
Atomic-Scale Insights into Carbon Dioxide Hydrogenation over Bimetallic Iron–Cobalt Catalysts: A Density Functional Theory Study

Department of Chemical Engineering, Izmir Institute of Technology, Urla, 35430 Izmir, Turkey
*
Author to whom correspondence should be addressed.
Catalysts 2023, 13(11), 1390; https://doi.org/10.3390/catal13111390
Submission received: 22 September 2023
/
Revised: 13 October 2023
/
Accepted: 17 October 2023
/
Published: 24 October 2023
(This article belongs to the Section Catalytic Reaction Engineering)
Abstract
:The conversion of carbon dioxide to fuels and chemicals is a promising long-term approach for mitigating CO2 emissions. Despite extensive experimental efforts, a fundamental understanding of the bimetallic catalytic structures that selectively produce the desired products is still lacking. Here, we report on a computational surface science approach into the effect of the Fe doping of Co(111) surfaces in relation to CO2 hydrogenation to C1 products. Our results indicate that Fe doping increases the binding strength of surface species but slightly decreases the overall catalytic activity due to an increase in the rate-limiting step of CO dissociation. FeCo(111) surfaces hinder hydrogenation reactions due to lower H coverages and higher activation energies. These effects are linked to the Lewis basic character of the Fe atoms in FeCo(111), leading to an increased charge on the adsorbates. The main effect of Fe doping is identified as the inhibition of oxygen removal from cobalt surfaces, which can be expected to lead to the formation of oxidic phases on bimetallic FeCo catalysts. Overall, our study provides comprehensive mechanistic insights related to the effect of Fe doping on the catalytic behavior and structural evolution of FeCo bimetallic catalysts, which can contribute to the rational design of bimetallic catalysts.

1. Introduction
Climate change, highly influenced by CO2 emissions, is a significant and ongoing challenge. The catalytic hydrogenation of CO2 to fuels and chemicals provides a promising long-term approach to mitigating the emission of CO2 into the atmosphere [1]. The Fischer–Tropsch synthesis is an industrially applied method for synthesizing hydrocarbons using synthesis gas (syngas), which is a mixture of CO and H2. In addition to syngas, a mixture of CO2, CO, and H2 can be used as the feed for the Fischer–Tropsch synthesis (FTS), which is called CO2-FTS. However, although active and selective catalysts are well known for use in FTS based on synthesis gas, optimizing the catalytic performance of CO2-FTS has proven challenging and, therefore, is an area of active academic research.
For FTS, although several transition metals, including Ru and Ni, have been investigated scientifically, Co- and Fe-based catalysts have been applied industrially for almost a century. Co-based catalysts are mainly used for converting synthesis gas with high H2 ratios, typically obtained from natural gas, to mainly paraffinic long-chain hydrocarbons. Co catalysts provide technical benefits in the form of high activity, high selectivity to paraffins, and low CO2 formation [2]. The other industrially proven catalytic metal for FTS, Fe, is typically used to convert synthesis gas with lower H2 ratios, such as that obtained from coal due to its water–gas shift (WGS) activity, which allows for the in situ modification of H2 pressures under operating conditions. Fe-based catalysts are cheaper compared to those that are Co-based, more resistant to poisons, and offer a more diverse range of products, including olefins and oxygenates, in addition to a mixture of short- and long-chain paraffins. Under applied FTS operating conditions, the active phase of cobalt catalysts is widely accepted as metallic Co nanoparticles, while for iron catalysts, a complex phase of mixed oxides and carbides is deemed responsible [3].
Although the structure and activity relationships are relatively well understood for Co-based FTS based on synthesis gas due to extensive research efforts; switching from the use of CO to CO2 within synthesis gas leads to important changes in terms of selectivity. Co-based catalysts that have a high selectivity to long-chain paraffins under a CO atmosphere mainly produce CH4, an undesired by-product, under CO2 pressure [4]. This shift in selectivity for cobalt nanoparticles was experimentally hypothesized to be related the ratio of the surface CO-to-H coverage on cobalt nanoparticles [4], which was later supported by Density Functional Theory (DFT) [5], combined experimental and modeling analyses [6], and microkinetic modeling [7] based on DFT calculations. Various research efforts have focused on modifying the selectivity of Co-based catalysts for CO2 hydrogenation, using structural and electronic modifications and including the use of promoters [8], the optimization of the support [9], and utilizing Co-based bimetallic catalysts.
Bimetallic iron–cobalt (FeCo) catalysts have been experimentally reported to modify selectivity towards decreased methane and increased CO, olefins, oxygenates, and multi-carbon products for CO2-FTS. Although these selectivity improvements are important, the contradictory results regarding increased selectivity to different products is problematic. For example, Satthawong et al. investigated the effect of a K promoter on the olefin selectivity of bimetallic FeCo catalysts [10]. In this study, alumina-supported FeCo bimetallic catalysts synthesized by adding a small amount of Co (with an atomic ratio of 0.17 to the total metal) to Fe-based catalysts were examined for CO2-FTS, which were carried out at 300 °C and 11 bar conditions. While olefin selectivity is not observed in bimetallic catalysts without K, it has been observed that as the K content is increased, olefins become the main product, methane selectivity decreases from around 50% to 10%, and the total selectivity of the multi-carbon (C2+ and C5+) products can be increased by up to 69%. A 5% increase in CO selectivity was observed.
The observed changes in the selectivity of the catalyst due to the addition of K were associated with the binding energies of the CO2 and H adsorbates based on TPD measurements. Based on these findings, the authors speculated that Co and K promoters increased the reverse WGS (RWGS) activity of the catalyst and olefin selectivity due to the decrease in H concentration, while the formation of olefins increased due to higher CO2 and CO coverage on the surface. However, as the study did not include a surface characterization or mechanistic insights, their hypothesis could not be verified for the specific active phase formed on the bimetallic catalyst.
Recently, Wang et al. [11] found that Co doping could modify the performance of K-promoted Fe-based bimetallic catalysts, by increasing activity while also increasing the selectivity to CH4, C2–C4 olefins, and C5+ hydrocarbons. In order to obtain a mechanistic explanation of the observed behavior, the authors investigated CO2, CO, and H2 adsorption energies on pure and Co-doped Fe and K-Fe oxide/carbide surfaces using DFT modeling for the proposed active phases of Fe-rich bimetallic FeCo catalysts. Their results pointed out that Co doping increases the binding strength of CO2 and CO, while weakening H2 adsorption on iron oxide surfaces. However, for iron carbide surfaces, negligible effects were observed for CO2 and CO bonding due to cobalt doping, while H2 adsorption became stronger. Together, these effects were hypothesized to lead to long-chain hydrocarbon selectivity, via increased CO and H coverages on carbides, mainly due to the effect of the K promoter.
An interesting research area that offers possibilities for selectivity optimization is the combined use of (bi)metallic active sites with zeolites. Several advances in catalytic performance have been obtained with this approach for CO2 conversion, such as a high selectivity to gasoline/jet fuel [12], olefins [13,14], and aromatics [15]. Zeolites can provide unique advantages for catalytic transformation when combined with metallic particles, as outlined elegantly in a recent review [16]. Among these are the introduction of nano-confinement effects, the control of acidic/basic sites, the stable control of metallic particle sizes, the tuning of the hydrophobicity/hydrophilicity of surface sites, and the adjustable proximity of multifunctional metal/Si-Al sites [16]. These advantages result from the unique structural features of zeolites, such as their controllable pore size, acidity, and framework topologies, which can bring unprecedented performance to catalytic nanoparticles in terms of their selectivity, stability, and activity. In relation to CO2 hydrogenation, Dong et al. [13] utilized FeCo bimetallic catalysts supported by ZIF-67, a zeolite-like imidazole metal–organic framework, to obtain high light-olefin selectivity at a high CO2 conversion. Guo et al. [14] examined the selectivity of K-, Ce-, or L-doped Y-zeolite-supported Co@Fe catalysts to long-chain olefins. In their tests performed at 380 °C and 10 bar, and the highest C4+ olefin selectivity was obtained with the K promoter. In catalyst characterization studies, the surface structure of the catalyst was examined by XPS, while surface acidity was examined by NH3-TPD and surface basicity by CO2-TPD. As a result of these investigations, it was observed that adding Co to Fe-based catalysts increased the strong acid and base sites on the surface. In addition, through XPS measurements, it was observed that cobalt atoms were located on the surface as metallic and oxidic structures, and the authors suggested that these structures could play a role in C=O splitting and C–C coupling by working together with Fe5C2 active phases [14]. However, the conclusion of the authors linking decreased CO coverage to increased olefin production is in contrast to the hypothesis provided by Satthawong et al. [10] on bimetallic FeCo catalysts.
Although it is obvious that zeolite supported metallic particles can offer unique advantages in catalytic performance, the identification of the fundamental factors, or the descriptors behind the observed advantages, which can stem from pore size, electronic, and/or mass transfer effects, can be challenging. In addition, a key difficulty in zeolite-based catalysts is the absence of synthesis protocols that could allow for the reproducible production of the controlled zeolite characteristics at the atomic scale [16]. Furthermore, the understanding of the catalytic phase, structure, and selectivity relationships of bimetallic phases without the effect of supports/promoters, as investigated in our study, is also an essential step towards the design of zeolite-based bi-metallic catalysts.
In contrast to the mentioned studies that mainly investigate the effect of Co doping on Fe-based bimetallic catalysts, a limited number of studies also focused on the effect of Fe doping on the CO2 hydrogenation performance of Co-based catalysts. One of the studies revealing comprehensive relationships between structural characterization and product selectivity on cobalt-based Fe-doped bimetallic catalysts is by Gnanamani et al. [17]. The authors examined unsupported Co-doped Fe and Fe-doped Co catalysts with varying Fe–Co ratios for CO2-FTS in the temperature range of 220–270 °C and under 10 bar pressure, concluding that Fe doping reduced the activity and H2 consumption rate with respect to monometallic Co-based catalysts. It was also observed that when Fe was added to Co-based catalysts at only a 10% atomic ratio, C2–C5 (paraffin) selectivity increased from 4% to 20%, and with higher (50%) Fe additions, and that CO and oxygenate (mainly in the form of methanol and acetic acid) selectivity increased while CH4 selectivity decreased. Since it was observed that these positive changes in selectivity decreased when the Fe ratio was increased by more than 50%, the optimum bimetallic catalyst composition was determined to be 50%Co–50%Fe.
As a result of experimental characterizations, it was observed that the catalyst structure depends on the gas atmosphere used during the activation of the catalysts. While the FeCo bimetallic alloy and small amounts of metallic Fe and Co structures were mainly observed in the catalysts activated with H2, quite complex phases were observed in the catalysts activated with CO. It was determined that the phases observed in the bulk structure of the catalyst included Fe3O4, CoFe2O4, Fe5C2, FeCo, Co2C, CoO, Co, and Fe metals and their mixed forms. In particular, monometallic Co catalysts were in mainly metallic fcc-Co and partially reduced CoO phases. With the addition of small amounts of Fe (10% atomic), the oxidic cobalt phases diminished, while mixed FeCo oxidic and carbidic phases started to appear. The authors [17] therefore concluded that the addition of iron enhanced the reduction and carbidization of cobalt catalysts.
Ischenko et al. [18] also investigated the effect of Fe doping of Co nanoparticles related to CO2 methanation at 30–450 °C and 0.1 MPa. They concluded that Fe doping reduced the activity of cobalt nanoparticles for methanation while slightly (up to 8%) shifting the selectivity to CO. Based on their surface characterizations utilizing XPS, they also concluded that Fe doping resulted in the formation of surface FeOOH species, in addition to the metallic Co, CoO, and FeCo alloy phases on nanoparticle surfaces. They mentioned that the formation of surface FeOOH phases could be correlated with lower desorption rates of CO2, CO, and H2O from the bimetallic FeCo catalysts.
Sandupatla et al. [19] reported on the effect of Fe doping on the CO2 hydrogenation performance of alumina supported cobalt catalysts at 250 °C and 1 bar pressure. They mentioned that 10% atomic doping of Fe doping increases the methane yield while decreasing CO yield. As a result of the experimental characterizations, a metallic FeCo alloy was observed, and Fe doping was proposed to promote the reduction of cobalt oxide to the metallic phase while also promoting the formation of mixed iron-cobalt oxide phases. With the DFT calculations, it was found that the direct dissociation energy of CO2 increased from 34 kJ·mol−1 to 47 kJ·mol−1 as a result of the addition of 0.11 ML Fe on the Co(111) surface, which was linked to the lowered CO yield on FeCo catalysts compared to cobalt. The increased methane yield obtained in the study are in contrast to the results reported by Ischenko et al. [18], while the DFT modeling of CO2 dissociation reaction alone is not sufficient to explain the higher methane yields obtained on FeCo catalysts.
Recently, more in-depth reports were published related to the structure–activity relationships of Co-rich bimetallic FeCo catalysts, combining experimental observations with DFT modeling. Zhang et al. [20] reported a Na-promoted FeCo catalyst, which could selectively convert CO2 to jet fuels in the C8–C16 range at 240 °C and 3 bar. Based on experimental data and DFT modeling, they concluded that FeCo alloy in the metallic phase could be the active phase for the high selectivity to the desired hydrocarbons, as it hinders methane formation (CH hydrogenation) and promotes CH coupling when promoted by Na species. In particular, they compared metallic Co(10–11), CoO(100) and FeCo(110) alloy surfaces for CH hydrogenation and CH–CH coupling reactions. Their results showed that although the FeCo alloy surface favors CH hydrogenation over coupling compared to metallic Co, in the presence of Na, the FeCo surface promotes coupling over hydrogenation. In order to explain the high CH coverage necessary for the long-chain hydrocarbon selectivity, the postulated that Fe oxide species formed on the bimetallic surface could facilitate the RWGS formation and increase CO and CH coverages on the surface. It is also mentioned in their study that no carbidic phases of Fe or Co were detected on bimetallic nanoparticles. Despite these valuable and in-depth explanations, it is unclear from their study how Fe doping affects CO and CH formation from CO2, how Fe or mixed FeCo oxidic phases form on the bimetallic surface, and how paraffins are preferred over olefinic products.
Similarly, Hwang et al. [21] synthesized a K-promoted FeCo catalyst derived from N-coordinated Co single-atom carbon that had high C5+ selectivity and low CH4 selectivity for CO2 conversion at 300 °C and 2.5 MPa. Their spectroscopic investigations revealed that the catalysts were present in both carbidic and oxidic phases during the reaction. Based on DFT modeling, they reported extensive results for direct CO2 dissociation, O hydrogenation, carbon hydrogenation, and CH coupling over a wealth of surfaces, including pure Co and Fe, FeCo alloy, Fe-doped Co oxide/carbide and Co-doped Fe oxide/carbide surfaces. They concluded that mixed oxide surfaces promote RWGS reaction (assumed to be represented by direct CO2 dissociation and OH formation) compared to monometallic oxides, while mixed carbidic FeCo surfaces are likely to be responsible for the FTS reaction (assumed to be represented by carbon hydrogenation vs. CH coupling). They noted that although the FeCo alloy in the metallic phase may also be responsible for C–C coupling, it may not be selective to higher hydrocarbons as it has similar ΔG values for carbon hydrogenation and C–C coupling. However, the lack of full mechanistic pathways on CO2 dissociation, oxygen removal, and methane formation makes it hard to compare the performance of different catalytic surfaces investigated in the study.
Based on the discussed studies in the literature, it can be concluded that there is no consensus over the active sites that are formed on FeCo bimetallic catalysts (metallic, carbidic, or oxidic). Furthermore, a detailed mechanistic understanding related to CO2 and CO dissociation (direct or H-assisted), oxygen removal, and the formation of different products, such as CO or oxygenates, due to Fe doping is lacking. In this study, we compare the full mechanistic pathways from the reactants CO2 and H2 to water and C1 products, including CH4, CO, and CH3OH, on monometallic Co(111) and bimetallic FeCo(111) surfaces. We investigate all mechanistic pathways to take into account the modifications on preferred pathways on Co(111) due to the change of electronic structure by Fe doping. Our results indicate that Fe doping, which is segregated to the topmost Co(111) layers due to a stronger interaction with CO, yields higher CO and lower H coverages on cobalt surfaces during CO2 hydrogenation, leading to the decreased hydrogenation ability of the surface. While the bimetallic FeCo(111) surface is not found to be responsible for CO or CH3OH formation, the main of effect of Fe doping is found to inhibit oxygen removal from the FeCo(111) surface, possibly leading to the conversion of the metallic FeCo surfaces to mixed oxidic FeCo surfaces. Our findings provide atomic-scale insights into the performance of metallic FeCo phases and their structural evolution, which can contribute to the design of high-performance bimetallic catalysts for CO2 conversion.
2. Results and Discussion
2.1. Adsorption of Species Involved in CO2 Hydrogenation to Single Carbon Products on Co(111) and FeCo(111) Surfaces
Surface coverages of intermediates can have a significant effect on the evolution of active sites and the catalytic performance in CO2 hydrogenation. Therefore, we investigated the adsorption of major surface intermediates involved in the production of C1 species from CO2 hydrogenation, including H, C, O, OH, CO, CO2, CH, CH2, CH3, CH4, H2O, HCO, COOH, HCOO, H2CO, H3CO, and H3COH, on Co(111) and FeCo(111) surfaces. The adsorption sites investigated on Co(111) and FeCo(111) surfaces are shown in Figure S1. The most stable geometries for surface species on Co(111) and FeCo(111) surfaces are shown in Figure 1, while the adsorption energies are listed in Table 1.
Table 1 demonstrates that the introduction of 1 ML Fe on the Co(111) surface led to increased adsorption energies of all the chemisorbed and physisorbed surface species, with the exception of physisorbed H2 and CH4. The adsorption energies of species calculated on Co(111) are in line with the previous studies as shown in Table S2 [5,22,23,24,25]. The comparison of the physisorbed species show that Fe doping increased the binding strength of oxygenated species like CO2 and H2O without affecting the hydrogenated species of H2 and CH4. In particular, H2O adsorption is significantly promoted, and the adsorption mode shifts from physisorption to weak chemisorption. The effect of Fe on chemisorbed species also shows a similar trend. Oxygenated species, including CO groups (CO and HxCO), have much higher adsorption energies (31 to 116%) on FeCo(111) compared to the hydrogenated species, such as OH and CHx, which have relatively minor (10 to 19%) increases. As indicated in Table 1, the differences in the relative change of adsorption strength for these different species are not due to changes in adsorption sites. From Figure 1, it is shown that the effect of Fe on adsorption sites is limited as adsorbates continue to occupy the same top and hollow sites on FeCo(111) as on Co(111).
These results show that CO2 and CO coverage would increase on Fe-doped Co(111) surfaces, while the coverage of hydrogen and hydrogenated species would decrease. Therefore, Fe doping results in less hydrogenating surface conditions on the Co(111) surface. This analysis supports and provides an explanation for the observation by Gnanamani et al. [17] that Fe doping results in decreased H2 consumption and methane selectivity.
Fe doping is also expected to contribute to the modification of elementary reaction kinetics on the Co(111) surface. In order to understand how Fe doping affects the complex reaction network of CO2 hydrogenation on the Co(111) surface, we investigated the elementary reactions involved in CO2 hydrogenation to major C1 products.
2.2. H2 Dissociation
As indicated by van Helden et al. [26], H2 dissociation on cobalt surfaces takes place as two elementary reactions in series. First, H2 has to migrate from a far physisorbed state to a close physisorbed state. In the second step, H2 dissociation takes place from this activated precursor state. Our calculations also indicate that H2 dissociation takes place in two consecutive steps on both Co(111) and FeCo(111) surfaces, as shown in Figure 2.
As can be seen in Figure 2, the physisorption of hydrogen molecule on the Co(111) surface proceeds with an activation energy of 26 kJ·mol−1 and a reaction energy of 13 kJ·mol−1 (endothermic). The physisorption of hydrogen molecule on the FeCo(111) surface proceeds with an activation energy of 14 kJ·mol−1 and a reaction energy of −9 kJ·mol−1 (exothermic). The reaction energy of H2 physisorption on Co(111) is higher by 12 kJ·mol−1 than that on Fe-doped Co(111), which indicates that the incorporation of Fe in Co(111) favors H2 physisorption. In the second step that involves the dissociation of molecular H2 that is physisorbed closely on the catalytic surface, the addition of Fe on Co(111) decreases the activation energy of H2 dissociation by 20 kJ·mol−1 by making the reaction energy more exothermic (by 30 kJ·mol−1). The configurations of the IS, TS, and FS structures on Co(111) and FeCo(111) are given in Figure 3.
The configurations shown in Figure 3 indicate that Fe incorporation does not lead to a change in the H2 dissociation mechanism, and that the reaction configurations are similar on both surfaces. Therefore, the effect of Fe doping for H2 dissociation on Co(111) can be explained to be due to the stronger adsorption of hydrogen atoms on the surface, which increases the exothermicity and in turn decreases the activation energy for dissociation.
These results show that the introduction of Fe promotes the physisorption and dissociation of H2 on Co(111) surface. These results are rather surprising given the higher hydrogenation ability of cobalt surfaces compared to iron facets. However, it should be kept in mind that H2 dissociation and surface hydrogen coverage is also significantly affected by coadsorbed CO on the catalytic surface [6,27]. Therefore, we further discuss the effect of Fe doping on the surface CO-to-H ratio after analyzing CO2 and CO dissociation reactions.
2.3. CO2 Dissociation
The dissociation of carbon dioxide on the cobalt surface can occur via direct or H-assisted dissociation routes [23,24]. The direct dissociation of CO2 is also referred to as the redox mechanism, while in the H-assisted dissociation, CO2 dissociation via COOH formation is known as the carboxylate, and via HCOO formation is known as the formate mechanism. We investigated the effect of Fe doping on these mechanistic pathways on Co(111). Energy profiles for CO2 dissociation on Co(111) vs. FeCo(111) are shown in Figure 4.
- (a)
- Direct CO2 Dissociation
Figure 4a illustrates that the direct CO2 dissociation on Co(111) proceeds with an activation energy of 61 kJ·mol−1 and a reaction energy of −77 kJ·mol−1, while on FeCo(111), it has an activation energy of 17 kJ·mol−1 and a reaction energy of −152 kJ·mol−1. Based on the increase in exothermicity of the reaction and decrease in the activation energy, the results indicate that the presence of 1 ML Fe coverage on the Co(111) surface promotes direct CO2 dissociation, leading to higher rates of CO and O formation on the FeCo(111) surface. Configurations of the structures associated with direct CO2 dissociation on Co(111) and FeCo(111) are shown in Figure 5.
- (b)
- H-assisted CO2 Dissociation
In the hydrogen-assisted pathway, CO2 dissociation takes place after the formation of COOH (carboxylate) or HCOO (formate) intermediates. Energy profiles for H-assisted CO dissociation via the carboxylate mechanism on Co(111) vs. FeCo(111) are shown in Figure 5b.
Figure 4b shows that the activation energy for COOH formation increased from 94 kJ·mol−1 for Co(111) to 149 kJ·mol−1 for FeCo(111). This increase in the activation energy is also consistent with the change in reaction energy from −18 kJ·mol−1 (exothermic) for Co(111) to 41 kJ·mol−1 (endothermic) for FeCo(111). A comparison of the results for Co(111) and FeCo(111) shows that H-assisted CO2 dissociation can take place on Co(111), with an activation energy of 94 kJ·mol−1, although with a slower rate compared to direct dissociation (Ea of 61 kJ·mol−1) if the activation energies are comparable with H-assisted dissociation via formate mechanism. However, Fe doping completely inhibits CO2 dissociation via the carboxylate mechanism as the sizeable activation energy of 149 kJ·mol−1 will not take place under typical temperatures, 210 to 240 °C, used for CO2 hydrogenation on cobalt catalysts. Configurations of the structures associated with CO2 dissociation via carboxylate mechanism on Co(111) and FeCo(111) are shown in Figure 6.
In the H-assisted CO2 dissociation pathway, CO2 can be hydrogenated to formate (HCOO) species and further dissociate to HCO and O in the so-called formate mechanism. Energy profiles for the H-assisted CO2 dissociation via the formate mechanism on Co(111) vs. FeCo(111) are shown in Figure 5c.
The results outlined in Figure 4 point out to important mechanistic implications for CO2 dissociation due to Fe doping on Co(111). On Co(111), H-assisted CO2 dissociation have activation energies of 94 and 71 kJ·mol−1 for carboxylate and formate mechanisms. Compared to the direct dissociation energy of 61 kJ·mol−1, these results indicate that CO2 dissociation is not expected to take place on Co(111) via the H-assisted carboxylate mechanism. CO2 dissociation would mainly proceed via the direct dissociation pathway on Co(111) due to having the lowest activation energy among the three dissociation pathways, while H-assisted CO dissociation via the formate mechanism would also contribute to the reaction due to having a comparable activation energy with the direct dissociation and the high H concentration that is expected to be present on Co(111) under experimental conditions. These findings are also in line with a recent computational study performed on Co(0001) [24]. On FeCo(111), the formate mechanism is still preferable compared to carboxylate mechanism due to its reasonable activation energy of 51 kJ·mol−1. In contrast to the carboxylate pathway, Fe doping results in the reduction in activation energy for both HCOO formation and dissociation by 20 and 6 kJ·mol−1 respectively for formate mechanism. Configurations of the structures associated with CO2 dissociation via formate mechanism on Co(111) and FeCo(111) are shown in Figure 7.
An investigation of the TS structures shown in Figure 7 reveals that on FeCo(111), CO2 species are located closer to the surfaces, compared to the case on Co(111). This difference, which is mainly due to stronger interactions between the catalytic surface and CO2, CO, and O species, can be expected to result in the reduced activation energies for HCOO formation on FeCo(111) compared to Co(111). Despite the promotion of CO2 dissociation via formate mechanism on FeCo(111), the difference between direct CO2 dissociation and H-assisted CO2 dissociation is increased from 10 kJ·mol−1 on Co(111) to 34 kJ·mol−1 on FeCo(111) surface. Therefore, an evaluation made by considering direct and H-assisted CO2 dissociation mechanisms indicates that although the CO2 dissociation takes place predominantly by the redox mechanism and minorly by formate mechanism on Co(111), it would take place predominantly by direct CO2 dissociation on FeCo(111). Further hydrogenation of HCOO or COOH species to HCOOH (formic acid) is not investigated in this study, as H-assisted CO2 dissociation does not take place on the FeCo(111) surface and as there is a consensus in the experimental literature that HCOOH formation does not take place during CO2 hydrogenation on cobalt catalysts [3]. In summary, our findings indicate that Fe doping inhibits H-assisted CO2 dissociation on Co(111) while promoting the direct CO2 dissociation.
2.4. CO Dissociation
Energy profiles for CO dissociation on Co(111) vs. FeCo(111) are shown in Figure 8.
- (a)
- Direct CO Dissociation
Similar to the case of CO2 dissociation, CO dissociation can take place on the catalytic surface via direct or H-assisted pathways. Energy profiles for the direct CO dissociation on Co(111) vs. FeCo(111) are shown in Figure 8a.
Figure 8a shows that the direct dissociation of CO on the FeCo(111) surface is more exothermic than on the Co(111) surface, with a significant decrease in the activation energy from 267 kJ·mol−1 to 173 kJ·mol−1. Despite the significant decrease in the activation energy, direct CO dissociation reaction is not expected on FeCo(111) surfaces, as on Co(111) surfaces, since the 173 kJ·mol−1 activation energy is not feasible under typical low temperature CO2 hydrogenation conditions used for cobalt catalysts. Therefore, despite significant increase in the exothermicity of the reaction, Fe doping does not induce direct CO dissociation on Co(111) surfaces. Configurations of the structures associated with direct CO dissociation on Co(111) and FeCo(111) are shown in Figure 9.
- (b)
- H-assisted CO Dissociation
In the H-assisted dissociation mechanism, CO is first hydrogenated to HCO. The dissociation of CO is feasible from the HCO intermediate, as well as via further hydrogenation to H2CO and successive dissociation. The hydrogenation of CO to COH is not investigated in our study, as it is reported to be hindered by both thermodynamic and kinetic limitations [25,28]. Energy profiles for the H-assisted CO dissociation on Co(111) vs. FeCo(111) are shown in Figure 9b.
The results shown in Figure 8b point out that HCO formation is slowed down due to a slight (3 kJ·mol−1) increase in the activation energy, while its dissociation is promoted due to a decrease in the activation energy. Combining these findings on the effect of Fe doping on CO2 and CO dissociation mechanisms, it can be inferred that CO2 dissociation will proceed mainly via the direct pathway, while CO dissociation will occur via the H-assisted pathway on both Co(111) and FeCo(111) surfaces. Configurations of the structures associated with H-assisted CO dissociation on Co(111) and FeCo(111) are shown in Figure 10.
As Fe doping hinders the hydrogenation of both CO2 and CO while promoting CO formation via direct CO2 dissociation, this leads to increased CO coverages on the FeCo(111) surface compared to that of Co(111), while HCO coverage is reduced due to slower kinetics of formation, and a faster dissociation. Overall, these results point out three important effects of Fe doping on the activation of CO2 on cobalt surfaces. Firstly, Fe doping would promote the formation of a CO-rich, hydrogen-deficient adsorbate pool on the catalytic surface, as indicated both by the modification of adsorption energies and the kinetics of CO2/CO dissociation. Secondly, as it increases the activation energy of the rate-limiting step, HCO formation, Fe doping would decrease the overall activity of cobalt surfaces for CO2 activation. Thirdly, Fe doping results in a significantly high (173 kJ·mol−1) CO adsorption energy, while the activation energy for its H-assisted dissociation stays much lower at 114 kJ·mol−1. This ratio would inhibit the desorption of CO from the bimetallic FeCo(111). Therefore, FeCo(111) surface is not the active phase that leads to increased CO selectivity, which was observed in bimetallic FeCo catalysts in the literature [17].
2.5. CH4 Formation
Methane is reported to form on cobalt surfaces via the sequential hydrogenation of C or CH species that are formed as a result of CO2/CO dissociation [5,29]. Energy profiles for CHx hydrogenation on Co(111) and FeCo(111) are given in Figure 11.
The results indicate that Fe doping hinders the hydrogenation of CHx species by making the reaction steps more endothermic and increasing the activation energies by ~20 kJ·mol−1. The rate-limiting step for CH4 formation on Co(111), CH3 hydrogenation, remains the same on FeCo(111). Despite the increase in activation energies, the rate-limiting step of CH3 hydrogenation is still feasible on the FeCo(111) surface, as its activation energy of 110 kJ·mol−1 would still be overcome at typical low-temperature CO2 hydrogenation conditions. Therefore, combined with the effect of Fe on the adsorbates on Co(111), which makes the surface more hydrogen-deficient, these results point out that Fe doping would decrease the selectivity to CH4 on cobalt surfaces. Configurations of the structures associated with CH4 formation on Co(111) and FeCo(111) are shown in Figure 12.
As C is mainly removed as CH4 on Co(111) and FeCo(111) based on our analysis, these results can also be used to obtain a basic understanding of coke formation on Co and FeCo catalysts. Fundamentally, coke formation occurs due to a higher rate of carbon build-up on catalytic surfaces compared to its removal. Our results based on CO2 and CO activation indicate that the rate of carbon build-up is slightly reduced on FeCo(111) surfaces compared to Co(111) due to a minor increase in the rate-limiting step of HCO formation. Carbon removal via methane formation can also be expected to occur sufficiently on FeCo(111) as on Co(111), although with slower kinetics, as the activation energy of 110 kJ·mol−1 for the rate-limiting step of methane formation can be overcome under CO2-FTS conditions. Therefore, our results do not indicate that Fe doping results in a strong driving force for coke formation on FeCo(111) surfaces compared to Co(111). However, coke formation is a complex process and can also be significantly affected by structural and temperature effects [23]. Therefore, a more in-depth study including microkinetic modelling and surface/subsurface carbon build-up would be necessary to fully compare the coke formation on Co vs. FeCo catalysts, which is beyond the scope of this study.
2.6. H2O Formation
Water is a major product in CO2/CO hydrogenation on cobalt catalysts, as most oxygen is removed as H2O. Furthermore, based on recent experimental surface science investigations [30,31] and computational analyses [27], it is proposed as the rate-limiting step in CO-based FTS. In particular, OH formation is reported to be the elementary step with a high activation energy of ~120 kJ·mol−1 [31] that limits the rate of H2O formation. After OH is formed on the catalytic surface, H2O formation is reported to proceed via the coupling of OH species instead of OH hydrogenation due to OH coupling having a much lower activation energy compared to OH hydrogenation. Therefore, understanding how Fe doping effects OH formation on Co(111) is important as it can result in major effects, related to the catalytic activity and selectivity. Energy profiles for OH and H2O formation are given in Figure 13, while configurations of the structures associated with H2O formation on Co(111) and FeCo(111) are shown in Figure 14.
Figure 13a shows that OH formation on FeCo(111) is severely inhibited, as Fe doping increases its activation energy from 124 to 168 kJ·mol−1. This important result indicates that O cannot be removed from the bimetallic FeCo(111) surface as water, as the high activation energy would prevent the reaction from taking place under typical reaction conditions for CO2 hydrogenation on cobalt catalysts. The inhibition of OH formation may have critical consequences, such as surface oxidation as a result of O accumulation on the surface or the formation of O-containing oxygenate products. Therefore, we also investigate the formation of methanol, as a model compound for C1 oxygenates, on Co(111) and FeCo(111).
2.7. H3COH Formation
Methanol can be formed on cobalt surfaces via the hydrogenation of H2CO, which is formed as a result of CO hydrogenation, or the coupling of O with CH2 and CH3 species and further hydrogenation. Our results on Co(111) and FeCo(111) surfaces indicated that H-assisted CO dissociation takes place on Co(111) via H2CO formation, while on FeCo(111), H2CO formation is inhibited, as HCO dissociation occurs with 45 kJ·mol−1, compared to H2CO formation energy with 72 kJ·mol−1, as shown in Figure 8. However, the coupling of CHx species with O can provide a feasible pathway for oxygenate formation and also allow for the removal of oxygen from the catalytic surface. Therefore, we investigated the energetics of these pathways of methanol formation, as shown in Figure 15.
The results shown in Figure 15 indicate that on Co(111), H2CO formation via CH2 and O coupling can occur with a relatively high activation energy of 124 kJ·mol−1, while the formation of H3CO via CH3 and O coupling is not feasible due to a high activation energy of 150 kJ·mol−1. Although H2CO can be further hydrogenated to H3CO on Co(111) with a low activation energy, the formation of methanol is not possible on Co(111), due to a formidable activation energy of 177 kJ·mol−1. The structures associated with successive hydrogenation of HCO to CH3OH on Co(111) and FeCo(111) surfaces are shown in Figure 16.
These findings are in line with a computational analysis related to methanol steam reforming on cobalt [32] and the experimental literature explain that oxygenates are not part of the product spectrum for CO2 hydrogenation on cobalt catalysts [3]. On FeCo(111), the formation of both H2CO and H3CO via CHx and O coupling reactions are inhibited due to high activation energies of >150 kJ·mol−1. The formation of methanol via CH3 and OH coupling is not investigated as it was reported to have a high activation energy on Co(111) [32], and our calculations indicate that H2CO formation is kinetically hindered on FeCo(111). These results point out that Fe doping does not induce the formation of oxygenates on the metallic Co(111) surfaces. The kinetic parameters of all elementary reactions investigated in our study are summarized in Table 2 and Figure 17.
The activation energies outlined in Table 2 are in line with the literature, as demonstrated in Table S3 [22,25,32,33]. The summary of the kinetics parameters provided in Table 2 allows us to draw a comprehensive analysis related to the effect of Fe doping on Co(111) surfaces related to the catalytic performance of bimetallic FeCo catalysts for CO2 hydrogenation.
In terms of activity, on the bimetallic FeCo(111) surface, CO2 dissociation rates would increase due to a lower activation energy for the direct dissociation pathway. This would lead to higher CO coverages on the FeCo(111), compared to Co(111) surface. Further activation of CO to atomic C can only proceed via H-assisted dissociation, first yielding HCO and finally CH and O species. Although increased adsorption energies of C and O result in ~90 kJ·mol−1 lower activation energy on Co(111), the direct CO dissociation pathway is not possible on FeCo(111) due to an unsurmountable activation energy of 173 kJ·mol−1. Despite the increased affinity of the FeCo(111) surface to CO2 and CO adsorption, and an increased rate of direct CO2 dissociation, Fe doping would still decrease the overall activity of the cobalt surface for CO2 hydrogenation due to a slight increase in the activation energy for the rate-determining step of HCO formation.
In terms of selectivity, the bimetallic FeCo(111) surface demonstrates an altered performance with respect to hydrogenation reactions. In general, the hydrogenation activity of a catalytic surface depends on the hydrogen coverage and the kinetics of elementary hydrogenation reactions. As it was mentioned in both experimental [4,6] and computational [27] studies, the CO coverage is the key factor that determines the coverage of atomic H on the surface due to repulsive interactions between coadsorbed CO and H. Therefore, although there is a reduction of ~20 kJ·mol−1 for the activation energy of H2 dissociation on the FeCo(111) surface due to a significantly stronger CO adsorption and an increased rate of CO formation via CO2 dissociation, it can be expected that surface hydrogen coverage will be less on FeCo(111) compared to Co(111). Therefore, it can be concluded that FeCo(111) surfaces will have an increased CO-to-H ratio, resulting in decreased selectivity to methane. The diminished hydrogenation capacity of the FeCo(111) surface is also observed via increased activation energies for CO2 and CO hydrogenation, and most significantly reflected by the inhibition of OH formation and therefore water removal on FeCo(111). The shift of the selectivity behavior towards less hydrogenated products was observed experimentally on unpromoted and alkali-promoted FeCo bimetallic catalysts [17]. However, in the experimental studies, the catalytic phase responsible for this effect and the impact of alkali promoters could not be directly identified. Our calculations show that the hydrogenation activity already decreases on the metallic phase of the FeCo catalyst, without the need to form the carbidic and oxidic phases.
The most profound effect of Fe doping on cobalt catalysts is observed with respect to oxygen removal from the bimetallic FeCo(111) surface. On monometallic catalysts, oxygen removal takes place mainly as H2O formation, with slow kinetics due to a high activation energy for OH formation. On the bimetallic FeCo(111) surface, OH formation is inhibited due to a very high activation energy of 160 kJ·mol−1, in parallel with the decreased hydrogenation ability of the bimetallic surface. Furthermore, atomic O can also not be removed as methanol (i.e., oxygenates), as it is kinetically inhibited by unfeasible activation energies of >180 kJ·mol−1. These results show that there would be an accumulation of atomic oxygen on FeCo(111) due to CO2 and CO dissociation. In turn, this would lead to the formation of an iron oxide or mixed iron–cobalt oxide phase on the outer layers of the catalytic FeCo nanoparticles. This phenomena was also observed in CO-based FTS on bimetallic FeCo catalysts [34]. Gnanamani et al. [17] also observed the formation of iron oxides and carbides, and the diminishing of monometallic cobalt oxides on FeCo nanoparticles. In line with these experimental findings, our analysis indicates that the main effect of Fe on cobalt catalysts would therefore be the induction of the evolution for the catalytically active phase from metallic to oxidic Fe or FeCo structures. Therefore, our results indicate that the selectivity to CO or oxygenates observed on FeCo bimetallic catalysts is not associated with the incorporation of Fe in the metallic cobalt phase, but rather related to surface iron oxide and/or carbide phases that are promoted by the Fe doped cobalt surfaces.
2.8. Effect of Fe-Induced Charge Modification on Adsorbates and Elementary Reactions on Bimetallic FeCo(111) Surface
In order to understand how Fe doping alters the electronic charge of the species adsorbed onto the FeCo(111) surface, Bader charge analysis was performed and compared to the adsorbates on Co(111) and FeCo(111) surfaces, as shown in Table 3.
As outlined in Table 3, all adsorbates on Co(111) and FeCo(111) surfaces withdraw electronic charge from the catalytic surface, which is in line with the literature [5,6]. The results show that Fe doping results in significant charge accumulation on adsorbates. Therefore, the fundamental reason behind the increased adsorption energies of reactants and intermediates on FeCo(111) surface can be attributed to the additional charge on surface species due to the Lewis basic character of Fe atoms doped in Co(111). The charge density differences between surfaces covered with CO and pure surfaces are further analyzed to understand the electron donating properties of Fe atoms, as shown in Figure 18.
Figure 18 indicates that for surfaces covered with CO, Co(111) donates electrons to CO, while in the case of FeCo(111), Fe atoms donate a higher amount of electrons to CO, resulting in increased charge accumulation of the adsorbed CO molecule, while transferring some charge also to the underlying cobalt layers in FeCo(111). An analysis of the charges transferred from Fe atoms to the neighboring Co atoms vs. adsorbates species, as shown in Table S5, confirm that for all surface species, Fe atoms donate charges to both Co atoms and the adsorbate species. In order to obtain a more fundamental picture of the electronic differences between Co(111) and FeCo(111) surfaces, the Projected Density of States were compared for both surfaces, as shown in Figure 19.
As shown in Figure 19, the Fermi level of the topmost surface layer of Co atoms at the Co(111) surface is calculated as 0.9 eV, while the d-band center is located at −0.9 eV. For the bimetallic surface, the Fermi level of the Fe atoms located at the topmost layer of the FeCo(111) surface is located at 0.39 eV, while the d-band center is calculated as 0.40 eV. These values show that Fe doping at the outer layers of the Co(111) surface results in an upward shift of the d-band center towards the Fermi level. Based on the Hammer-Norskov model [35,36], such upward shifts were found to correlate with the increases in adsorption strengths for surface species on various transition-metal surfaces. Furthermore, the comparison of the PDOS in Figure 19 also shows that the d-band becomes narrower for FeCo(111) compared to Co(111). The narrowing of the d-band would also result in higher localization of the d-electrons, leading to a higher reactivity of the surface atoms and thus increased electron transfer from the surface to the adsorbates. These insights indicated by the comparison of PDOS profiles indicate that the higher adsorption strengths due to the increased electron transfer on FeCo(111) are due the upward shift of the d-band center and narrowing of the d-band compared to the Co(111) surface. These results are also in line with the results shown in Table 3, where higher electron densities were obtained on adsorbates on FeCo(111) and Co(111), together with the stronger binding of these species.
The results obtained for FeCo(111) with adsorbates complementing our findings show that Fe atoms have a Lewis basic character for both clean and adsorbate-covered Co(111) surfaces. In fact, on the FeCo(111) surface, Fe acts as an electron donor for both the adsorbate and the other cobalt layers of the slab. As illustrated in Table S4, the first layer composed of Fe atoms on the FeCo(111) catalyst surface exhibits a significant positive charge compared to the first layer of Co(111) catalyst surface. Furthermore, when the Bader charge values on the top two layers of the monometallic and bimetallic cobalt catalysts are compared with the charges of adsorbates, it can be seen that the first layer of the Co(111) surface not only charges the adsorbates but also takes charge from the other layers on the catalyst. In contrast, the Fe atoms on the FeCo(111) surface charge both the adsorbates and other layers on the catalyst. These findings indicate that Fe plays a distinct role as an electron donor on the FeCo(111) surface, leading the increased binding energies of surface species on FeCo(111) compared to Co(111).
The increased charge distribution due to Fe doping on Co(111) can also provide an explanation for the observed changes in activation energies on FeCo(111). Our findings indicate that Fe doping increases the activation energies for hydrogenation reactions on Co(111). Previously, charge transfer effects were also observed related to the effect of Pt on H2O formation and the effect of sulfur on CH4 formation on Co(111) surfaces. Govender et al. [37] showed that Pt promotes the formation of oxygen hydrogenation to OH, the rate determining step in H2O formation on cobalt surfaces, by withdrawing electrons and therefore decreasing the charge density of the Co(111) surface. Similarly, Daga and Kizilkaya [5] showed that sulfur also plays a similar effect in a more pronounced way, by significantly decreasing the charge density on Co(111), leading to an increased H/CO ratio on the surface and resulting in decreased activation energies for the hydrogenation of atomic C and H. Our results point out that for Fe doping on Co(111), a consistent effect is observed, although the charge transfer is in the opposite direction compared to the effect of Pt or S on Co(111). A synthesis of our results and the findings in the literature allow us to identify a fundamental descriptor that can be used to modify the activity of cobalt catalysts in terms of hydrogenation: Increased charge density on cobalt hinders the hydrogenation reactions while promoting the activity for CO2 activation and increasing CO coverage.
Altered charge distribution by Fe also affects the activity of the cobalt surface for CO2 activation, but in a contrasting way compared to hydrogenation. As the adsorption energies of coadsorbed CO and O, the final state of direct CO2 dissociation, are significantly increased, the FeCo(111) surface has a lowered activation energy for the reaction, compared to Co(111). A similar finding was also recently reported in an experimental study, where the effect of charge transfer on the CO2 photo-reduction on Co3O4 surfaces was investigated. Yang et al. [38] showed that due to the increased charge density on cobalt oxide surfaces, CO2 adsorption and its dissociation to CO were promoted. Our results also show that Fe doping would lead to increased CO2 and CO coverages on the FeCo(111) surface, due to increased binding strengths of the species and faster rates of CO2 dissociation, which are caused by the Lewis basic character of the Fe atoms located on FeCo(111) surfaces.
3. Computational Methodology
3.1. Computational Model
Bimetallic FeCo nanoparticles have complex structures due to the variations in the concentration of Fe and Co components, chemical phases, nanoparticles shape and size effects, and the facets exposed. In particular, it is important to note the differences in nanoparticles that are based on either Fe or Co as the dominant component. There is a consensus in the literature that Fe-based nanoparticles that are utilized in CO or CO2 hydrogenation transform to the active iron oxide and/or iron carbide phases under reaction conditions. Therefore, FeCo bimetallic nanoparticles that have higher Fe contents are generally represented by iron oxide or iron carbide facets [3], that are typically modelled by Fe-based phases doped by Co atoms in the literature [11]. On the other hand, cobalt catalysts are typically found in their metallic form, and cobalt nanoparticles that are smaller than 100 nm are reported to adopt the fcc crystal structure, exposing predominantly the most stable 111 facet [39].
It is experimentally reported that bimetallic FeCo nanoparticles form alloys that can have bulk Fe atomic concentrations of 25 to 75% within FT operating conditions of 200–350 °C [40]. Despite alloy formation at equal atomic shares, it was mentioned that for low Fe concentrations, FeCo bimetallic nanoparticles adopt the fcc phase of cobalt [41]. As the aim of this study is to investigate how Fe doping affects the catalytic performance of Co nanoparticles, we modelled the Co-rich bimetallic FeCo surfaces and the fcc-Co(111) surface, the most stable and abundant facet on fcc-Co nanoparticles.
Another important point to consider for the computational model is related to where the Fe doping takes place on the cobalt surfaces. Moran-Lopez and Wise [42] analyzed the surface composition of 50% (equal) atomic share FeCo(110) and FeCo(100) surfaces with a theoretical model based on short- and long-range order parameters and experimental characterization by AES. They concluded theoretically that the surface consists of 100% Fe, while AES results show 75% Fe enrichment at the topmost 10 Å of the surface. Similarly, Allie et al. [43] investigated the surface structure of an equal atomic share FeCo(100) alloy by AES and LEED. They concluded that the topmost layer of the surface is predominantly iron, which can be due to the high mobility of iron towards the surface, as induced by the restoring of the surface iron content after sputtering–annealing cycles. These studies show that under ultra-high vacuum conditions, the surface of a FeCo alloy is predominantly composed of Fe atoms.
Similar results related to the surface segregation of Fe species in FeCo bimetallic catalysts were also reported for FeCo nanoparticles under FTS operating conditions. Calderone et al. [41] synthesized Fe core and Co shell nanoparticle structures and used them under typical FTS conditions. Based on experimental characterizations, it was observed that the active phase in these catalysts was metallic cobalt located in the shell part. Additionally, it was proposed that Fe segregated to the upper layers of cobalt surface, and the resulting electronic modification was the probable cause for changes in catalytic selectivity in FeCo catalysts compared to the cobalt ones.
Ismail et al. [34] also synthesized bimetallic FeCo nanoparticles and utilized them in low- and high-temperature FTS conditions. It was mentioned that Fe and Co atoms were distributed homogenously in a spinel structure in the freshly synthesized structure. However, after reduction, it was found that nanoparticles converted to a FeCo alloy core and iron oxide shell, documenting the Fe segregation in bimetallic FeCo nanoparticles. Iron at the surface layers was proposed to be oxidized as it came in contact with air during its transfer to the TEM.
As these applied studies indicate, under gas atmosphere, the surface composition of nanoparticles is also determined based on their interactions with adsorbates. Specifically, it is well known that in bimetallic surfaces, the component which binds stronger to the major adsorbates is enriched under specific gas atmospheres [44]. In terms of CO and CO2-FTS, the adsorbate, which induces surface segregation, is mainly CO, as indicated by previous experimental [45] and computational [46] analyses on CoCu bimetallic surfaces. Therefore, it can be expected that the Fe enrichment of the FeCo surfaces would be due to the stronger interaction of Fe with CO, and/or atomic O, as observed in the analysis by Ismail et al. [34].
Based on these insights, we modelled a Fe-doped Co-rich bimetallic FeCo as an fcc-Co(111) surface, which is covered by varying coverages of the dopant Fe atoms at the topmost layer, as shown in Figure 20.
On this model, we investigated how the adsorption energies of CO and major atomic adsorbates change as a function of Fe coverage in order to assess Fe surface segregation in bimetallic FeCo surfaces. The results summarized in Table S1 indicate important trends related to adsorption strengths as surface Fe coverage increases. In particular, CO adsorption preferentially occurs at Fe atoms instead of Co, while CO binding strength significantly increases as Fe coverage at the topmost layer increases. For 0.33 and 1.00 ML Fe-doped Co(111) surfaces, the adsorption energies of all adsorbates demonstrated an increase, as detailed in Table S1.
When the 0.33 ML Fe-doped surface is examined, adsorption energy (Ead) increases in the range of 1–8% are observed in the adsorbates. Although the increase in the Ead values is relatively low, it is noteworthy to mention that the increase in Ead for atomic H is higher than that of the Ead of CO, implying a reduction in the CO/H concentration at the surface. In contrast, a distinct situation is observed for 1.00 ML Fe-doped FeCo(111) surface from other coverage ratios. Here, Ead for CO increased by 31%, and the Ead for atomic H increased by 9%, leading to a predicted elevation in the CO/H concentration on the surface. Moreover, there is a 9% increase in the Ead values of atomic C and O. Therefore, it can be concluded that there is a significant driving force for Fe segregation under CO and O adsorbate coverages, which is in line with the previous experimental observations for CuCo [43] and FeCo [34] bimetallic surfaces. Based on these findings, the optimum Fe contribution for bimetallic FeCo(111) surfaces was determined as 1.00 ML, that is the surface model in which the top layer of the surface is completely covered with Fe atoms. These results provide solid insights related to the surface enrichment of Fe for Co-rich fcc-FeCo(111) surfaces under CO2-FTS conditions and validate our surface model used in the investigation of bimetallic FeCo surfaces.
3.2. Computational Details
Periodic spin-polarized Density Functional Theory (DFT) calculations were performed using the Vienna Ab-initio Simulation Package (VASP) [47,48]. The exchange-correlation energy was calculated with the revised Perdew–Burke–Ernzerhof functional (revPBE) [49], including the non-local vdW–DF correlation [50,51,52,53]. The use of the vdW-DF functional allowed us to calculate the adsorption energy of CO2 accurately and also predict the experimentally observed adsorption site and energy for adsorbed CO on the cobalt surface [54]. The surfaces were cut from a bulk fcc-Co structure with a lattice parameter of 3.56 Å, optimized with the vdW-DF functional. The calculated lattice parameter is in line with the experimental value and the previous DFT calculated values [5].
The electron–ion interactions were modeled using the projector-augmented wave (PAW) method. Spin-polarized calculations were performed to account for the magnetic properties of cobalt with a plane wave cut-off energy of 500 eV. A 15 Å vacuum region was placed above the slabs to avoid electronic interactions in the z-direction. For the Brillouin-zone integration, a (3 × 3 × 1) Monkhorst-Pack k-points grid [55] was utilized. The surface terraces of face-centered cubic (fcc) Co nanoparticles and FeCo bimetallic nanoparticles in the metallic phase were modeled with Co(111) and FeCo(111) slabs with four atomic layers. The number of layers, the size of the k-point mesh, the height of the vacuum region, and the cut-off energy were optimized based on the adsorption energy of CO on Co(111) surface. A further increase in these parameters above the ones used in the calculations led to less than 5 kJ·mol−1 change in CO adsorption energy on Co(111). The optimization of adsorbate structures and reaction steps was performed on unit cells of the slabs with p(3 × 3) periodicity in the x and y directions, which corresponds to a 0.11 ML adsorbate coverage on catalytic surfaces. This coverage was utilized in previous DFT investigations on cobalt surfaces [23,54] that led to accurate results, which matched well with the experimental observations on cobalt surfaces.
The effect of the periodic boundary conditions were negligible on the results reported in this study, as the use of a p(4 × 4) unit cell of Co(111) resulted in the change of adsorption energies by less than 5 kJ·mol−1, as reported in one of our previous publications [5]. The effect of zero-point (ZPE) corrections were tested for the CH4 formation reaction, where the effect of ZPE would be the greatest as this reaction involves the species with the highest number of C–H bonds. Our analysis indicated that ZPE corrections change the activation energy of CH4 formation on Co(111) by 2 kJ·mol−1, and on FeCo(111) by 1 kJ·mol−1. As the aim of our study is to compare differences in energetics on Co(111) and FeCo(111) surfaces, this analysis indicated that ZPE corrections do not influence the results and conclusions obtained in our study. Therefore, ZPE corrections were not included in the results reported in this study.
The binding (adsorption) energies (Ead) of surface species (adsorbates) were reported with respect to their gas-phase structures, according to the following formula:
where Eslab_ad represents the energy of the optimized system of adsorbate on the slab, Eslab represents the energy of the clean slab (without the adsorbate), and Ead_vac represents the energy of the adsorbate molecule in a vacuum.
Ead = Eslab_ad − Eslab − Ead_vac
The reaction and activation energies were defined as follows:
ΔE = EFS − EIS
Ea = ETS − EIS
The transition states were optimized using the CI-NEB [56] method until the forces acting on the image with the highest energy were lower than 0.04 eV/Å. All transition states were further validated by vibrational frequency analysis, showing a single imaginary vibrational frequency. During the vibrational frequency analysis, the atoms were displaced from their equilibrium positions by 0.015 Å. Bader charge analysis [57,58,59] and Density of States (DOS) calculations were performed to investigate the electronic properties of Co(111) and FeCo(111) surfaces under vacuum conditions, and the charge transfer between catalytic surfaces and adsorbates.
4. Conclusions
The effect of Fe doping on cobalt surfaces was investigated using periodic DFT calculations, in order to obtain atomic scale insights related to the catalytic performance of bimetallic FeCo catalysts in the metallic phase for CO2 hydrogenation to C1 products. Bimetallic FeCo catalysts were modelled with a FeCo(111) surface, which represents the surface segregation of Fe atoms in bimetallic FeCo catalysts, based on the experimental literature and our results based on increased CO adsorption energies. From our analysis of the modification of the adsorption energies of surface species involved in CO2 hydrogenation and the activation energies of elementary reactions, we draw the following conclusions:
- (1)
- Fe doping leads to increased adsorption energies for all surface species, including CO2; CO; and atomic species, such as C, O, and H. However, with respect to the relative increases in adsorption energies, we find that Fe doping mainly leads to increased binding strengths for CO containing species, which can be projected to cause increased COx/H adsorbate ratios on the metallic FeCo(111) surface.
- (2)
- In terms of catalytic activity, FeCo(111) surface leads to decreased activation energies for direct CO2 dissociation, which yields increased surface CO and O coverages. However, due to a slight increase in the activation energy for HCO formation, the rate-limiting step for CO dissociation on FeCo(111) surfaces can be expected to have lower overall activity for CO2 hydrogenation, as previously observed in experimental studies.
- (3)
- In terms of selectivity, FeCo(111) surface hinders the formation of hydrogenated products due to increased activation energies and decreased H coverages. FeCo(111) surface neither promotes CH4 and methanol formation (due to high activation energies) nor CO formation (due to stronger adsorption), which indicates that the bimetallic FeCo surfaces does not play a role in the formation of these species, as experimentally observed on FeCo nanoparticles.
- (4)
- The fundamental cause of increased adsorption energies and increased activation energies for hydrogenation due to Fe doping is linked to the electron-donating ability of Fe atoms to both cobalt atoms and adsorbates. Therefore, the tuning of the electronic charge on the cobalt surface is projected to be a fundamental descriptor, which can adjust the balance between CO2 activation and hydrogenation reactions.
- (5)
- The main effect of the formation of a bimetallic FeCo(111) surface on the CO2 hydrogenation performance of cobalt surfaces is the inhibition of oxygen removal from the catalytic surface. Therefore, the introduction of Fe to cobalt surfaces in FeCo nanoparticles can be expected to lead the structural evolution of the bimetallic surfaces towards iron oxide and mixed iron–cobalt oxide phases, which in turn can lead to a different selectivity behavior of bimetallic FeCo nanoparticles.
Overall, our study provides the first atomic-level insights in the literature related to the effect of Fe doping on the catalytic behavior and structural evolution of FeCo bimetallic catalysts. These insights can be expected to contribute to the design of high-performance bimetallic catalysts for CO2 conversion.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13111390/s1, Table S1: The change in adsorption energies (kJ·mol−1) of H, O, C, and CO with the change in iron coverage of the surface, Figure S1: The adsorption sites investigated on Co(111) and FeCo(111) surfaces, Table S2: Comparison of the adsorption energies (kJ·mol−1) of the adsorbates examined on the surface of the Co(111) catalyst in the literature, Table S3: Comparison of the activation barriers (kJ·mol−1) and reaction energies (kJ·mol−1) of the reactions examined on the surface of the Co(111) catalyst in the literature, Table S4: Total Bader charges of the adsorbates and the first two layers of the slab for Co(111) and FeCo(111) surfaces, Table S5: Electronic charges on Co(111) vs. FeCO(111) surfaces under adsorbate coverages.
Author Contributions
Conceptualization, A.C.K.; methodology, A.C.K.; validation, A.C.K.; formal analysis, A.C.K.; investigation, D.T. and A.C.K.; resources, A.C.K.; data curation, A.C.K.; writing—original draft preparation, D.T. and A.C.K.; writing—review and editing, A.C.K.; visualization, D.T. and A.C.K.; supervision, A.C.K.; project administration, A.C.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Scientific and Technological Research Council of Turkey (TUBITAK), Grant No. 221Z342.
Data Availability Statement
Data are contained within this article and Supplementary Materials.
Acknowledgments
The numerical calculations reported in this paper were fully performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources).
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- De, S.; Dokania, A.; Ramirez, A.; Gascon, J. Advances in the Design of Heterogeneous Catalysts and Thermocatalytic Processes for CO2 Utilization. ACS Catal. 2020, 10, 14147–14185. [Google Scholar] [CrossRef]
- Van de Loosdrecht, J.; Botes, F.G.; Ciobica, I.M.; Ferreira, A.; Gibson, P.; Moodley, D.J.; Saib, A.M.; Visagie, J.L.; Weststrate, C.J.; Niemantsverdriet, J.W. Comprehensive Inorganic Chemistry II, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Puga, A.V. On the nature of active phases and sites in CO and CO2 hydrogenation catalysts. Catal. Sci. Technol. 2018, 8, 5681–5707. [Google Scholar] [CrossRef]
- Visconti, C.G.; Martinelli, M.; Falbo, L.; Fratalocchi, L.; Lietti, L. CO2 hydrogenation to hydrocarbons over Co and Fe-based Fischer-Tropsch catalysts. Catal. Today 2016, 277, 161–170. [Google Scholar] [CrossRef]
- Daga, Y.; Kizilkaya, A.C. Mechanistic Insights into the Effect of Sulfur on the Selectivity of Cobalt-Catalyzed Fischer–Tropsch Synthesis: A DFT Study. Catalysts 2022, 12, 425. [Google Scholar] [CrossRef]
- Jeske, K.; Kizilkaya, A.C.; López-Luque, I.; Pfänder, N.; Bartsch, M.; Concepción, P.; Prieto, G. Design of Cobalt Fischer-Tropsch Catalysts for the Combined Production of Liquid Fuels and Olefin Chemicals from Hydrogen-Rich Syngas. ACS Catal. 2021, 11, 4784–4798. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, B.; Broos, R.J.P.; Chen, W.; Filot, I.A.W.; Hensen, E.J.M. First-principles based microkinetic modeling of transient kinetics of CO hydrogenation on cobalt catalysts. Catal. Today 2020, 342, 131–141. [Google Scholar] [CrossRef]
- Owen, R.E.; O’Byrne, J.P.; Mattia, D.; Plucinski, P.; Pascu, S.I.; Jones, M.D. Cobalt catalysts for the conversion of CO2 to light hydrocarbons at atmospheric pressure. Chem. Commun. 2013, 49, 11683–11685. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, G.; Jiang, X.; Liu, Y.; Zhu, J.; Ding, F.; Liu, Z.; Guo, X.; Song, C. CO2 Hydrogenation on Unpromoted and M-Promoted Co/TiO2 Catalysts (M = Zr, K, Cs): Effects of Crystal Phase of Supports and Metal-Support Interaction on Tuning Product Distribution. ACS Catal. 2019, 9, 2739–2751. [Google Scholar] [CrossRef]
- Satthawong, R.; Koizumi, N.; Song, C.; Prasassarakich, P. Light olefin synthesis from CO2 hydrogenation over K-promoted Fe-Co bimetallic catalysts. Catal. Today 2015, 251, 34–40. [Google Scholar] [CrossRef]
- Wang, W.; Toshcheva, E.; Ramirez, A.; Shterk, G.; Ahmad, R.; Caglayan, M.; Cerrillo, J.L.; Dokania, A.; Clancy, G.; Shoinkhorova, T.B.; et al. Bimetallic Fe-Co catalysts for the one step selective hydrogenation of CO2 to liquid hydrocarbons. Catal. Sci. Technol. 2023, 13, 1527–1540. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Tan, L.; Zhang, P.; Peng, X.; Oruganti, A.; Yang, G.; Abe, H.; Wang, Y.; Tsubaki, N. Integrated tuneable synthesis of liquid fuels via Fischer–Tropsch technology. Nat. Catal. 2018, 6, 787–793. [Google Scholar] [CrossRef]
- Dong, Z.; Zhao, J.; Tian, Y.; Zhang, B.; Wu, Y. Preparation and Performances of ZIF-67-Derived FeCo Bimetallic Catalysts for CO2 Hydrogenation toLight Olefins. Catalysts 2020, 10, 455. [Google Scholar] [CrossRef]
- Guo, L.; Cui, Y.; Li, H.; Fang, Y.; Prasert, R.; Wu, J.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Selective formation of linear-alpha olefins (LAOs) by CO2 hydrogenation over bimetallic Fe/Co-Y catalyst. Catal. Commun. 2019, 130, 105759. [Google Scholar] [CrossRef]
- Liang, J.; Guo, L.; Gao, W.; Wang, C.; Guo, X.; He, Y.; Yang, G.; Tsubaki, N. Direct Conversion of CO2 to Aromatics over K−Zn−Fe/ZSM-5 Catalysts via a Fischer−Tropsch Synthesis Pathway. Ind. Eng. Chem. Res. 2022, 61, 10336–10346. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, J.; Corma, A. Applications of Zeolites to C1 Chemistry: Recent Advances, Challenges, and Opportunities. Adv. Mater. 2020, 32, 2002927. [Google Scholar] [CrossRef] [PubMed]
- Gnanamani, M.K.; Jacobs, G.; Hamdeh, H.H.; Shafer, W.D.; Liu, F.; Hopps, S.D.; Thomas, G.A.; Davis, B.H. Hydrogenation of Carbon Dioxide over Co-Fe Bimetallic Catalysts. ACS Catal. 2016, 6, 913–927. [Google Scholar] [CrossRef]
- Ischenko, O.V.; Dyachenko, A.G.; Saldan, I.; Lisnyak, V.V.; Diyuk, V.E.; Vakaliuk, A.V.; Yatsymyrskyi, A.V.; Gaidai, S.V.; Zakharova, T.M.; Makota, O.; et al. Methanation of CO2 on bulk Co–Fe catalysts. Int. J. Hydrogen Energy 2021, 46, 37860–37871. [Google Scholar] [CrossRef]
- Sandupatla, A.S.; Banerjee, A.; Deo, G. Optimizing CO2 hydrogenation to methane over CoFe bimetallic catalyst: Experimental and density functional theory studies. Appl. Surf. Sci. 2019, 485, 441–449. [Google Scholar] [CrossRef]
- Zhang, L.; Dang, Y.; Zhou, X.; Gao, P.; van Bavel, A.P.; Wang, H.; Li, S.; Shi, L.; Yang, Y.; Vovk, E.I.; et al. Direct conversion of CO2 to a jet fuel over CoFe alloy catalysts. Innovation 2021, 2, 100170. [Google Scholar] [CrossRef]
- Hwang, S.M.; Han, S.J.; Park, H.G.; Lee, H.; An, K.; Jun, K.W.; Kim, S.K. Atomically alloyed Fe-Co catalyst derived from a N-coordinated Co single-atom structure for CO2 hydrogenation. ACS Catal. 2021, 11, 2267–2278. [Google Scholar] [CrossRef]
- Gunasooriya, G.T.K.K.; Van Bavel, A.P.; Kuipers, H.P.C.E.; Saeys, M. Key Role of Surface Hydroxyl Groups in C-O Activation during Fischer-Tropsch Synthesis. ACS Catal. 2016, 6, 3660–3664. [Google Scholar] [CrossRef]
- Chen, S.; Zaffran, J.; Yang, B. Dry reforming of methane over the cobalt catalyst: Theoretical insights into the reaction kinetics and mechanism for catalyst deactivation. Appl. Catal. B Environ. 2020, 270, 118859. [Google Scholar] [CrossRef]
- Huang, H.; Yu, Y.; Zhang, M. Mechanistic insight into methane dry reforming over cobalt: A density functional theory study. Phys. Chem. Chem. Phys. 2020, 22, 27320–27331. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Q.; Wang, G.; Hou, B.; Jia, L.; Li, D. Mechanistic insight into the C2 hydrocarbons formation from Syngas on fcc-Co(111) surface: A DFT study. J. Phys. Chem. C 2016, 120, 9132–9147. [Google Scholar] [CrossRef]
- Van Helden, P.; Weststrate, C.J. Hydrogen Adsorption on Co Surfaces: A Density Functional Theory and Temperature Programmed Desorption Study. ACS Catal. 2012, 2, 1097–1107. [Google Scholar] [CrossRef]
- Zijlstra, B.; Broos, R.J.P.; Chen, W.; Bezemer, G.L.; Filot, I.A.W.; Hensen, E.J.M. The vital role of step-edge sites for both CO activation and chain growth on cobalt fischer-tropsch catalysts revealed through first-principles-based microkinetic modeling including lateral interactions. ACS Catal. 2020, 10, 9376–9400. [Google Scholar] [CrossRef]
- Yu, M.; Liu, L.; Wang, Q.; Jia, L.; Wang, J.; Li, D. Rediscovering Tuning Product Selectivity by an Energy Descriptor: CH4 Formation and C1-C1 Coupling on the FCC Co Surface. J. Phys. Chem. C 2020, 124, 11040–11049. [Google Scholar] [CrossRef]
- Pedersen, E.Ø.; Svenum, I.H.; Blekkan, E.A. Mn promoted Co catalysts for Fischer-Tropsch production of light olefins—An experimental and theoretical study. J. Catal. 2018, 361, 23–32. [Google Scholar] [CrossRef]
- Kizilkaya, A.C.; Niemantsverdriet, J.W.; Weststrate, C.J. Oxygen Adsorption and Water Formation on Co(0001). J. Phys. Chem. C 2016, 120, 4833–4842. [Google Scholar] [CrossRef]
- Weststrate, C.J.; Sharma, D.; Rodríguez, D.G.; Fredriksson, H.O.A.; Niemantsverdriet, J.W. Water Formation Kinetics on Co(0001) at Low and Near-Ambient Hydrogen Pressures in the Context of Fischer-Tropsch Synthesis. J. Phys. Chem. C 2023, 127, 3452–3461. [Google Scholar] [CrossRef]
- Luo, W.; Asthagiri, A. Density functional theory study of methanol steam reforming on Co(0001) and Co(111) surfaces. J. Phys. Chem. C 2014, 118, 15274–15285. [Google Scholar] [CrossRef]
- Ma, F.F.; Ma, S.H.; Jiao, Z.Y.; Dai, X.Q. Adsorption and decomposition of H2O on cobalt surfaces: A DFT study. Appl. Surf. Sci. 2016, 384, 10–17. [Google Scholar] [CrossRef]
- Ismail, A.S.M.; Casavola, M.; Liu, B.; Gloter, A.; Van Deelen, T.W.; Versluijs, M.; Meeldijk, J.D.; Stéphan, O.; De Jong, K.P.; De Groot, F.M.F. Atomic-Scale Investigation of the Structural and Electronic Properties of Cobalt-Iron Bimetallic Fischer-Tropsch Catalysts. ACS Catal. 2019, 9, 7998–8011. [Google Scholar] [CrossRef]
- Greeley, J.; Norskov, J.K.; Mavrikakis, M. Electronic structure and catalysis on metal surfaces. Annu. Rev. Phys. Chem. 2002, 53, 319–348. [Google Scholar] [CrossRef] [PubMed]
- Kitchin, J.R.; Norskov, J.K. Modification of the surface electronic and chemical properties of Pt(111) by subsurface 3d transition metals. J. Chem. Phys. 2004, 120, 10240–10246. [Google Scholar] [CrossRef] [PubMed]
- Govender, S.; Gambu, T.G.; van Heerden, T.; van Steen, E. Mechanistic pathways for oxygen removal on Pt-doped Co(111) in the Fischer-Tropsch reaction. Catal. Today 2020, 342, 142–151. [Google Scholar] [CrossRef]
- Yang, P.; Zhang, Q.; Yi, Z.; Wang, J.; Yang, H. Rational electronic control of carbon dioxide reduction over cobalt oxide. J. Catal. 2020, 387, 119–128. [Google Scholar] [CrossRef]
- Van Helden, P.; Ciobîc, I.M.; Coetzer, R.L.J. The size-dependent site composition of FCC cobalt nanocrystals. Catal. Today 2016, 261, 48–59. [Google Scholar] [CrossRef]
- Rochana, P.; Wilcox, J. A theoretical study of CO adsorption on FeCo(100) and the effect of alloying. Surf. Sci. 2011, 605, 681–688. [Google Scholar] [CrossRef]
- Calderone, V.R.; Shiju, N.R.; Curulla-ferrø, D.; Chambrey, S.; Khodakov, A.; Rose, A.; Thiessen, J.; Jess, A.; Rothenberg, G. De Novo Design of Nanostructured Iron–Cobalt Fischer–Tropsch Catalysts. Angewandte 2013, 52, 4397–4401. [Google Scholar] [CrossRef]
- Moran-Lopez, J.L. Surface Composition of the Ordered Binary Alloy System: Fe-Co. Appl. Surf. Sci. 1984, 19, 167–180. [Google Scholar] [CrossRef]
- Allié, G.; Lauroz, C.; Villemain, P. Leed and aes study of the (100) surface of a 50-50 iron-cobalt monocrystalline alloy. Surf. Sci. 1981, 104, 583–598. [Google Scholar] [CrossRef]
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics; John Wiley and Sons: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Eren, B.; Torres, D.; Karsl, O.; Liu, Z.; Wu, C.H.; Stacchiola, D.; Bluhm, H.; Somorjai, G.A.; Salmeron, M. Structure of Copper−Cobalt Surface Alloys in Equilibrium with Carbon Monoxide Gas. J. Am. Chem. Soc. 2018, 140, 6575–6581. [Google Scholar] [CrossRef] [PubMed]
- Collinge, G.; Kruse, N.; Mcewen, J. Role of Carbon Monoxide in Catalyst Reconstruction for CO Hydrogenation: First-Principles Study of the Composition, Structure, and Stability of Cu/Co (1012) as a Function of CO Pressure. J. Phys. Chem. C 2017, 121, 2181–2191. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, W. Comment on “generalized gradient approximation made simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Dion, M.; Rydberg, H.; Schröder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 2004, 92, 22–25. [Google Scholar] [CrossRef]
- Román-Pérez, G.; Soler, J.M. Efficient implementation of a van der waals density functional: Application to double-wall carbon nanotubes. Phys. Rev. Lett. 2009, 103, 096102. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Van der Waals density functionals applied to solids. Phys. Rev. B 2011, 83, 195131. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Chemical accuracy for the van der Waals density functional. J. Phys. Condens. Matter 2010, 22, 022201. [Google Scholar] [CrossRef] [PubMed]
- Gunasooriya, G.T.K.K.; Van Bavel, A.P.; Kuipers, H.P.C.E.; Saeys, M. CO adsorption on cobalt: Prediction of stable surface phases. Surf. Sci. 2015, 642, L6–L10. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. Climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
Figure 1.
The most stable sites for adsorbates on Co(111) and FeCo(111) surfaces. Gray, red, white, blue, and dark red spheres represent C, O, H, Co, and Fe atoms, respectively.
Figure 1.
The most stable sites for adsorbates on Co(111) and FeCo(111) surfaces. Gray, red, white, blue, and dark red spheres represent C, O, H, Co, and Fe atoms, respectively.
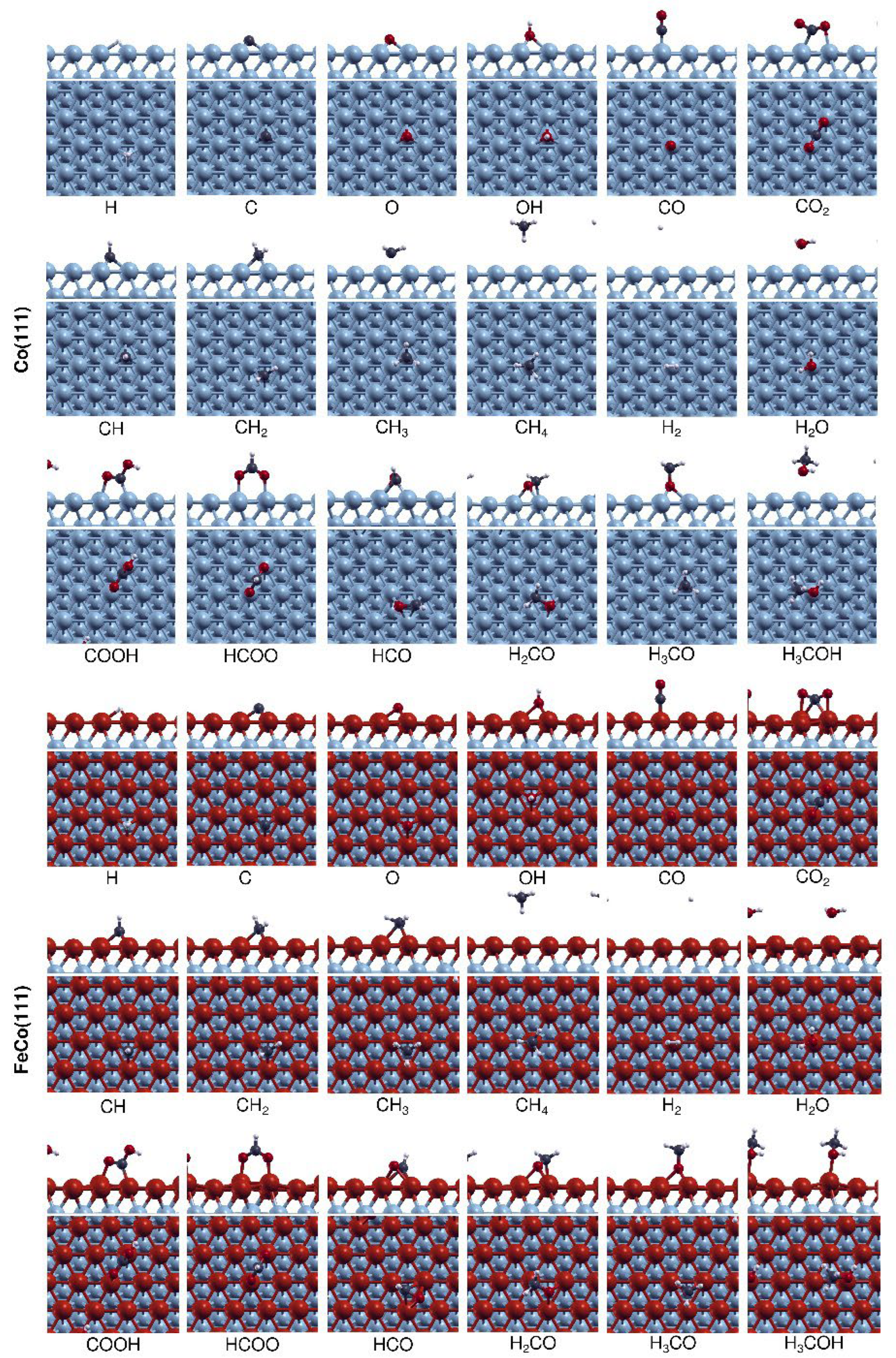
Figure 2.
Potential Energy Diagram (PED) for H2 dissociation on Co(111) and FeCo(111) surfaces.

Figure 3.
IS, TS and FS structures for H2 dissociation on Co(111) and FeCo(111).

Figure 4.
PED for CO2 dissociation via direct (a), carboxylate (b), and formate (c) mechanisms on Co(111) and FeCo(111) surfaces.
Figure 4.
PED for CO2 dissociation via direct (a), carboxylate (b), and formate (c) mechanisms on Co(111) and FeCo(111) surfaces.

Figure 5.
IS, TS, and FS structures for the direct CO2 dissociation on Co(111) and FeCo(111).

Figure 6.
IS, TS, and FS structures for H-assisted CO2 dissociation via the carboxylate mechanism on Co(111) and FeCo(111).
Figure 6.
IS, TS, and FS structures for H-assisted CO2 dissociation via the carboxylate mechanism on Co(111) and FeCo(111).
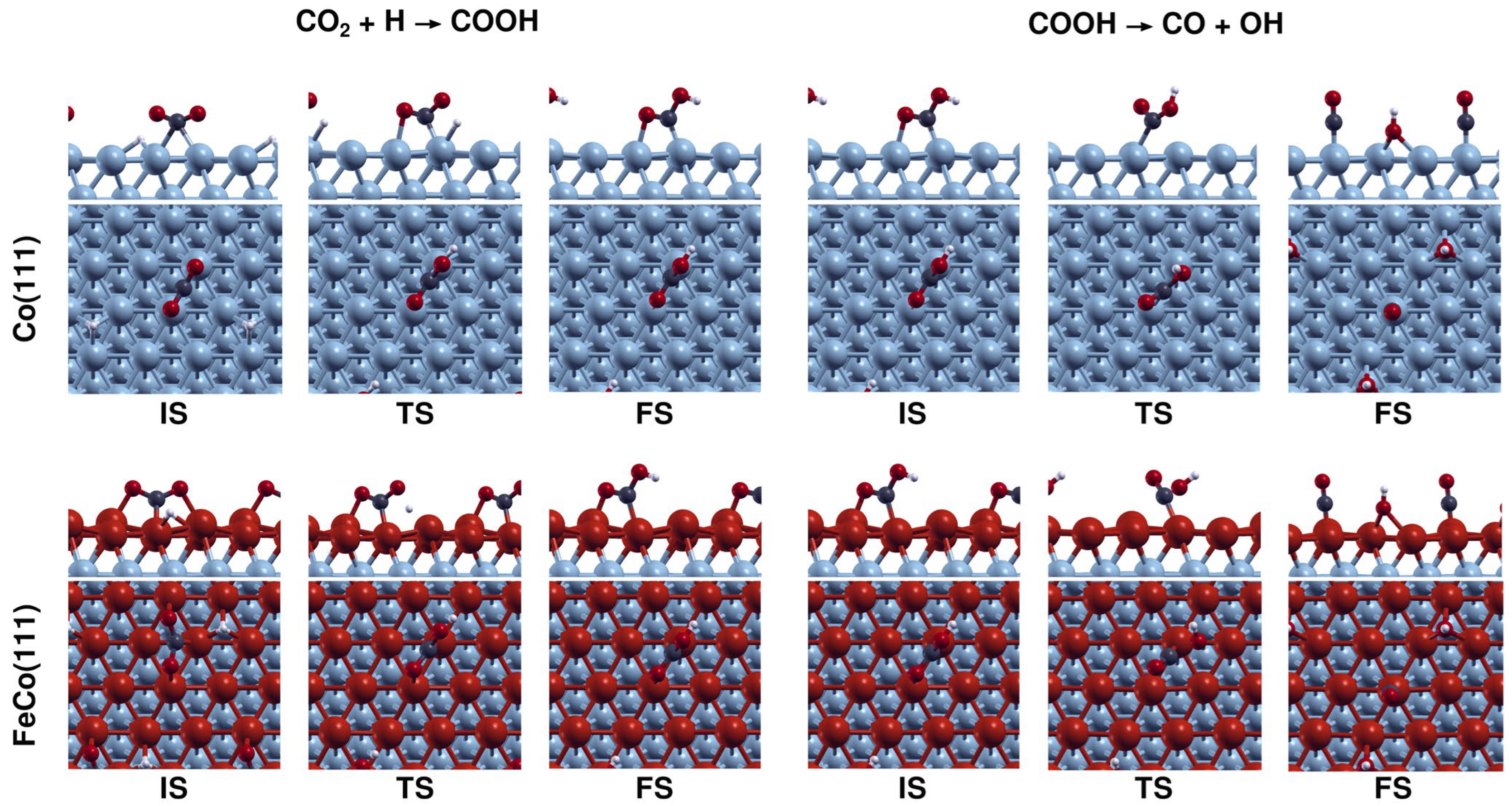
Figure 7.
IS, TS, and FS structures for H-assisted CO2 dissociation via the formate mechanism on Co(111) and FeCo(111).
Figure 7.
IS, TS, and FS structures for H-assisted CO2 dissociation via the formate mechanism on Co(111) and FeCo(111).
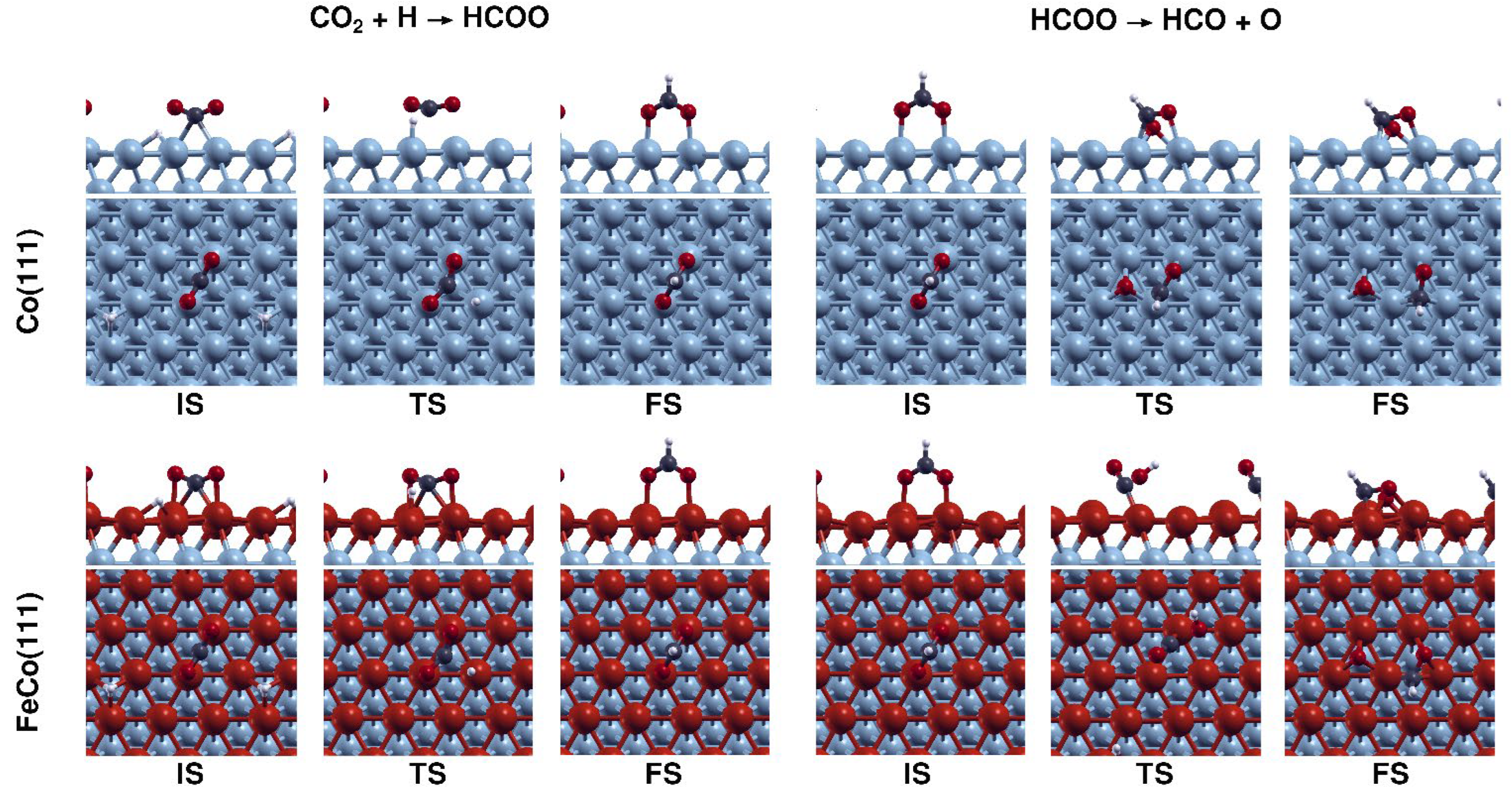
Figure 8.
PED for CO dissociation via various mechanisms, (a) direct, (b) H-assisted via HCO, and (c) H-assisted via H2CO, on Co(111) and FeCo(111) surfaces.
Figure 8.
PED for CO dissociation via various mechanisms, (a) direct, (b) H-assisted via HCO, and (c) H-assisted via H2CO, on Co(111) and FeCo(111) surfaces.
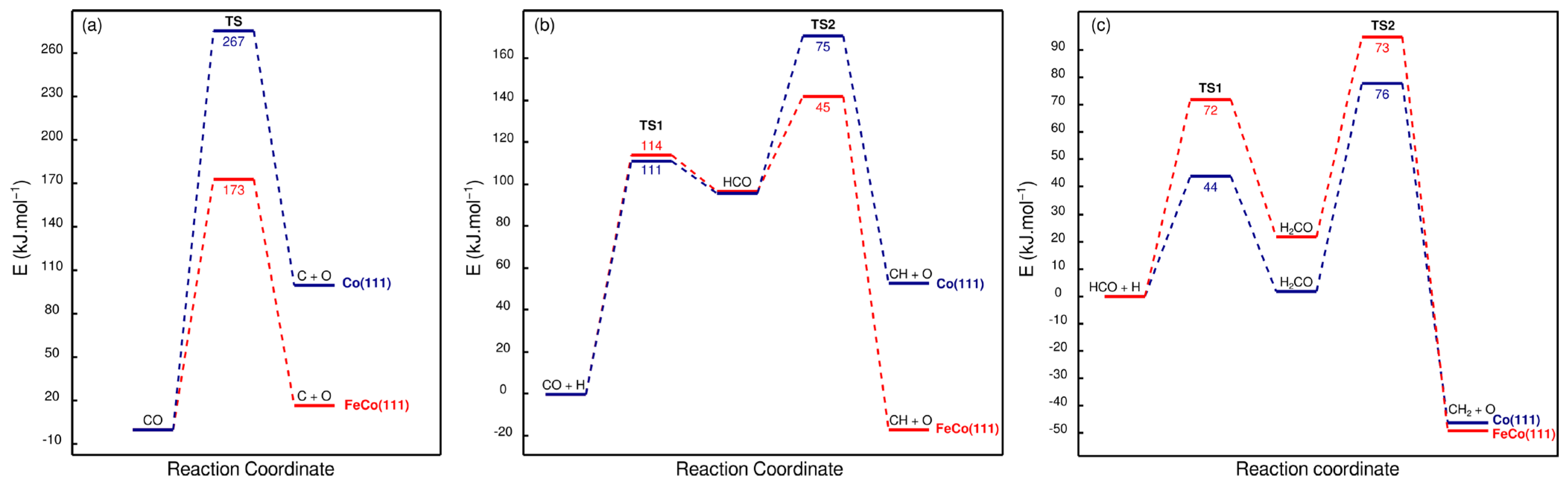
Figure 9.
IS, TS, and FS structures for the direct CO dissociation on Co(111) and FeCo(111).

Figure 10.
IS, TS, and FS structures for the H-assisted CO dissociation on Co(111) and FeCo(111).
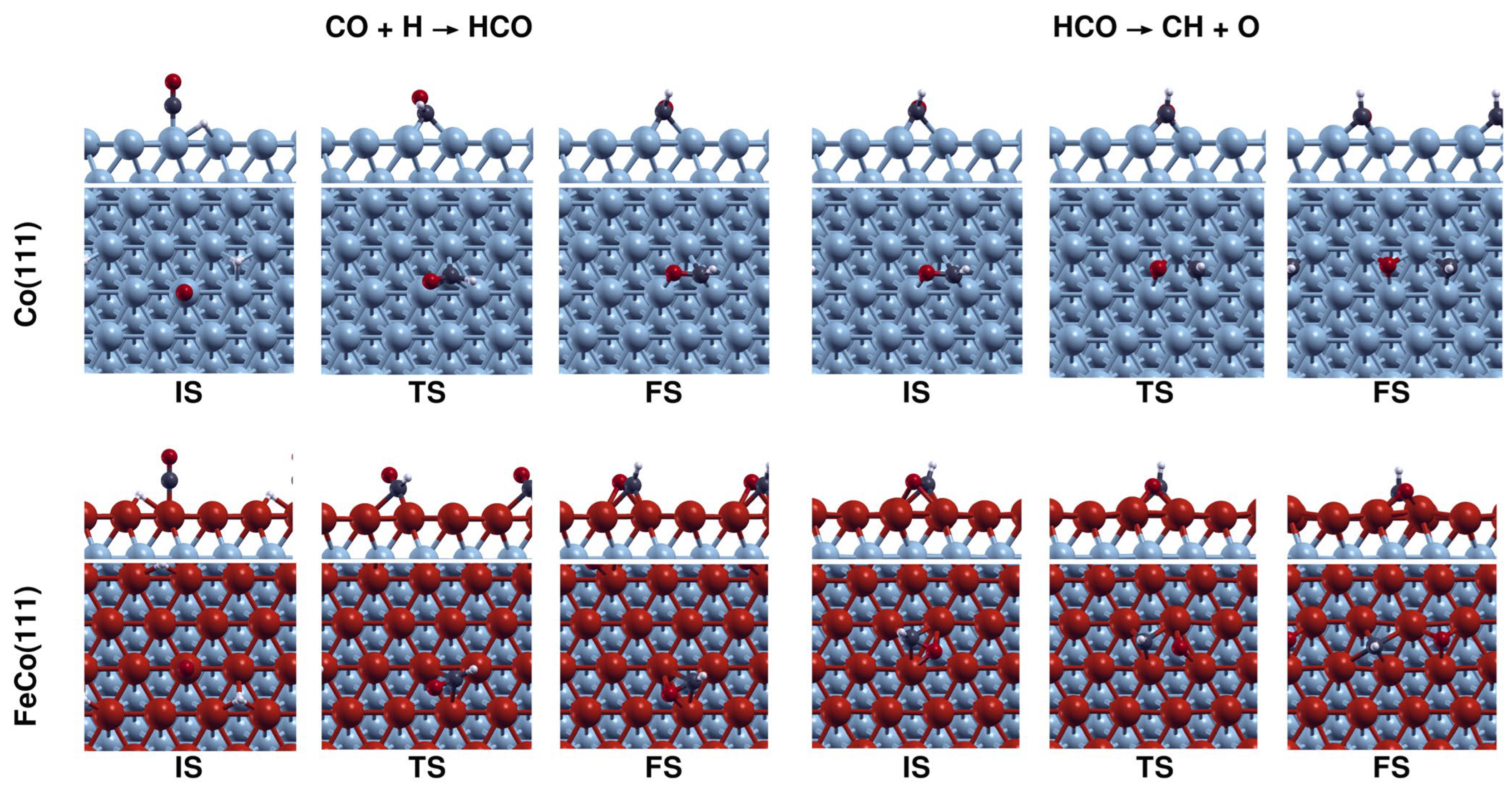
Figure 11.
PED for CH4 formation on Co(111) and FeCo(111) surfaces.
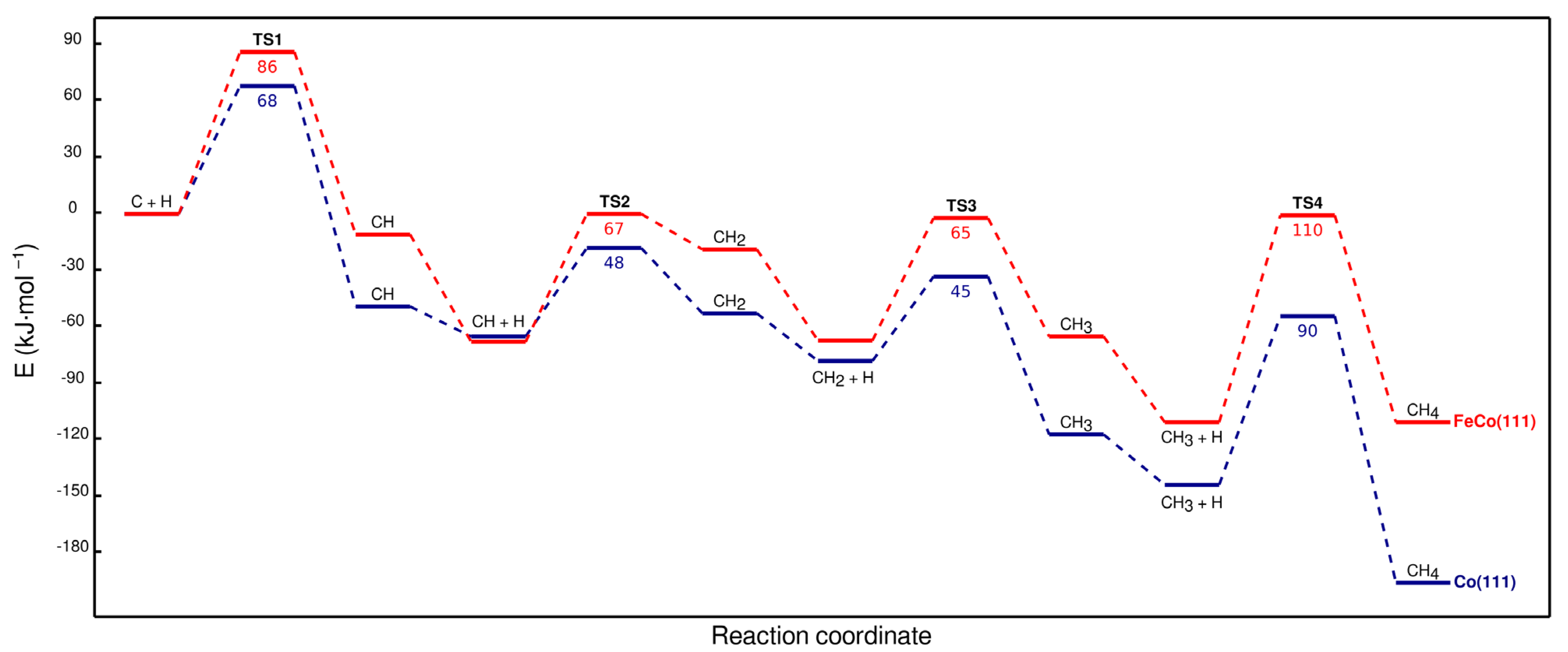
Figure 12.
IS, TS and FS structures for CH4 formation on Co(111) and FeCo(111) surfaces.

Figure 13.
PED for H2O formation on Co(111) and FeCo(111) surfaces: (a) OHx hydrogenation, (b) OH coupling.
Figure 13.
PED for H2O formation on Co(111) and FeCo(111) surfaces: (a) OHx hydrogenation, (b) OH coupling.
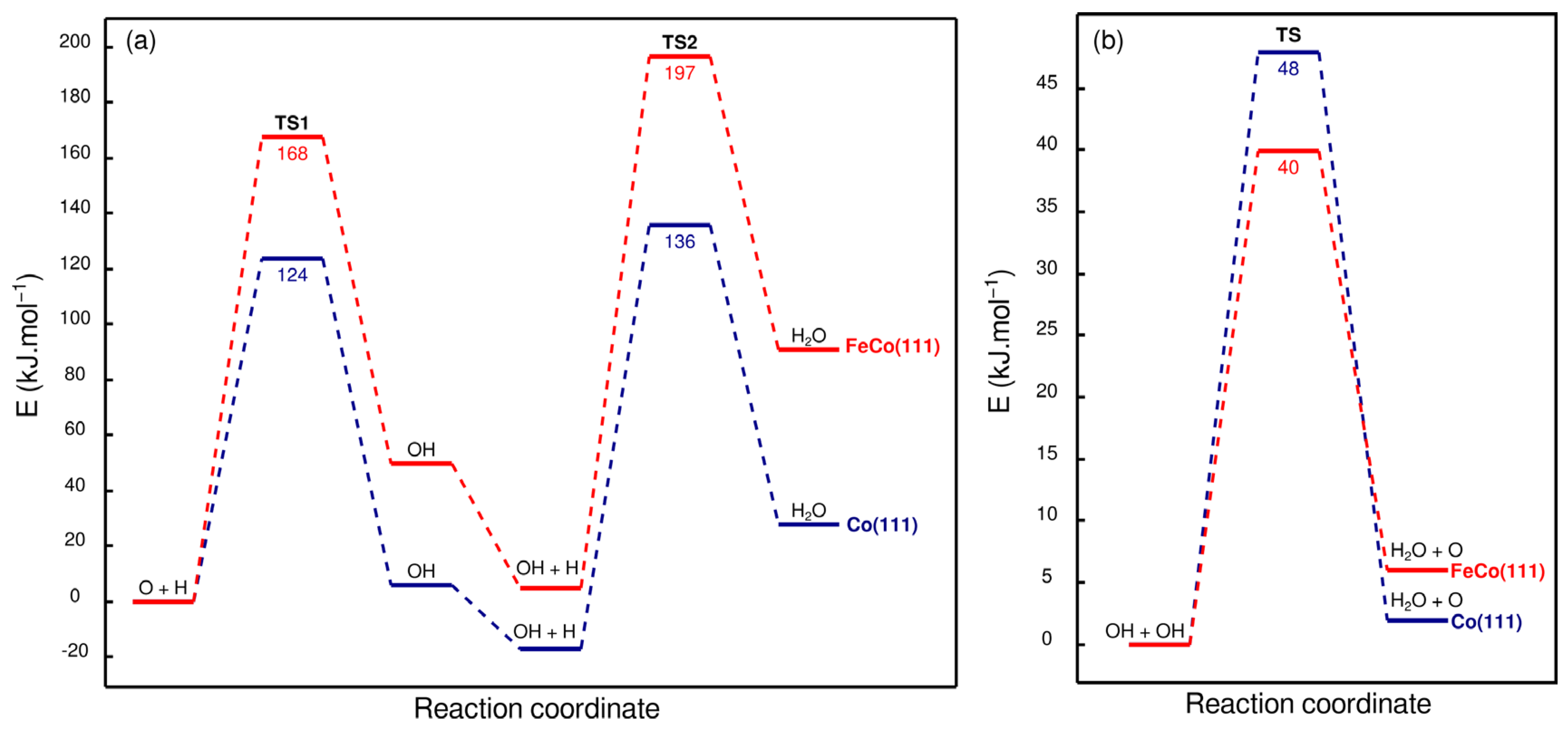
Figure 14.
IS, TS, and FS structures for H-assisted H2O formation on Co(111) and FeCo(111).
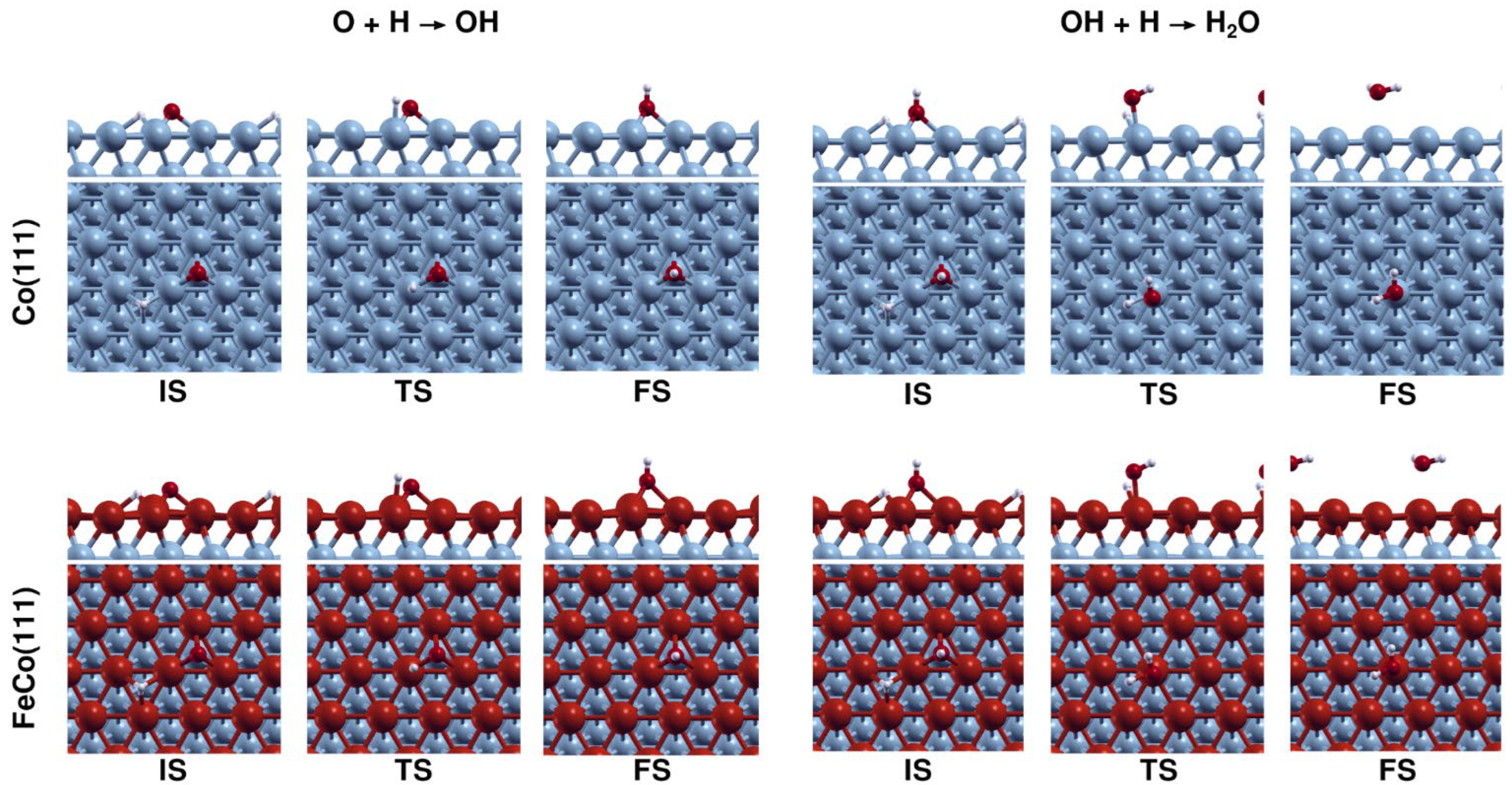
Figure 15.
PED for methanol formation on Co(111) vs. FeCo(111): (a) CH2 and O coupling, (b) CH3 and O coupling, and(c) H2CO hydrogenation to CH3OH.
Figure 15.
PED for methanol formation on Co(111) vs. FeCo(111): (a) CH2 and O coupling, (b) CH3 and O coupling, and(c) H2CO hydrogenation to CH3OH.

Figure 16.
IS, TS, and FS structures HCO hydrogenation to CH3OH on Co(111) and FeCo(111).

Figure 17.
Activation energies of CO2 hydrogenation on CO(111) and FeCo(111). * indicates that the species are adsorbed on the catalytic surface.
Figure 17.
Activation energies of CO2 hydrogenation on CO(111) and FeCo(111). * indicates that the species are adsorbed on the catalytic surface.

Figure 18.
Charge density difference plots for CO adsorbed on Co(111) and FeCo(111) surfaces.

Figure 19.
Projected Density of States (PDOS) for surface atoms on (a) Co(111) and (b) FeCo(111) surfaces.
Figure 19.
Projected Density of States (PDOS) for surface atoms on (a) Co(111) and (b) FeCo(111) surfaces.
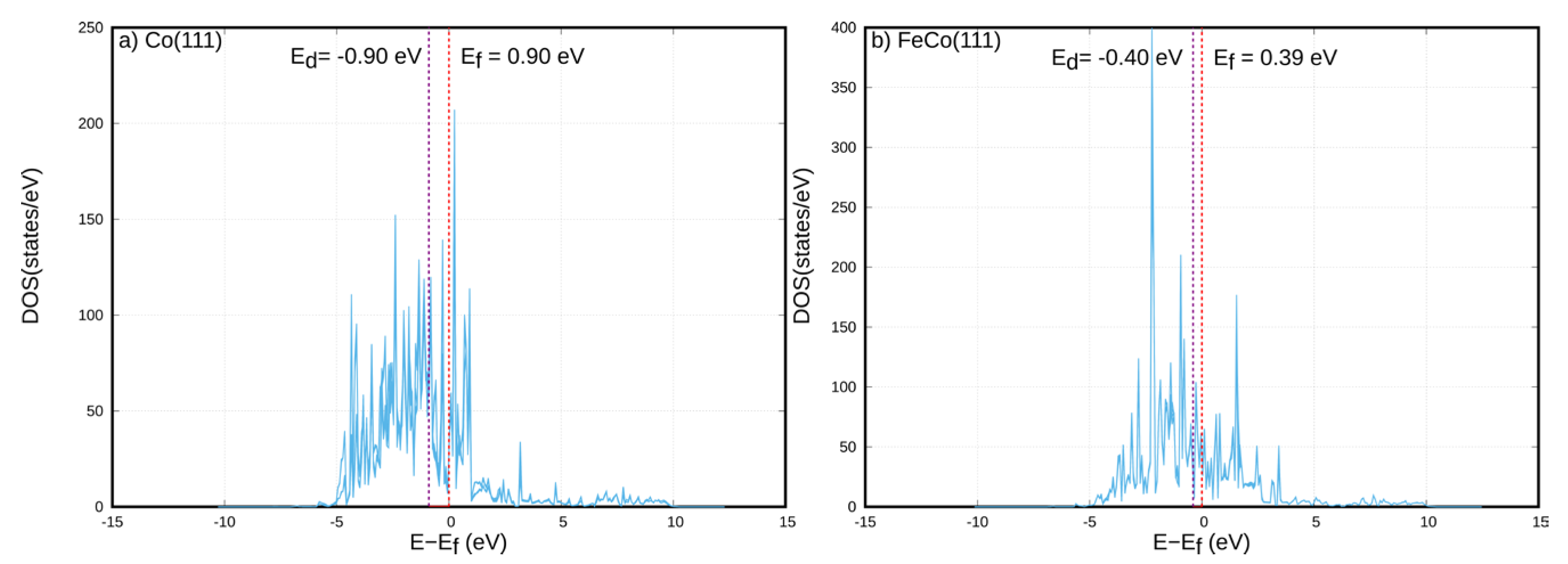
Figure 20.
Top and side views of Co(111) surfaces with various Fe coverages.
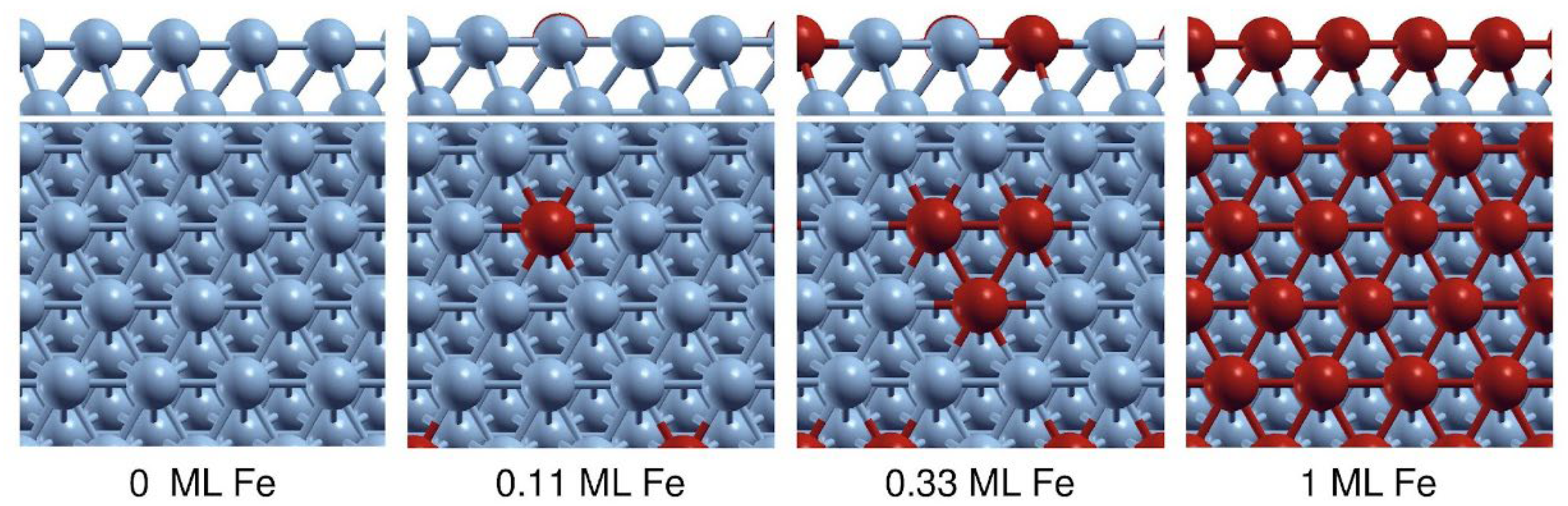

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated adsorption energies (kJ·mol−1) with adsorption sites of major intermediates involved in CO2 hydrogenation to C1 products on Co(111) and FeCo(111) surfaces.
Table 1.
Calculated adsorption energies (kJ·mol−1) with adsorption sites of major intermediates involved in CO2 hydrogenation to C1 products on Co(111) and FeCo(111) surfaces.
| Species | Co(111) | FeCo(111) | ΔE (%) |
|---|---|---|---|
| H | −278 (fcc) | −302 (fcc) | 9 |
| C | −671 (hcp) | −675 (hcp) | 9 |
| O | −564 (hcp) | −635 (fcc) | 13 |
| CO | −132 (top) | −173 (top) | 31 |
| CO2 | −19 (bridge via C) | −29 (bridge via C) | 53 |
| H2 | −6 (top) | −6 (top) | 0 |
| OH | −329 (hcp) | −368 (fcc) | 12 |
| CH | −561 (hcp) | −615 (fcc) | 10 |
| CH2 | −362 (fcc) | −413 (fcc) | 14 |
| CH3 | −166 (hcp) | −198 (fcc) | 19 |
| CH4 | −14 (top) | −14 (top) | 2 |
| H2O | −28 (top) | −47 (top) | 68 |
| HCO | −187 (bridge via O) | −255 (hcp via O) | 36 |
| COOH | −211 (bridge via C) | −257 (bridge via C) | 22 |
| HCOO | −311 (bridge via C) | −364 (bridge via C) | 17 |
| H2CO | −58 (fcc via O) | −125 (fcc via O) | 116 |
| H3CO | −284 (hcp) | −330 (fcc) | 16 |
| H3COH | −39 (top via O) | −62 (top via O) | 59 |
Table 2.
The activation energies (kJ·mol−1) and reaction energies (kJ·mol−1) of examined elementary reactions on Co(111) and FeCo(111) surfaces.
Table 2.
The activation energies (kJ·mol−1) and reaction energies (kJ·mol−1) of examined elementary reactions on Co(111) and FeCo(111) surfaces.
| No | Elementary Reaction | Ea | ΔE | ||
|---|---|---|---|---|---|
| Co(111) | FeCo(111) | Co(111) | FeCo(111) | ||
| R1 | H2 → H2 | 26 | 14 | 13 | −9 |
| R2 | H2 → H + H | 28 | 8 | −59 | −89 |
| R3 | CO2 → CO + O | 61 | 17 | −77 | −154 |
| R4 | CO2 + H → COOH | 94 | 149 | −18 | 41 |
| R5 | COOH → CO + OH | 53 | 74 | −112 | −142 |
| R6 | CO2 + H → HCOO | 71 | 51 | −74 | −55 |
| R7 | HCOO → HCO + O | 107 | 101 | 52 | 6 |
| R8 | CO → C + O | 267 | 173 | 104 | 17 |
| R9 | CO + H → HCO | 111 | 114 | 96 | 97 |
| R10 | HCO → CH + O | 75 | 45 | −43 | −114 |
| R11 | O + H → OH | 124 | 168 | 6 | 50 |
| R12 | OH + H → H2O | 153 | 178 | 44 | 86 |
| R13 | OH + OH → H2O | 48 | 40 | 2 | 6 |
| R14 | C + H → CH | 68 | 86 | −49 | −11 |
| R15 | CH + H → CH2 | 48 | 67 | 12 | 48 |
| R16 | CH2 + H → CH3 | 45 | 65 | −39 | 2 |
| R17 | CH3 + H → CH4 | 90 | 110 | −52 | 0 |
| R18 | HCO + H → H2CO | 44 | 72 | 2 | 22 |
| R19 | CH2 + O → H2CO | 124 | 150 | 48 | 86 |
| R20 | H2CO → CH2 + O | 76 | 73 | −48 | −71 |
| R21 | H2CO + H → H3CO | 42 | 51 | −83 | −50 |
| R22 | CH3 + O → H3CO | 141 | 166 | −4 | 49 |
| R23 | H3CO + H → H3COH | 160 | 177 | 56 | 94 |
| R24 | CH3 + OH → H3COH | 196 | 238 | 42 | 89 |
Table 3.
Total Bader charges (q) on adsorbates on Co(111) and FeCo(111) surfaces, along with the calculated difference (Δq).
Table 3.
Total Bader charges (q) on adsorbates on Co(111) and FeCo(111) surfaces, along with the calculated difference (Δq).
| Adsorbate | q on Co(111) (e−) | q on FeCo(111) (e−) | Δq (%) |
|---|---|---|---|
| H | −0.41 | −0.49 | 20 |
| C | −0.90 | −1.10 | 22 |
| O | −1.05 | −1.14 | 9 |
| CO | −0.39 | −0.49 | 26 |
| CO2 | −0.65 | −0.85 | 31 |
| H2 | −0.01 | −0.01 | 0 |
| OH | −0.64 | −0.70 | 9 |
| CH | −0.74 | −0.92 | 24 |
| CH2 | −0.63 | −0.77 | 22 |
| CH3 | −0.43 | −0.50 | 16 |
| CH4 | −0.02 | −0.03 | 50 |
| H2O | −0.02 | −0.30 | 1400 |
| HCO | −0.76 | −0.97 | 28 |
| COOH | −1.02 | −1.30 | 27 |
| HCOO | −0.57 | −0.74 | 30 |
| H2CO | −0.69 | −0.80 | 16 |
| H3CO | −0.63 | −0.68 | 8 |
| H3COH | −0.04 | −0.03 | −25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tuncer, D.; Kizilkaya, A.C. Atomic-Scale Insights into Carbon Dioxide Hydrogenation over Bimetallic Iron–Cobalt Catalysts: A Density Functional Theory Study. Catalysts 2023, 13, 1390. https://doi.org/10.3390/catal13111390
AMA Style
Tuncer D, Kizilkaya AC. Atomic-Scale Insights into Carbon Dioxide Hydrogenation over Bimetallic Iron–Cobalt Catalysts: A Density Functional Theory Study. Catalysts. 2023; 13(11):1390. https://doi.org/10.3390/catal13111390
Chicago/Turabian StyleTuncer, Dilan, and Ali Can Kizilkaya. 2023. "Atomic-Scale Insights into Carbon Dioxide Hydrogenation over Bimetallic Iron–Cobalt Catalysts: A Density Functional Theory Study" Catalysts 13, no. 11: 1390. https://doi.org/10.3390/catal13111390
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.