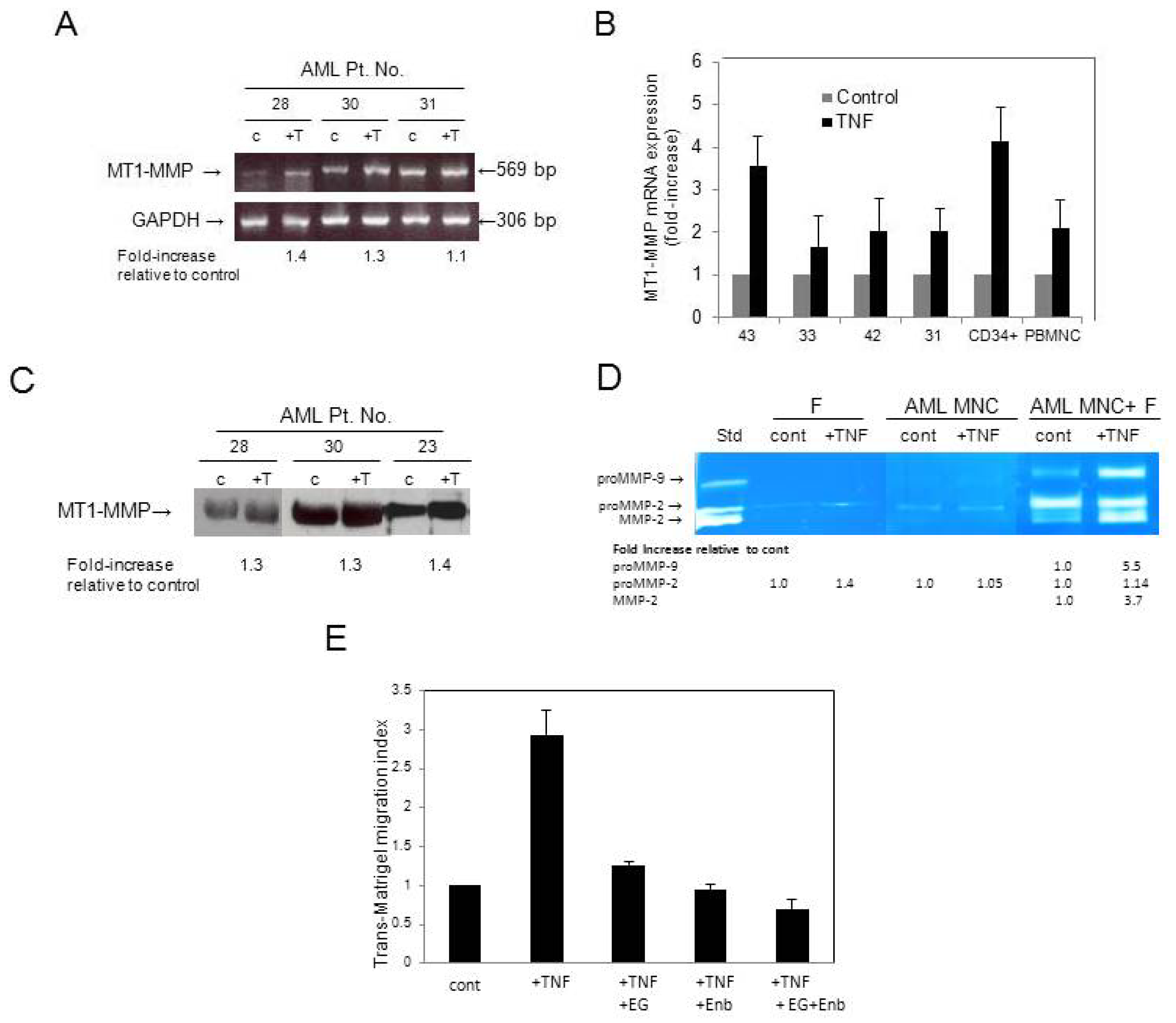
3.4. AML Cells Secrete TNF-α Which Upregulates MT1-MMP Expression and trans-Matrigel Migration
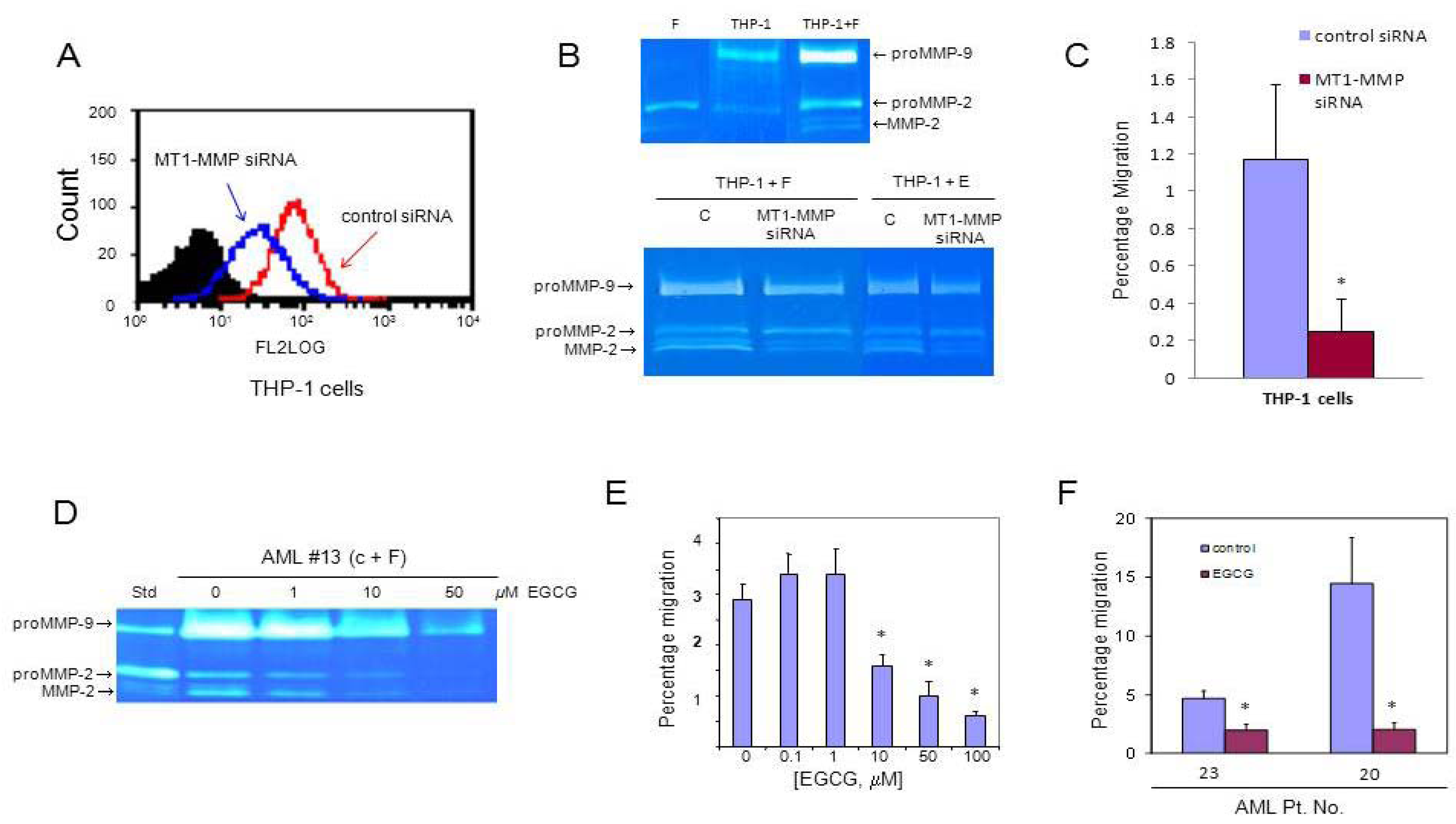


4. Discussion
5. Conclusions
Conflict of Interest
Acknowledgments
References
- Zitka, O.; Kukacka, J.; Krizkova, S.; Huska, D.; Adam, V.; Masarik, M.; Prusa, R.; Kizek, R. Matrix metalloproteinases. Curr. Med. Chem. 2010, 17, 3751–3768. [Google Scholar]
- Roy, R.; Yang, J.; Moses, M.A. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J. Clin. Oncol. 2009, 27, 5287–5297. [Google Scholar]
- Deryugina, E.I.; Quigley, J.P. Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: Contrasting, overlapping and compensatory functions. Biochim. Biophys. Acta 2010, 1803, 103–120. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Bourboulia, D.; Stetler-Stevenson, W.G. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Sem. Cancer Biol. 2010, 20, 161–168. [Google Scholar] [CrossRef]
- Butler, G.S.; Overall, C.M. Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry 2009, 48, 10830–10845. [Google Scholar]
- Rodriguez, D.; Morrison, C.J.; Overall, C.M. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 2010, 1803, 39–54. [Google Scholar] [CrossRef]
- Clark, I.M.; Singler, T.E.; Sampieri, C.L.; Edwards, D.R. The regulation of matrix metalloproteinases and their inhibitors. Int. J. Biochem. Cell Biol. 2008, 40, 1362–1378. [Google Scholar] [CrossRef] [Green Version]
- Janowska-Wieczorek, A.; Marquez, L.A.; Nabholtz, J.M.; Cabuhat, M.L.; Montaño, J.; Chang, H.; Rozmus, J.; Russell, J.A.; Edwards, D.R.; Turner, A.R. Growth factors and cytokines upregulate gelatinase expression in bone marrow CD34(+) cells and their transmigration through reconstituted basement membrane. Blood 1999, 93, 3379–3390. [Google Scholar]
- Strongin, A.Y.; Collier, I.; Bannikov, G.; Marmer, B.L.; Grant, G.A.; Goldberg, G.I. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 1995, 270, 5331–5338. [Google Scholar]
- Sato, H.; Takino, T. Coordinate action of membrane-type matrix metalloproteinase-1 (MT1-MMP) and MMP-2 enhances pericellular proteolysis and invasion. Cancer Sci. 2010, 101, 843–847. [Google Scholar] [CrossRef]
- Holmbeck, K.; Bianco, P.; Yamada, S.; Birkedal-Hansen, H. MT1-MMP, a tethered collagenase. J. Cell. Physiol. 2004, 200, 11–19. [Google Scholar] [CrossRef]
- Itoh, Y.; Seiki, M. MT1-MMP: A potent modifier of pericellular microenvironment. J. Cell. Physiol. 2006, 206, 1–8. [Google Scholar] [CrossRef]
- Strongin, A.Y. Proteolytic and non-proteolytic roles of membrane type-1 matrix metalloproteinase in malignancy. Biochim. Biophys. Acta 2010, 1803, 133–141. [Google Scholar] [CrossRef]
- Poincloux, R.; Lizarraga, F.; Chavrier, P. Matrix invasion by tumour cells: A focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 2009, 122, 3015–3024. [Google Scholar] [CrossRef]
- Velders, G.A.; Fibbe, W.E. Involvement of proteases in cytokine-induced hematopoietic stem cell mobilization. Ann. NY Acad. Sci. 2005, 1044, 60–69. [Google Scholar] [CrossRef]
- Lee, H.M.; Wu, W.; Wysoczynski, M.; Liu, R.; Zuba-Surma, E.K.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. Impaired mobilization of hematopoietic stem/progenitor cells in C5-deficient mice supports the pivotal involvement of innate immunity in this process and reveals novel promobilization effects of granulocytes. Leukemia 2009, 23, 2052–2062. [Google Scholar]
- Shirvaikar, N.; Marquez-Curtis, L.A.; Shaw, A.R.; Turner, A.R.; Janowska-Wieczorek, A. MT1-MMP association with membrane lipid rafts facilitates G-CSF-induced hematopoietic stem/progenitor cell mobilization. Exp. Hematol. 2010, 38, 823–835. [Google Scholar]
- Shirvaikar, N.; Marquez-Curtis, L.A.; Janowska-Wieczorek, A. Hematopoietic stem cell mobilization and homing after transplantation: The role of MMP-2, MMP-9, and MT1-MMP. Biochem. Res. Int. 2012, 2012, 685267. [Google Scholar]
- Vagima, Y.; Avigdor, A.; Goichberg, P.; Shivtiel, S.; Tesio, M.; Kalinkovich, A.; Golan, K.; Dar, A.; Kollet, O.; Petit, I.; et al. T1-MMP and RECK are involved in human CD34+ progenitor cell retention, egress, and mobilization. J. Clin. Invest. 2009, 119, 492–503. [Google Scholar]
- Matsuzaki, A.; Janowska-Wieczorek, A. Unstimulated human acute myelogenous leukemia blasts secrete matrix metalloproteinases. J. Cancer Res. Clin. Oncol. 1997, 123, 100–106. [Google Scholar] [CrossRef]
- Janowska-Wieczorek, A.; Marquez, L.A.; Matsuzaki, A.; Hashmi, H.R.; Larratt, L.M.; Boshkov, L.M.; Turner, A.R.; Zhang, M.C.; Edwards, D.R.; Kossakowska, A.E. Expression of matrix metalloproteinases (MMP-2 and -9) and tissue inhibitors of metalloproteinases (TIMP-1 and -2) in acute myelogenous leukaemia blasts: Comparison with normal bone marrow cells. Br. J. Haematol. 1999, 105, 402–411. [Google Scholar]
- Ries, C.; Loher, F.; Zang, C.; Ismair, M.G.; Petrides, P.E. Matrix metalloproteinase production by bone marrow mononuclear cells from normal individuals and patients with acute and chronic myeloid leukemia or myelodysplastic syndromes. Clin. Cancer Res. 1999, 5, 1115–1124. [Google Scholar]
- Janowska-Wieczorek, A.; Matsuzaki, A.; Marquez, L.A. The hematopoietic microenvironment: Matrix metalloproteinases in the hematopoietic microenvironment. Hematology 2000, 4, 515–527. [Google Scholar]
- Marquez-Curtis, L.A.; Dobrowsky, A.; Montaño, J.; Turner, A.R.; Ratajczak, J.; Ratajczak, M.Z.; Janowska-Wieczorek, A. Matrix metalloproteinase and tissue inhibitors of metalloproteinase secretion by haematopoietic and stromal precursors and their production in normal and leukaemic long-term marrow cultures. Br. J. Haematol. 2001, 115, 595–604. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Estey, E.; Wen, S.; Pierce, S.; Kantarjian, H.; Albitar, M.; Kurzrock, R. The prognostic significance of cytokine levels in newly diagnosed acute myeloid leukemia and high-risk myelodysplastic syndromes. Cancer 2008, 113, 1605–1613. [Google Scholar] [CrossRef]
- Mayani, H.; Guilbert, L.J.; Clark, S.C.; Belch, A.R.; Janowska-Wieczorek, A. Composition and functional integrity of the in vitro hemopoietic microenvironment in acute myelogenous leukemia: Effect of macrophage colony-stimulating factor. Exp. Hematol. 1992, 20, 1077–1084. [Google Scholar]
- Son, B.-R.; Marquez-Curtis, L.A.; Kucia, M.; Wysoczynski, M.; Turner, A.R.; Ratajczak, J.; Ratajczak, M.Z.; Janowska-Wieczorek, A. Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-met axes and involves matrix metalloproteinases. Stem Cells 2006, 24, 1254–1264. [Google Scholar]
- Janiak, M.; Hashmi, H.R.; Janowska-Wieczorek, A. Use of the Matrigel-based assay to measure the invasiveness of leukemic cells. Exp. Hematol. 1994, 22, 559–565. [Google Scholar]
- Bartolome, R.A.; Molina-Ortiz, I.; Samaniego, R.; Sanchez-Mateos, P.; Bustelo, X.R.; Teixido, J. Activation of Vav/Rho GTPase signaling by CXCL12 controls membrane-type matrix metalloproteinase-dependent melanoma cell invasion. Cancer Res. 2006, 66, 248–258. [Google Scholar]
- Yamakawa, S.; Asai, T.; Uchida, T.; Matsukawa, M.; Akizawa, T.; Oku, N. (-)-Epigallocatechin gallate inhibits membrane-type 1 matrix metalloproteinase, MT1-MMP, and tumor angiogenesis. Cancer Lett. 2004, 210, 47–55. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Travaglino, E.; Benatti, C.; Malcovati, L.; Della Porta, M.G.; Gallì, A.; Bonetti, E.; Rosti, V.; Cazzola, M.; Invernizzi, R. Biological and clinical relevance of matrix metalloproteinases 2 and 9 in acute myeloid leukaemias and myelodysplastic syndromes. Eur. J. Haematol. 2008, 80, 216–226. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Z.; Li, Z.; Cen, J. The essential roles of matrix metalloproteinase-2, membrane type 1 metalloproteinase and tissue inhibitor of metalloproteinase-2 in the invasive capacity of acute monocytic leukemia SHI-1 cells. Leuk. Res. 2010, 34, 1083–1090. [Google Scholar] [CrossRef]
- Sabeh, F.; Ota, I.; Holmbeck, K.; Birkedal-Hansen, H.; Soloway, P.; Balbin, M.; Lopez-Otin, C.; Shapiro, S.; Inada, M.; Krane, S.; et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol. 2004, 167, 769–781. [Google Scholar]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion and angiogenesis. Cancer Res. 2009, 69, 1517–1526. [Google Scholar]
- Friedl, P.; Wolf, K. Tube travel: The role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008, 68, 7247–7249. [Google Scholar] [CrossRef]
- Rowe, R.G.; Weiss, S.J. Navigating ECM barriers at the invasive front: The cancer cell-stroma interface. Ann. Rev. Cell Dev. Biol. 2009, 25, 567–595. [Google Scholar]
- Gonzalo, P.; Moreno, V.; Galvez, B.G.; Arroyo, A.G. MT1-MMP and integrins: Hand-to-hand in cell communication. Int. Union Biochem. Mol. Biol. 2010, 36, 248–254. [Google Scholar]
- Ayala, F.; Dewar, R.; Kieran, M.; Kalluri, R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia 2009, 23, 2233–2241. [Google Scholar] [CrossRef]
- Gearing, A.J.; Beckett, P.; Christodoulou, M.; Churchill, M.; Clements, J.M.; Crimmin, M.; Davidson, A.H.; Drummond, A.H.; Galloway, W.A.; Gilbert, R.; et al. Matrix metalloproteinases and processing of pro-TNF-alpha. J. Leukoc. Biol. 1995, 57, 774–777. [Google Scholar]
- Manicone, A.M.; McGuire, J.K. Matrix metalloproteinases as modulators of inflammation. Semin. Cell Dev. Biol. 2008, 19, 34–41. [Google Scholar]
- Clutterbuck, A.L.; Asplin, K.E.; Harris, P.; Allaway, D.; Mobasheri, A. Targeting matrix metalloproteinases in inflammatory conditions. Curr. Drug Targets 2009, 10, 1245–1254. [Google Scholar] [CrossRef]
- Keith, T.; Araki, Y.; Ohyagi, M.; Hasegawa, M.; Yamamoto, K.; Kurata, M.; Nakagawa, Y.; Suzuki, K.; Kitagawa, M. Regulation of angiogenesis in the bone marrow of myelodysplastic syndromes transforming to overt leukaemia. Br. J. Haematol. 2007, 137, 206–215. [Google Scholar]
- Tsimberidou, A.M.; Giles, F.J. TNF-alpha targeted therapeutic approaches in patients with hematologic malignancies. Exp. Rev. Anticancer Ther. 2002, 2, 277–286. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Kruger, A.; Kates, R.E.; Edwards, D.R. Avoiding spam in the proteolytic internet: Future strategies for anti-metastatic MMP inhibition. Biochim. Biophys. Acta 2010, 1803, 95–102. [Google Scholar]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Marquez-Curtis, L.A.; Shirvaikar, N.; Turner, A.R.; Mirza, I.; Surmawala, A.; Larratt, L.M.; Janowska-Wieczorek, A. Membrane Type-1 Matrix Metalloproteinase Expression in Acute Myeloid Leukemia and Its Upregulation by Tumor Necrosis Factor-α. Cancers 2012, 4, 743-762. https://doi.org/10.3390/cancers4030743
Marquez-Curtis LA, Shirvaikar N, Turner AR, Mirza I, Surmawala A, Larratt LM, Janowska-Wieczorek A. Membrane Type-1 Matrix Metalloproteinase Expression in Acute Myeloid Leukemia and Its Upregulation by Tumor Necrosis Factor-α. Cancers. 2012; 4(3):743-762. https://doi.org/10.3390/cancers4030743
Chicago/Turabian StyleMarquez-Curtis, Leah A., Neeta Shirvaikar, A. Robert Turner, Imran Mirza, Amir Surmawala, Loree M. Larratt, and Anna Janowska-Wieczorek. 2012. "Membrane Type-1 Matrix Metalloproteinase Expression in Acute Myeloid Leukemia and Its Upregulation by Tumor Necrosis Factor-α" Cancers 4, no. 3: 743-762. https://doi.org/10.3390/cancers4030743
APA StyleMarquez-Curtis, L. A., Shirvaikar, N., Turner, A. R., Mirza, I., Surmawala, A., Larratt, L. M., & Janowska-Wieczorek, A. (2012). Membrane Type-1 Matrix Metalloproteinase Expression in Acute Myeloid Leukemia and Its Upregulation by Tumor Necrosis Factor-α. Cancers, 4(3), 743-762. https://doi.org/10.3390/cancers4030743