
Cyclic Carbonates through the Photo-Induced Carboxylative Cyclization of Allylic Alcohol with CO2: A Comprehensive Kinetic Study of the Reaction Mechanism by In Situ ATR-IR Spectroscopy

Abstract
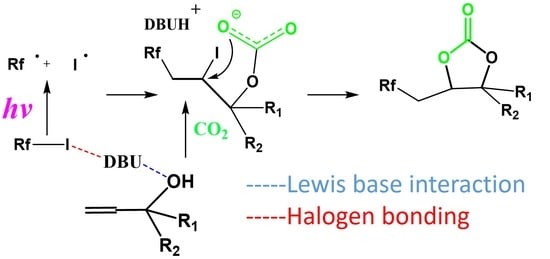
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
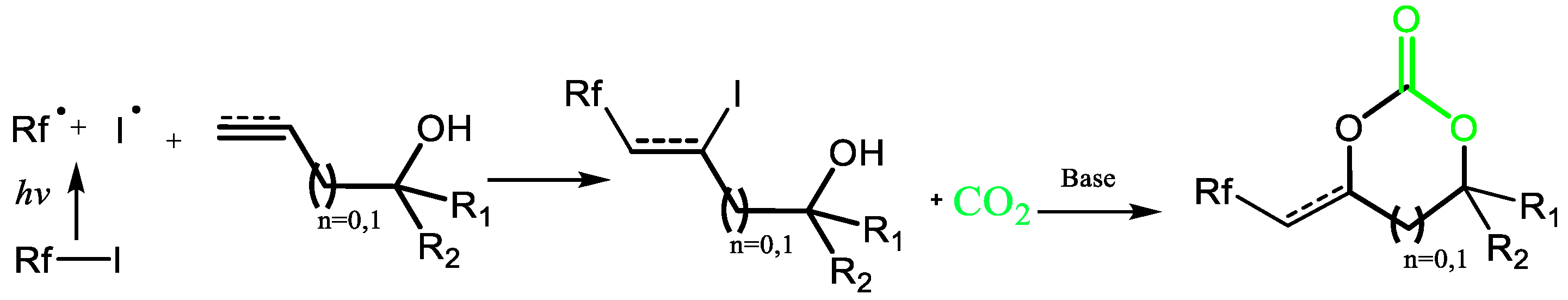
1. Introduction
2. Results and Discussion
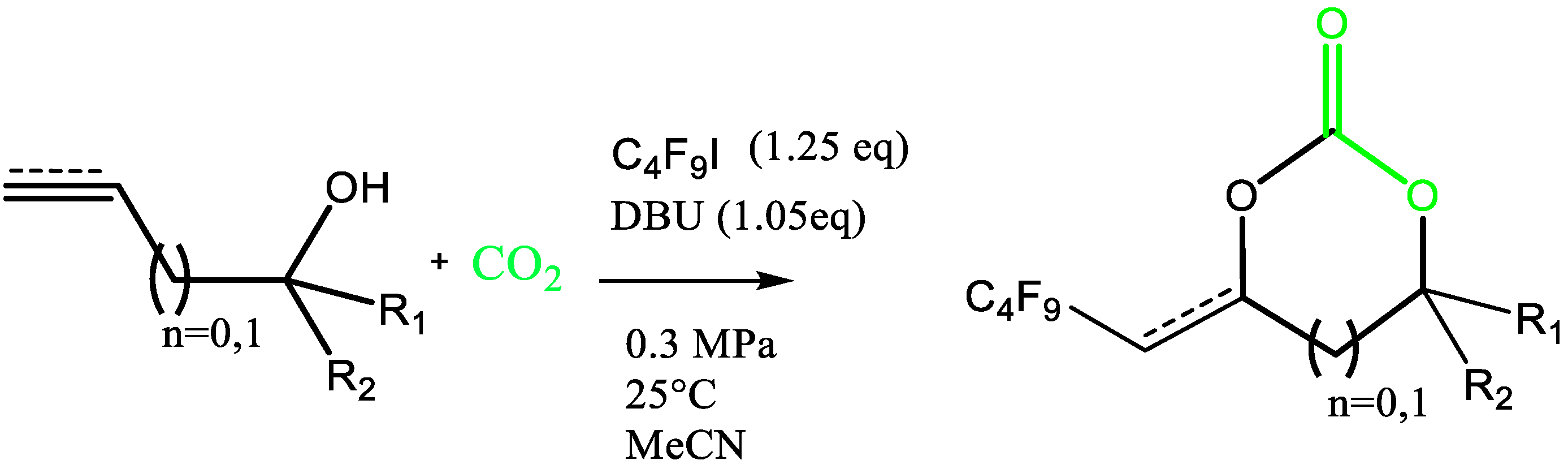
2.1. Synthesis of Perfluorobutyl Cyclic Carbonate 1 from the Coupling of CO2 with Allylic Alcohol and Perfluorobutyliodide: Mechanistic Study
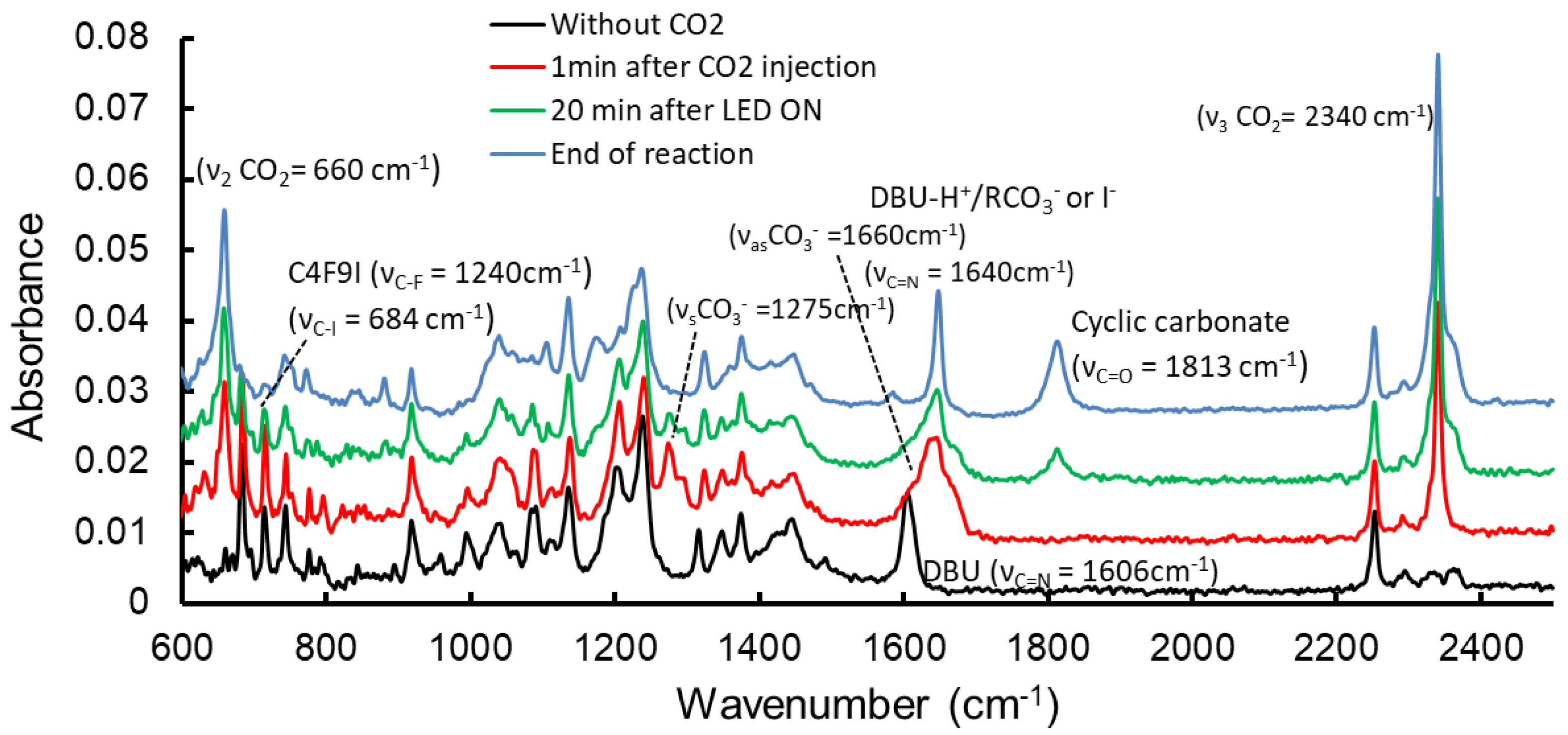
2.1.1. Model Reaction Using Allyl Alcohol, DBU and C4F9I
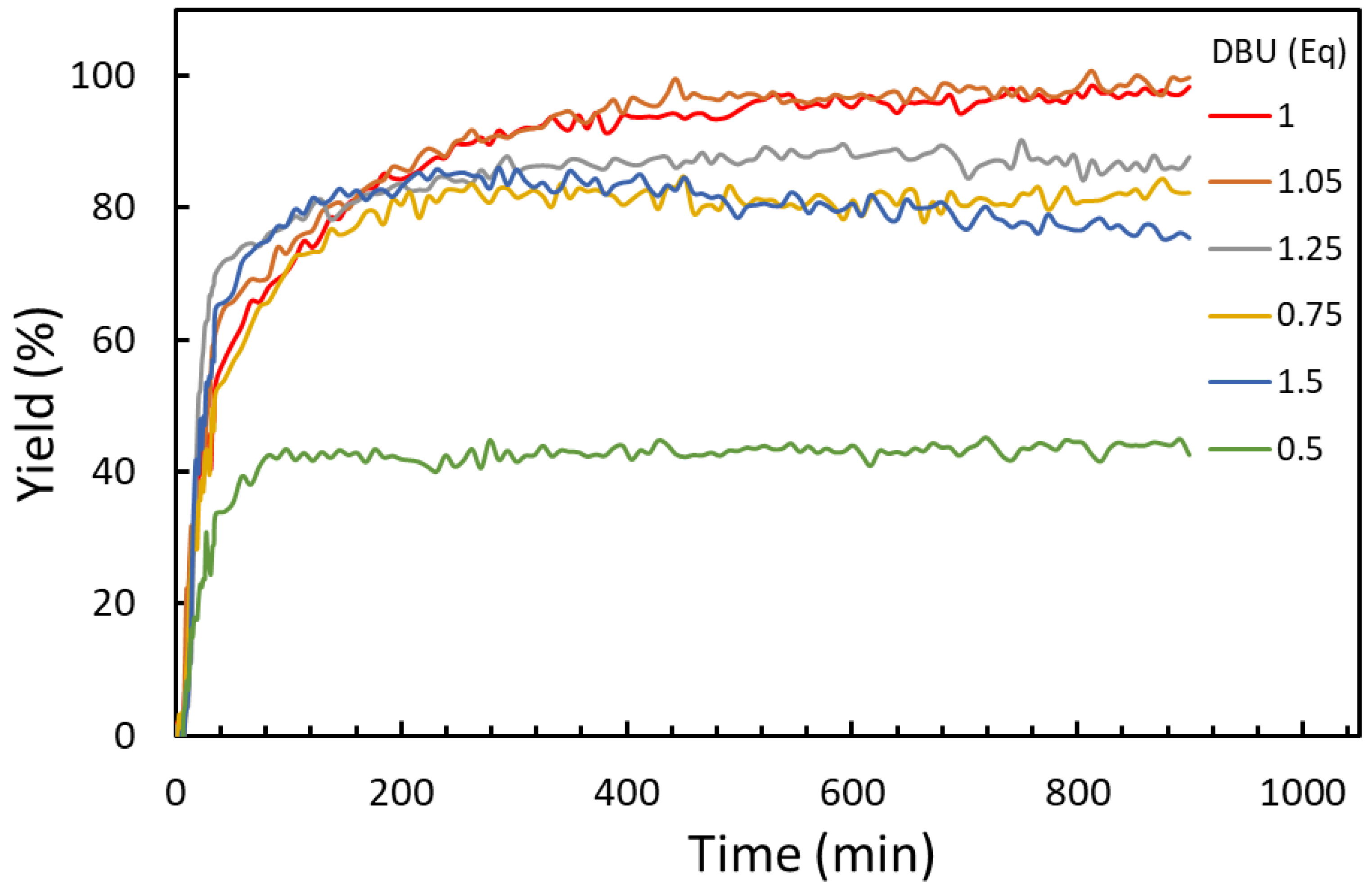
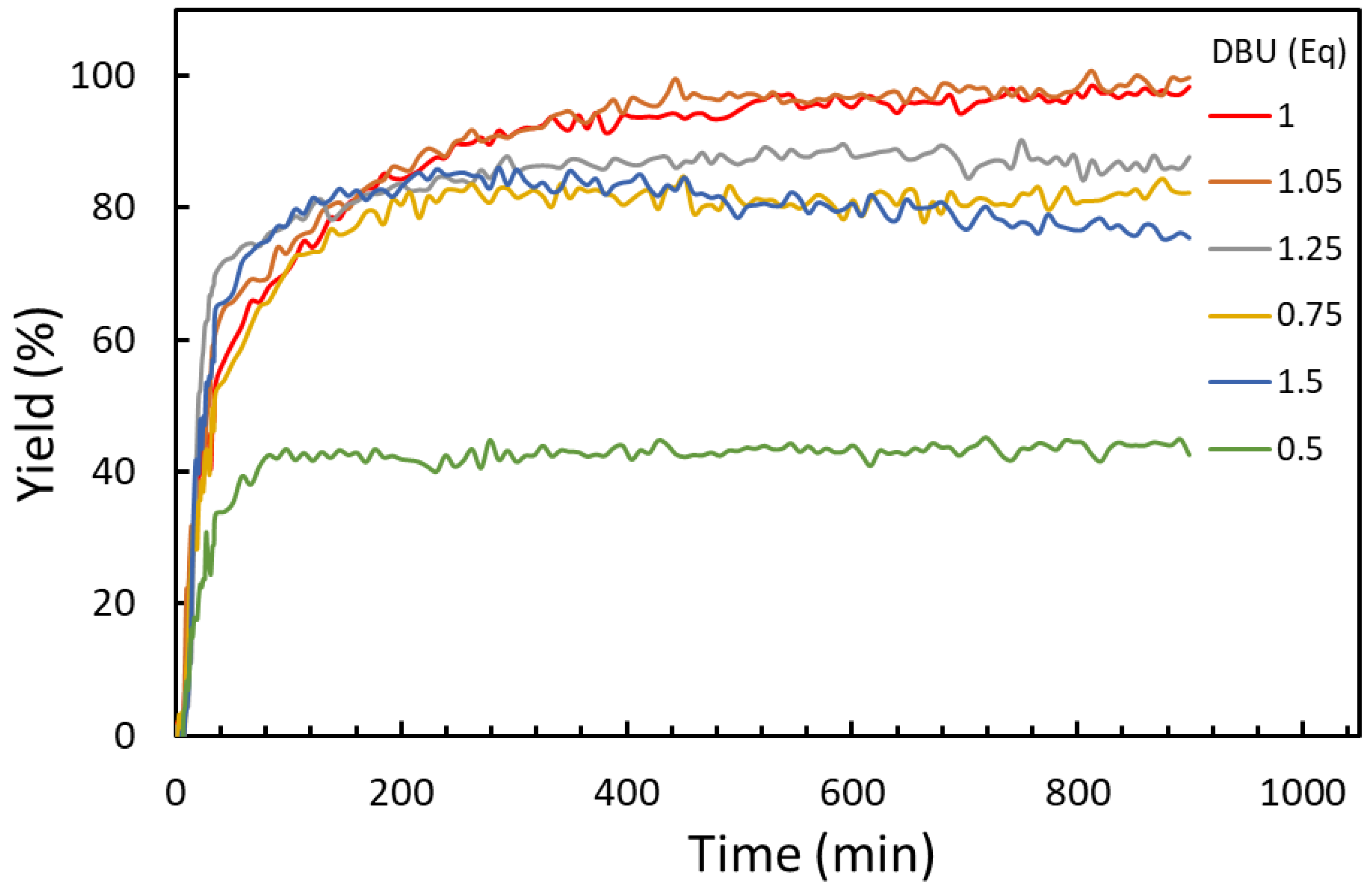
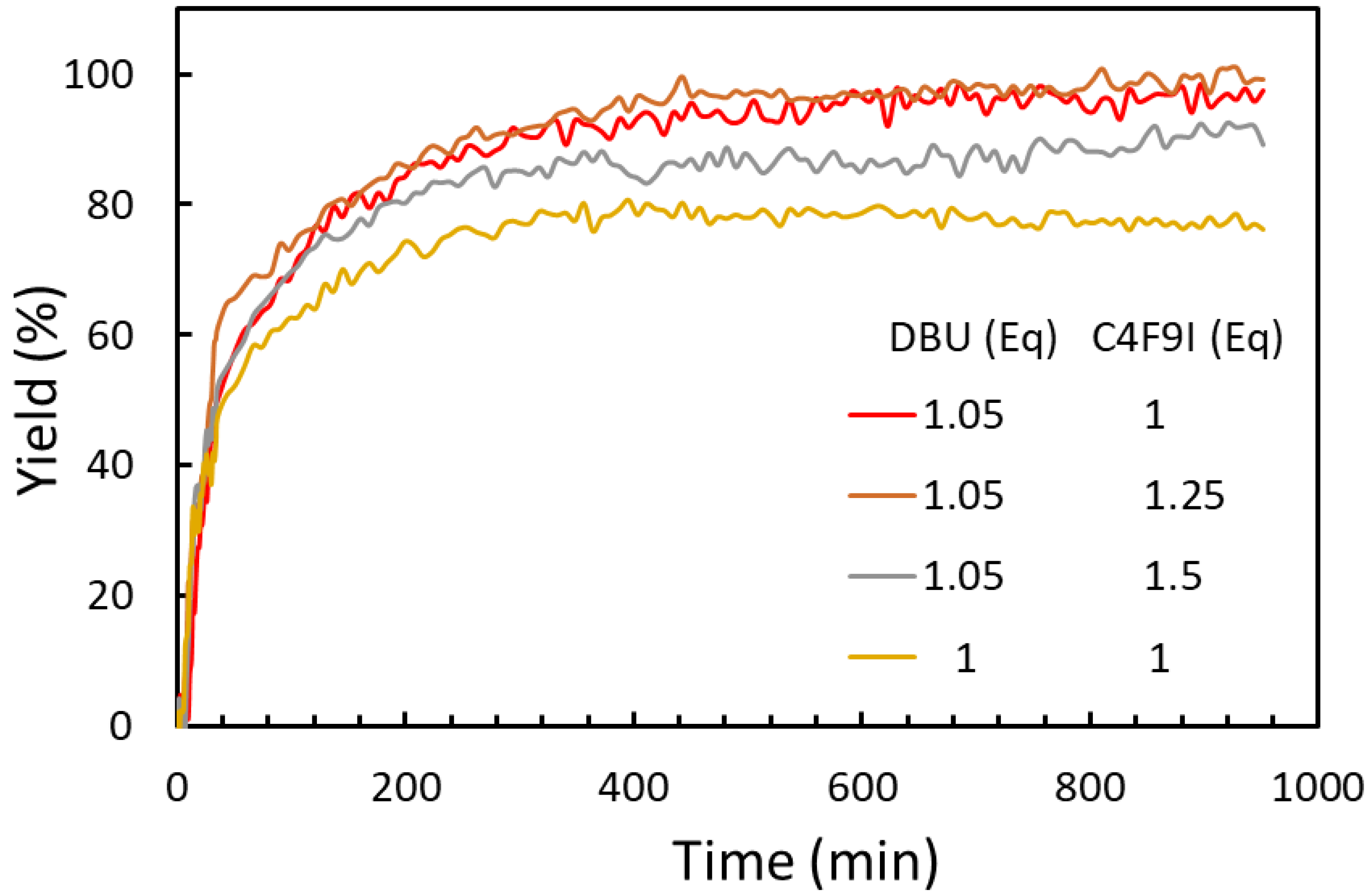
2.1.2. Effect of Reagent Stoichiometry
2.1.3. Halogen Bonding Effect
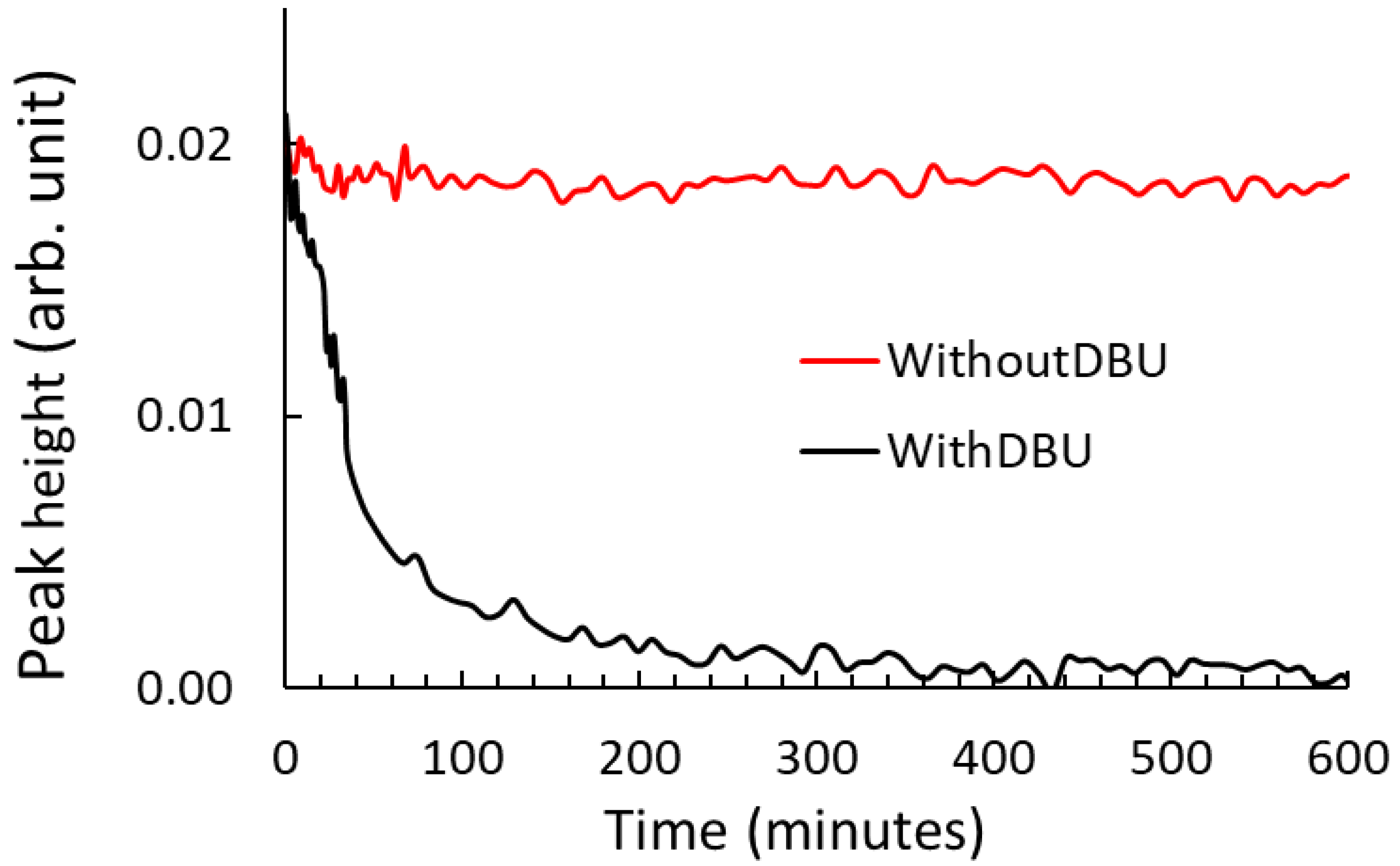
2.1.4. Lewis Base Effect
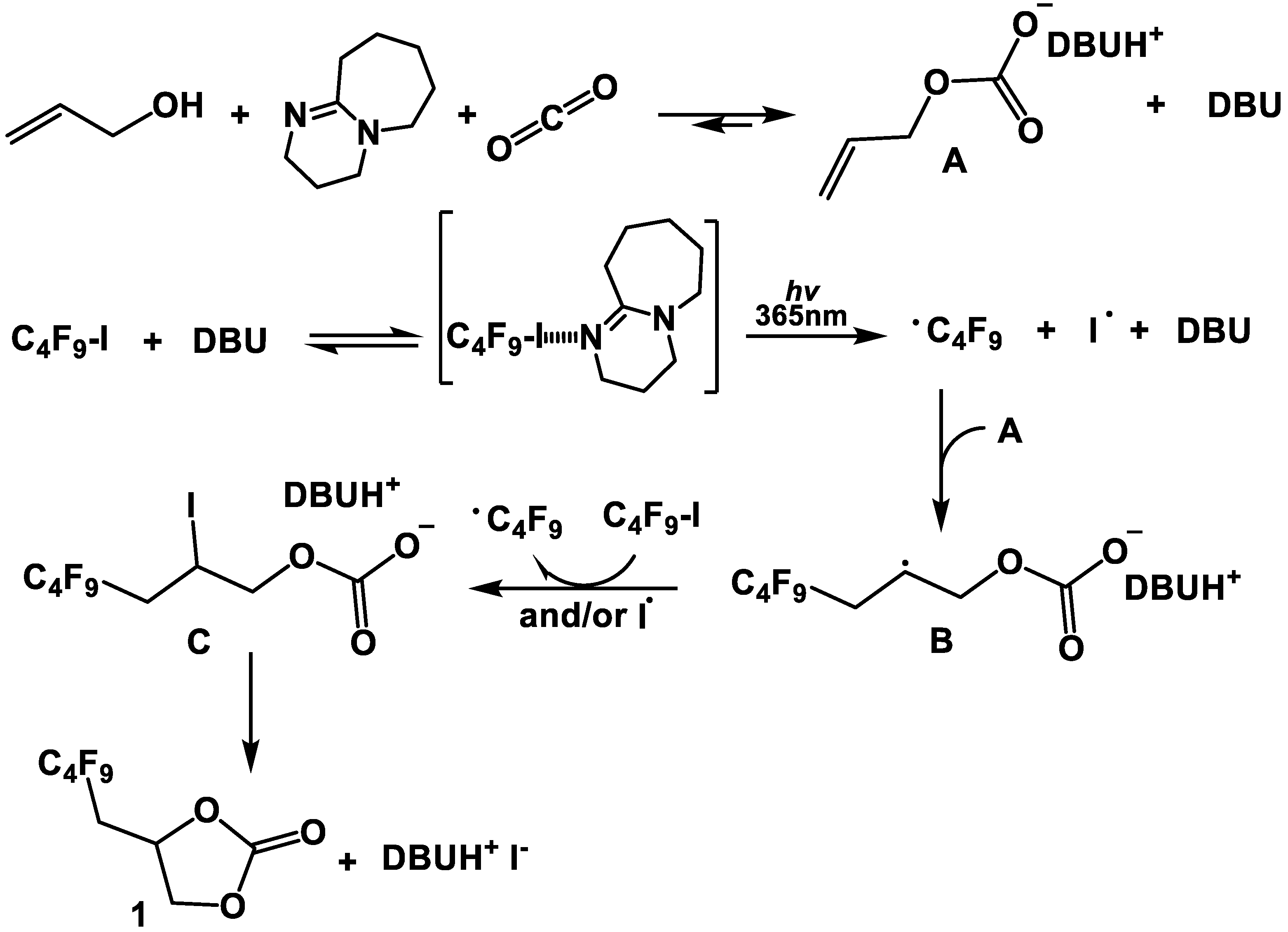
2.1.5. Mechanism
2.2. Influence of Experimental Parameters
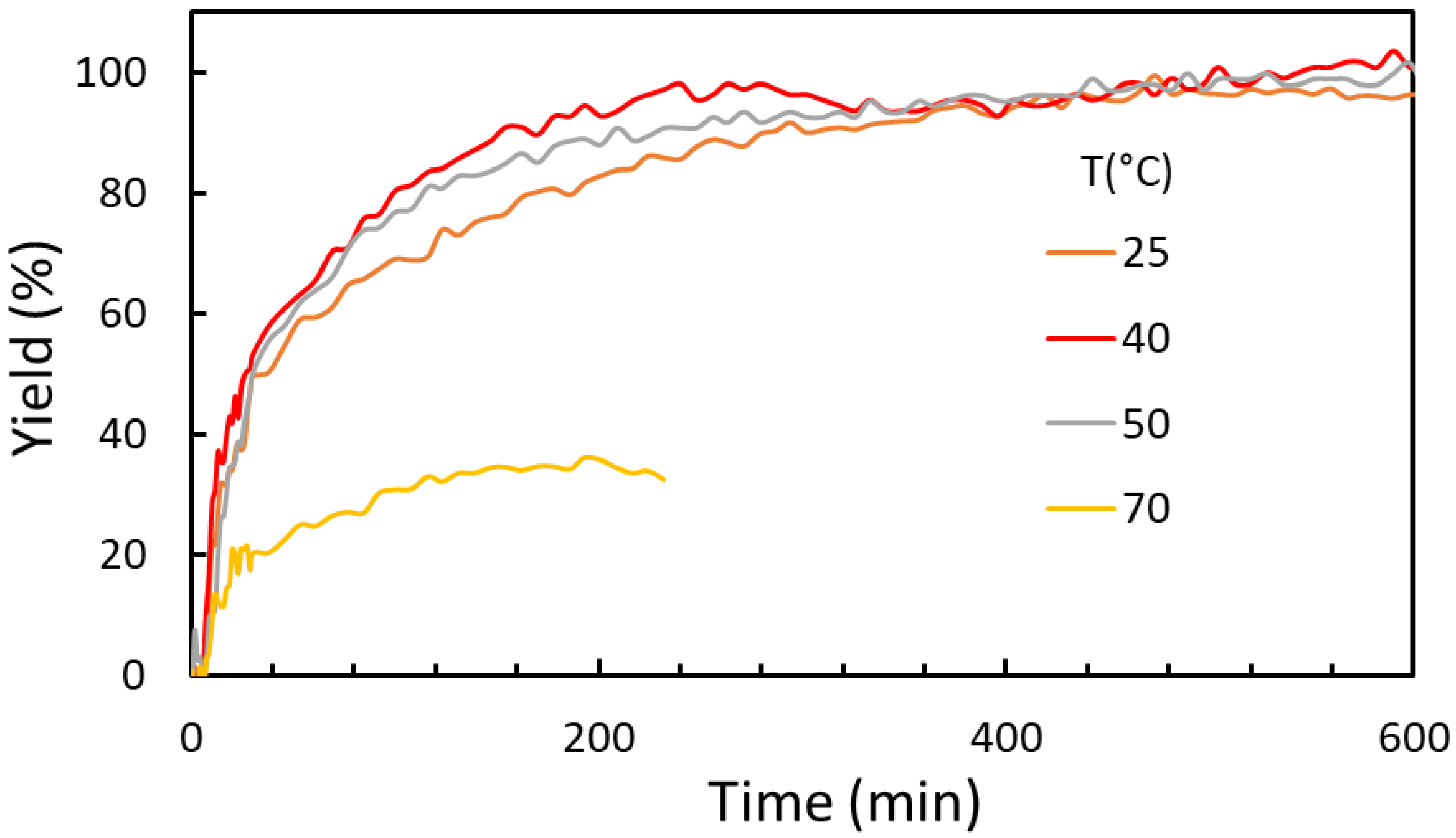
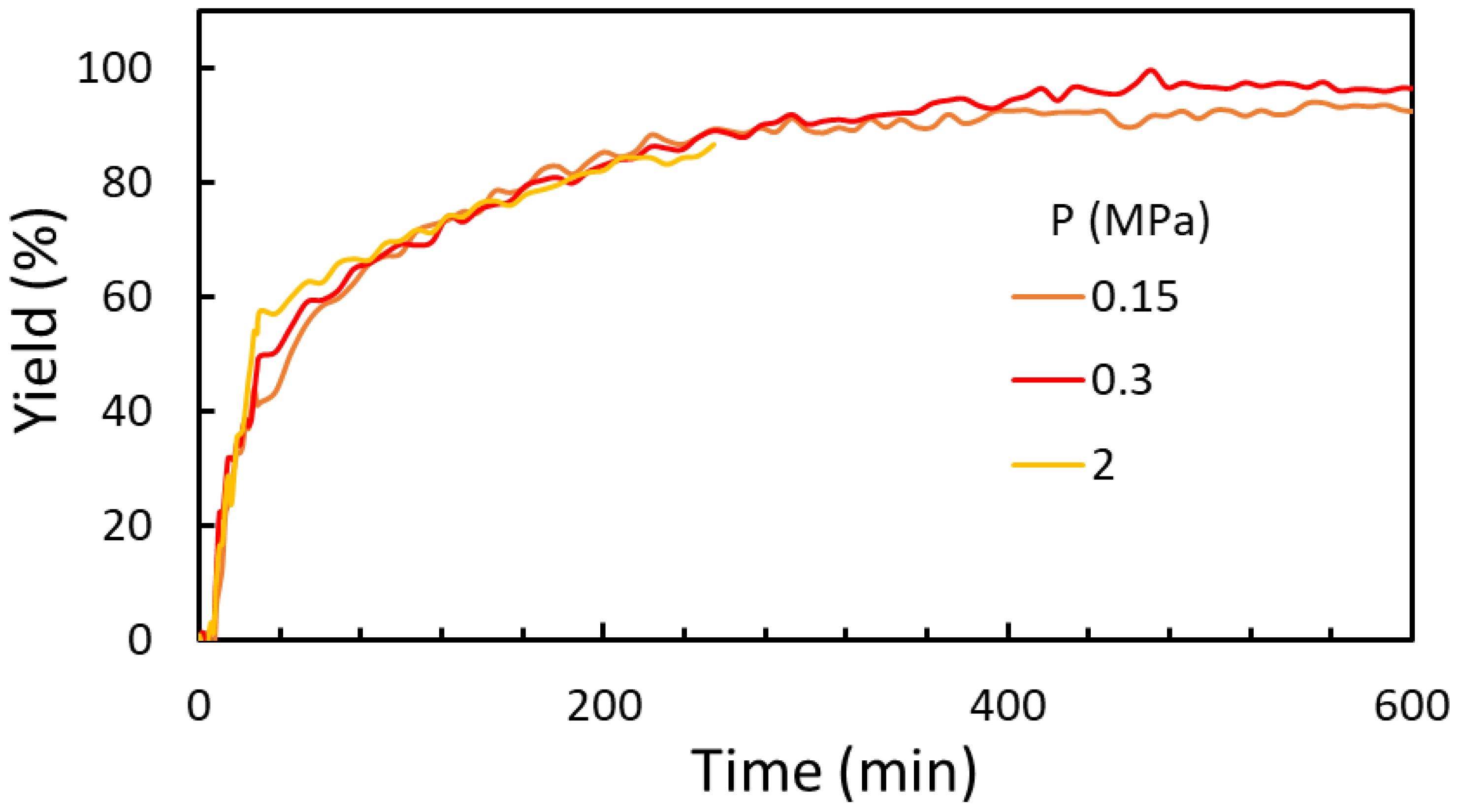
2.2.1. Temperature and Pressure Effect
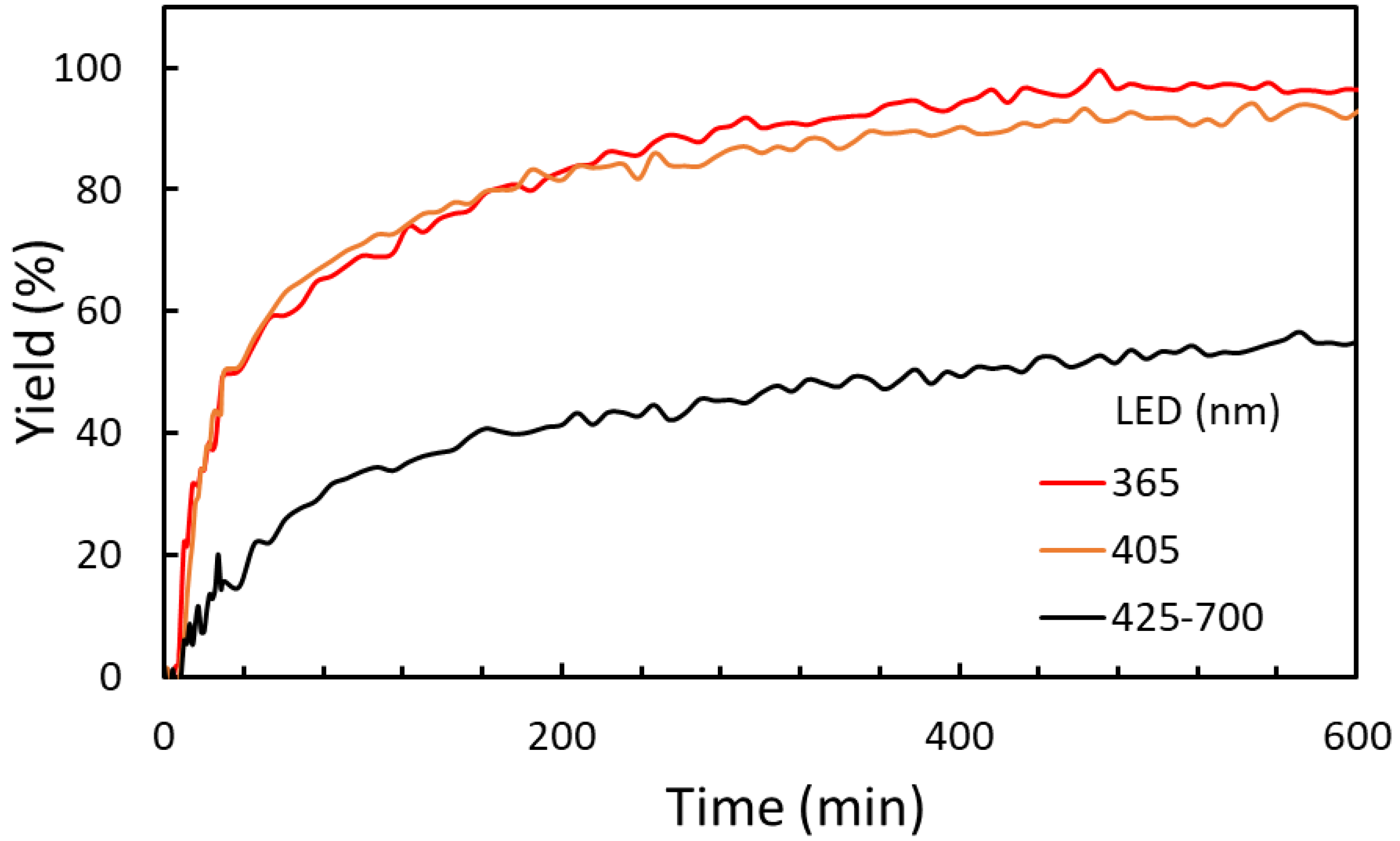
2.2.2. Irradiation Wavelength Effect
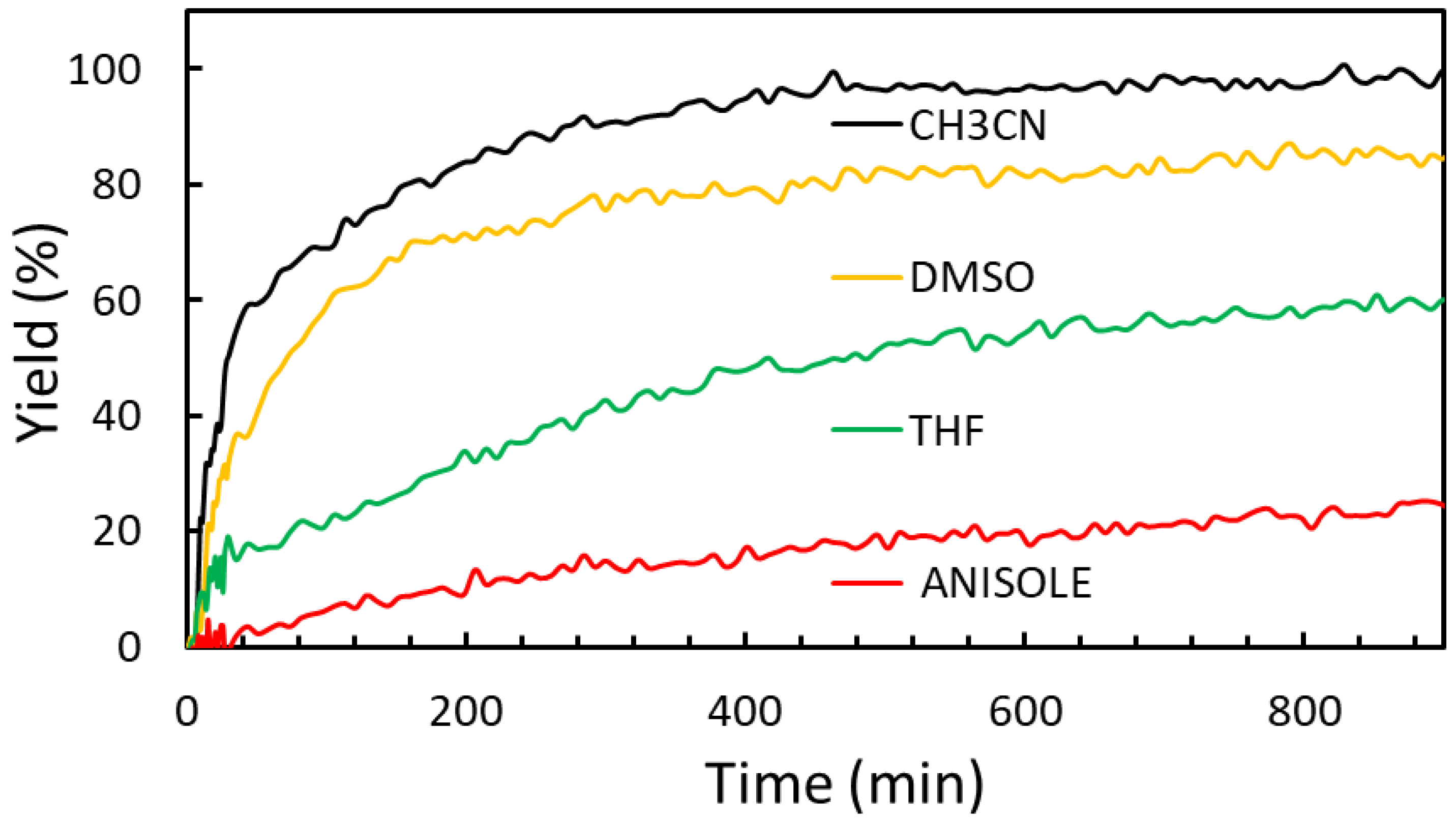
2.2.3. Solvent Effect
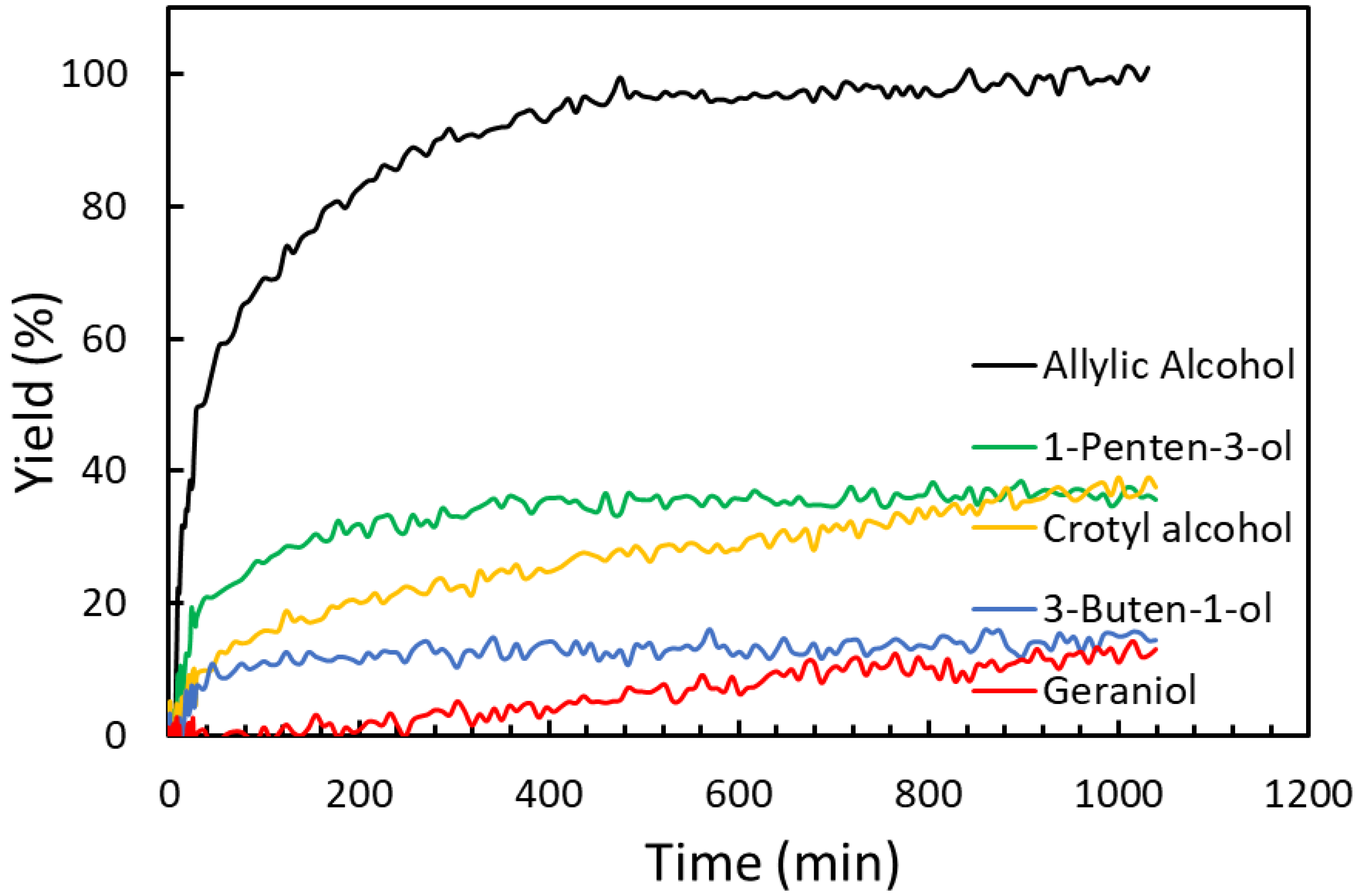
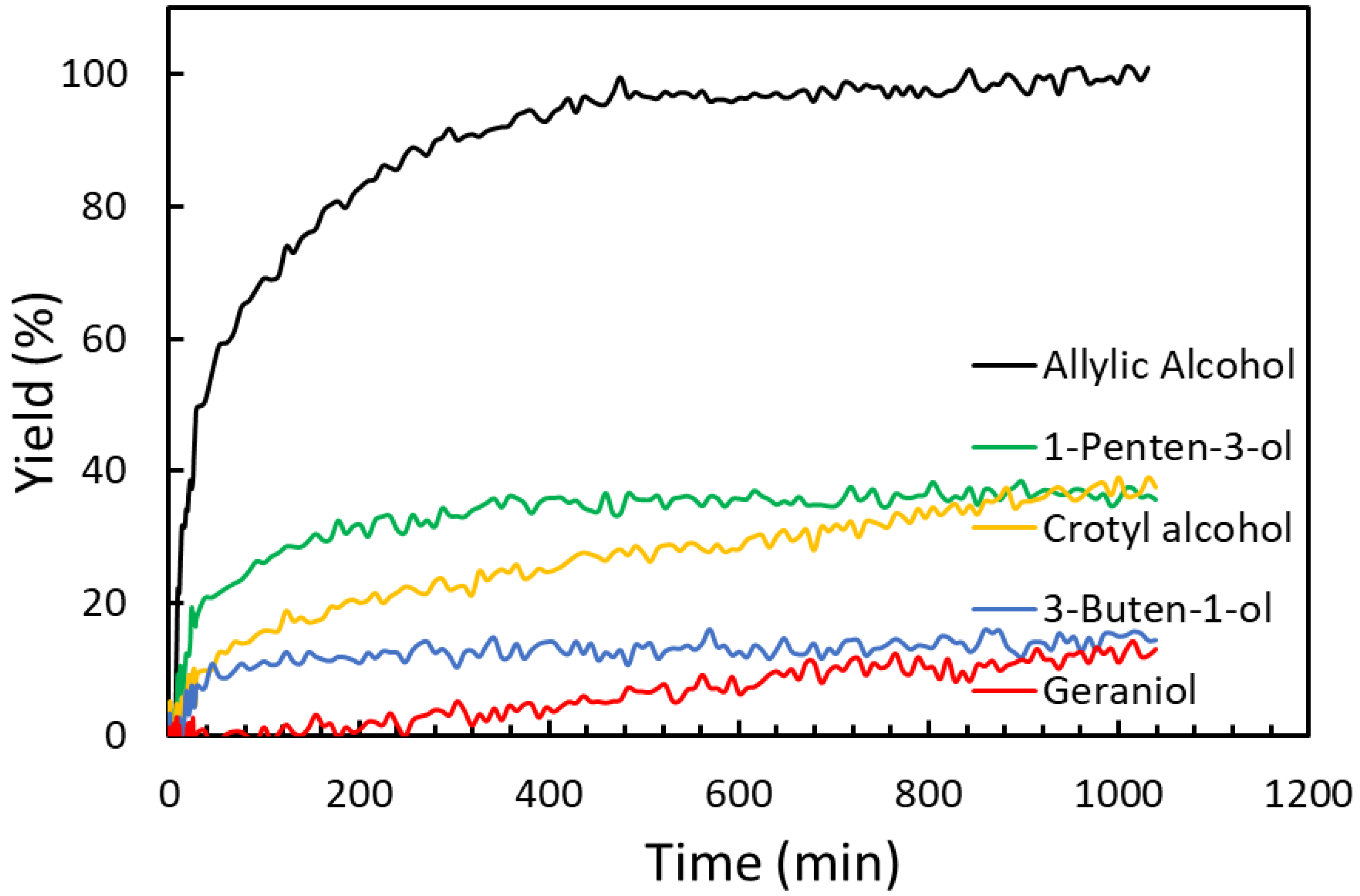
2.3. Substrate Scope
3. Materials and Methods
3.1. Material
3.2. In Situ Kinetics Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- North, M.; Pasquale, R.; Young, C. Synthesis of cyclic carbonates from epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Cokoja, M.; Wilhelm, M.E.; Anthofer, M.H.; Herrmann, W.A.; Kühn, F.E. Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide by Using Organocatalysts. ChemSuschem 2015, 8, 2436–2454. [Google Scholar] [CrossRef] [PubMed]
- Gennen, S.; Alves, M.; Méreau, R.; Tassaing, T.; Gilbert, B.; Detrembleur, C.; Jerome, C.; Grignard, B. Fluorinated Alcohols as Activators for the Solvent-Free Chemical Fixation of Carbon Dioxide into Epoxides. ChemSuschem 2015, 8, 1845–1849. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.; Grignard, B.; Gennen, S.; Mereau, R.; Detrembleur, C.; Jerome, C.; Tassaing, T. Organocatalytic promoted coupling of carbon dioxide with epoxides: A rational investigation of the cocatalytic activity of various hydrogen bond donors. Catal. Sci. Technol. 2015, 5, 4636–4643. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Mereau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Organocatalyzed coupling of carbon dioxide with epoxides for the synthesis of cyclic carbonates: Catalyst design and mechanistic studies. Catal. Sci. Technol. 2017, 7, 2651–2684. [Google Scholar] [CrossRef]
- Shaikh, A.-A.G.; Sivaram, S. Organic carbonates. Chem. Rev. 1996, 96, 951–976. [Google Scholar] [CrossRef]
- Tempelaar, S.; Mespouille, L.; Coulembier, O.; Dubois, P.; Dove, A.P. Synthesis and post-polymerisation modifications of aliphatic poly(carbonate)s prepared by ring-opening polymerisation. Chem. Soc. Rev. 2013, 42, 1312–1336. [Google Scholar] [CrossRef]
- Grignard, B.; Gennen, S.; Jérôme, C.; Kleij, A.W.; Detrembleur, C. Advances in the use of CO2 as a renewable feedstock for the synthesis of polymers. Chem. Soc. Rev. 2019, 48, 4466–4514. [Google Scholar] [CrossRef]
- Maisonneuve, L.; Lamarzelle, O.; Rix, E.; Grau, E.; Cramail, H. Isocyanate-Free Routes to Polyurethanes and Poly(hydroxy Urethane)s. Chem. Rev. 2015, 115, 12407–12439. [Google Scholar] [CrossRef]
- Grignard, B.; Thomassin, J.M.; Gennen, S.; Poussard, L.; Bonnaud, L.; Raquez, J.M.; Dubois, P.; Tran, M.P.; Park, C.B.; Jerome, C.; et al. CO2-blown microcellular non-isocyanate polyurethane (NIPU) foams: From bio- and CO2-sourced monomers to potentially thermal insulating materials. Green Chem. 2016, 18, 2206–2215. [Google Scholar] [CrossRef]
- Gennen, S.; Grignard, B.; Tassaing, T.; Jérôme, C.; Detrembleur, C. CO2-Sourced α-Alkylidene Cyclic Carbonates: A Step Forward in the Quest for Functional Regioregular Poly(urethane)s and Poly(carbonate)s. Angew. Chem. Int. Ed. 2017, 56, 10394–10398. [Google Scholar] [CrossRef]
- Monie, F.; Grignard, B.; Thomassin, J.-M.; Mereau, R.; Tassaing, T.; Jerome, C.; Detrembleur, C. Chemo- and Regioselective Additions of Nucleophiles to Cyclic Carbonates for the Preparation of Self-Blowing Non-Isocyanate Polyurethane Foams. Angew. Chem. Int. Ed. 2020, 59, 17033–17041. [Google Scholar] [CrossRef]
- Martín, C.; Fiorani, G.; Kleij, A.W. Recent Advances in the Catalytic Preparation of Cyclic Organic Carbonates. ACS Catal. 2015, 5, 1353–1370. [Google Scholar] [CrossRef]
- Rintjema, J.; Guo, W.; Martin, E.; Escudero-Adan, E.C.; Kleij, A.W. Highly Chemoselective Catalytic Coupling of Substituted Oxetanes and Carbon Dioxide. Chemistry 2015, 21, 10754–10762. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Boyaval, A.; Méreau, R.; De Winter, J.; Gerbaux, P.; Detrembleur, C.; Tassaing, T.; Jérôme, C. Organocatalytic Coupling of CO2 with Oxetane. ChemSuschem 2017, 10, 1128–1138. [Google Scholar] [CrossRef]
- Besse, V.; Camara, F.; Voirin, C.; Auvergne, R.; Caillol, S.; Boutevin, B. Synthesis and applications of unsaturated cyclocarbonates. Polym. Chem. 2013, 4, 4545–4561. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, Z.; Yu, B.; Zhang, H.; Xu, H.; Hao, L.; Han, B.; Liu, Z. Task-specific ionic liquid and CO2-cocatalysed efficient hydration of propargylic alcohols to [small alpha]-hydroxy ketones. Chem. Sci. 2015, 6, 2297–2301. [Google Scholar] [CrossRef]
- Song, Q.-W.; He, L.-N. Advances in CO2 Capture, Sequestration, and Conversion; American Chemical Society: Washington, DC, USA, 2015; Volume 1194, Chapter 2; pp. 47–70. [Google Scholar]
- Rintjema, J.; Kleij, A.W. Substrate-Assisted Carbon Dioxide Activation as a Versatile Approach for Heterocyclic Synthesis. Synthesis 2016, 48, 3863–3878. [Google Scholar] [CrossRef]
- Boyaval, A.; Méreau, R.; Grignard, B.; Detrembleur, C.; Jerome, C.; Tassaing, T. Organocatalytic Coupling of CO2 with a Propargylic Alcohol: A Comprehensive Mechanistic Study. ChemSuschem 2017, 10, 1241–1248. [Google Scholar] [CrossRef]
- Li, M.; Abdolmohammadi, S.; Hoseininezhad-Namin, M.S.; Behmagham, F.; Vessally, E. Carboxylative cyclization of propargylic alcohols with carbon dioxide: A facile and Green route to α-methylene cyclic carbonates. J. CO2 Util. 2020, 38, 220–231. [Google Scholar] [CrossRef]
- Huang, J.; Jehanno, C.; Worch, J.C.; Ruipérez, F.; Sardon, H.; Dove, A.P.; Coulembier, O. Selective Organocatalytic Preparation of Trimethylene Carbonate from Oxetane and Carbon Dioxide. ACS Catal. 2020, 10, 5399–5404. [Google Scholar] [CrossRef]
- Méreau, R.; Grignard, B.; Boyaval, A.; Detrembleur, C.; Jerome, C.; Tassaing, T. Tetrabutylammonium Salts: Cheap Catalysts for the Facile and Selective Synthesis of α-Alkylidene Cyclic Carbonates from Carbon Dioxide and Alkynols. ChemCatChem 2018, 10, 956–960. [Google Scholar] [CrossRef]
- Grignard, B.; Ngassamtounzoua, C.; Gennen, S.; Gilbert, B.; Méreau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Boosting the Catalytic Performance of Organic Salts for the Fast and Selective Synthesis of α-Alkylidene Cyclic Carbonates from Carbon Dioxide and Propargylic Alcohols. ChemCatChem 2018, 10, 2584–2592. [Google Scholar] [CrossRef]
- Dabral, S.; Bayarmagnai, B.; Hermsen, M.; Schießl, J.; Mormul, V.; Hashmi, A.S.K.; Schaub, T. Silver-Catalyzed Carboxylative Cyclization of Primary Propargyl Alcohols with CO2. Org. Lett. 2019, 21, 1422–1425. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Reyes, A.; Saxl, T.; Stein, P.M.; Rudolph, M.; Rominger, F.; Asiri, A.M.; Hashmi, A.S.K. Expanded Ring NHC Silver Carboxylate Complexes as Efficient and Reusable Catalysts for the Carboxylative Cyclization of Unsubstituted Propargylic Derivatives. ChemSuschem 2021, 14, 2367–2374. [Google Scholar] [CrossRef]
- Cervantes-Reyes, A.; Farshadfar, K.; Rudolph, M.; Rominger, F.; Schaub, T.; Ariafard, A.; Hashmi, A.S.K. Copper-catalysed synthesis of α-alkylidene cyclic carbonates from propargylic alcohols and CO2. Green Chem. 2021, 23, 889–897. [Google Scholar] [CrossRef]
- Kindermann, N.; Jose, T.; Kleij, A.W. Synthesis of Carbonates from Alcohols and CO2. Top Curr Chem. 2017, 375, 15. [Google Scholar] [CrossRef]
- Tamura, M.; Honda, M.; Nakagawa, Y.; Tomishige, K. Direct conversion of CO2 with diols, aminoalcohols and diamines to cyclic carbonates, cyclic carbamates and cyclic ureas using heterogeneous catalysts. J. Chem. Technol. Biotechnol. 2014, 89, 19–33. [Google Scholar] [CrossRef]
- Hosseinian, A.; Farshbaf, S.; Mohammadi, R.; Monfared, A.; Vessally, E. Advancements in six-membered cyclic carbonate (1,3-dioxan-2-one) synthesis utilizing carbon dioxide as a C1 source. RSC Adv. 2018, 8, 17976–17988. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Gruszka, W.; Hulla, M.; Das, S.; Dyson, P.J. Synthesis of cyclic carbonates from diols and CO2 catalyzed by carbenes. Chem. Commun. 2016, 52, 10787–10790. [Google Scholar] [CrossRef]
- Brege, A.; Grignard, B.; Méreau, R.; Detrembleur, C.; Jerome, C.; Tassaing, T. En Route to CO2-Based (a)Cyclic Carbonates and Polycarbonates from Alcohols Substrates by Direct and Indirect Approaches. Catalysts 2022, 12, 124. [Google Scholar] [CrossRef]
- Honda, M.; Tamura, M.; Nakagawa, Y.; Tomishige, K. Catalytic CO2 conversion to organic carbonates with alcohols in combination with dehydration system. Catal. Sci. Technol. 2014, 4, 2830–2845. [Google Scholar] [CrossRef]
- Honda, M.; Tamura, M.; Nakao, K.; Suzuki, K.; Nakagawa, Y.; Tomishige, K. Direct Cyclic Carbonate Synthesis from CO2 and Diol over Carboxylation/Hydration Cascade Catalyst of CeO2 with 2-Cyanopyridine. ACS Catal. 2014, 4, 1893–1896. [Google Scholar] [CrossRef]
- Reithofer, M.R.; Sum, Y.N.; Zhang, Y. Synthesis of cyclic carbonates with carbon dioxide and cesium carbonate. Green Chem. 2013, 15, 2086–2090. [Google Scholar] [CrossRef]
- Mcguire, T.M.; López-Vidal, E.M.; Gregory, G.L.; Buchard, A. Synthesis of 5- to 8-membered cyclic carbonates from diols and CO2: A one-step, atmospheric pressure and ambient temperature procedure. J. CO2 Util. 2018, 27, 283–288. [Google Scholar] [CrossRef]
- Gregory, G.L.; Ulmann, M.; Buchard, A. Synthesis of 6-membered cyclic carbonates from 1,3-diols and low CO2 pressure: A novel mild strategy to replace phosgene reagents. RSC Adv. 2015, 5, 39404–39408. [Google Scholar] [CrossRef]
- Kitamura, T.; Inoue, Y.; Maeda, T.; Oyamada, J. Convenient synthesis of ethylene carbonates from carbon dioxide and 1,2-diols at atmospheric pressure of carbon dioxide. Synth. Commun. 2015, 46, 39–45. [Google Scholar] [CrossRef]
- Lim, Y.N.; Lee, C.; Jang, H.-Y. Metal-Free Synthesis of Cyclic and Acyclic Carbonates from CO2 and Alcohols. Eur. J. Org. Chem. 2014, 2014, 1823–1826. [Google Scholar] [CrossRef]
- Brege, A.; Méreau, R.; Mcgehee, K.; Grignard, B.; Detrembleur, C.; Jerome, C.; Tassaing, T. The coupling of CO2 with diols promoted by organic dual systems: Towards products divergence via benchmarking of the performance metrics. J. CO2 Util. 2020, 38, 88–98. [Google Scholar] [CrossRef]
- Baran, T.; Dibenedetto, A.; Aresta, M.; Kruczała, K.; Macyk, W. Photocatalytic Carboxylation of Organic Substrates with Carbon Dioxide at Zinc Sulfide with Deposited Ruthenium Nanoparticles. ChemPlusChem 2014, 79, 708–715. [Google Scholar] [CrossRef]
- Masuda, Y.; Ishida, N.; Murakami, M. Light-Driven Carboxylation of o-Alkylphenyl Ketones with CO2. J. Am. Chem. Soc. 2015, 137, 14063–14066. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-Y.; Cao, Y.; Liu, X.; Wang, N.; He, L.-N.; Li, S.-H. Photoinduced radical-initiated carboxylative cyclization of allyl amines with carbon dioxide. Green Chem. 2017, 19, 1240–1244. [Google Scholar] [CrossRef]
- Gui, Y.-Y.; Zhou, W.-J.; Ye, J.-H.; Yu, D.-G. Photochemical Carboxylation of Activated C(sp3)−H Bonds with CO2. ChemSuschem 2017, 10, 1337–1340. [Google Scholar] [CrossRef]
- Murata, K.; Numasawa, N.; Shimomaki, K.; Takaya, J.; Iwasawa, N. Construction of a visible light-driven hydrocarboxylation cycle of alkenes by the combined use of Rh(i) and photoredox catalysts. Chem. Commun. 2017, 53, 3098–3101. [Google Scholar] [CrossRef] [PubMed]
- Long Ngo, H.; Kumar Mishra, D.; Mishra, V.; Chien Truong, C. Recent advances in the synthesis of heterocycles and pharmaceuticals from the photo/electrochemical fixation of carbon dioxide. Chem. Eng. Sci. 2021, 229, 116142. [Google Scholar] [CrossRef]
- He, X.; Yao, X.Y.; Chen, K.H.; He, L.N. Metal-Free Photocatalytic Synthesis of exo-Iodomethylene 2-Oxazolidinones: An Alternative Strategy for CO2 Valorization with Solar Energy. ChemSuschem 2019, 12, 5081–5085. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Bhatt, S.; Soni, A.; Khatri, P.K.; Guha, A.K.; Saikia, L.; Jain, S.L. Visible-light driven reaction of CO2 with alcohols using a Ag/CeO2 nanocomposite: First photochemical synthesis of linear carbonates under mild conditions. Chem. Commun. 2023, 59, 1313–1316. [Google Scholar] [CrossRef]
- Heldebrant, D.J.; Jessop, P.G.; Thomas, C.A.; Eckert, C.A.; Liotta, C.L. The Reaction of 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) with Carbon Dioxide. J. Org. Chem. 2005, 70, 5335–5338. [Google Scholar] [CrossRef]
- Onwukamike, K.N.; Tassaing, T.; Grelier, S.; Grau, E.; Cramail, H.; Meier, M.A.R. Detailed Understanding of the DBU/CO2 Switchable Solvent System for Cellulose Solubilization and Derivatization. ACS Sustain. Chem. Eng. 2018, 6, 1496–1503. [Google Scholar] [CrossRef]
- Heldebrant, D.J.; Yonker, C.R.; Jessop, P.G.; Phan, L. Organic liquid CO2 capture agents with high gravimetric CO2 capacity. Energy Environ. Sci. 2008, 1, 487–493. [Google Scholar] [CrossRef]
- Grondin, J.; Aupetit, C.; Vincent, J.-M.; Méreau, R.; Tassaing, T. Visible-light induced photochemistry of Electron Donor-Acceptor Complexes in Perfluoroalkylation Reactions: Investigation of halogen bonding interactions through UV–Visible absorption and Raman spectroscopies combined with DFT calculations. J. Mol. Liq. 2021, 333, 115993. [Google Scholar] [CrossRef]
- Postigo, A. Electron Donor-Acceptor Complexes in Perfluoroalkylation Reactions. Eur. J. Org. Chem. 2018, 2018, 6391–6404. [Google Scholar] [CrossRef]
- Smith, C.A.; Cramail, H.; Tassaing, T. Insights into the Organocatalyzed Synthesis of Urethanes in Supercritical Carbon Dioxide: An In Situ FTIR Spectroscopic Kinetic Study. ChemCatChem 2014, 6, 1380–1391. [Google Scholar] [CrossRef]
- Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; Mcelroy, C.R.; Abou-Shehada, S.; Dunn, P.J. CHEM21 selection guide of classical- and less classical-solvents. Green Chem. 2016, 18, 288–296. [Google Scholar] [CrossRef]
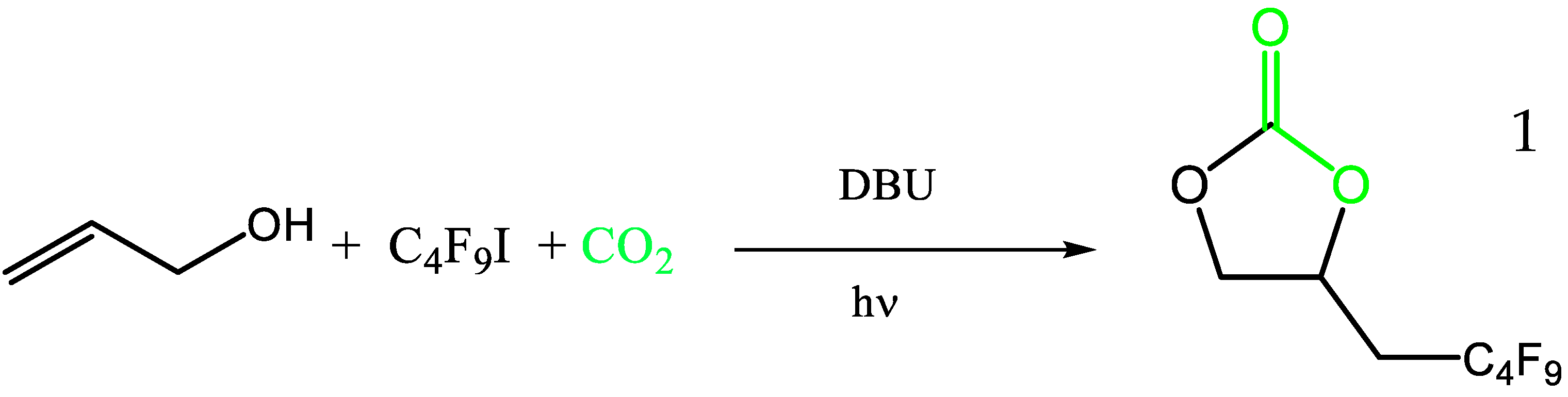



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grondin, J.; Aupetit, C.; Vincent, J.-M.; Tassaing, T. Cyclic Carbonates through the Photo-Induced Carboxylative Cyclization of Allylic Alcohol with CO2: A Comprehensive Kinetic Study of the Reaction Mechanism by In Situ ATR-IR Spectroscopy. Catalysts 2023, 13, 939. https://doi.org/10.3390/catal13060939
Grondin J, Aupetit C, Vincent J-M, Tassaing T. Cyclic Carbonates through the Photo-Induced Carboxylative Cyclization of Allylic Alcohol with CO2: A Comprehensive Kinetic Study of the Reaction Mechanism by In Situ ATR-IR Spectroscopy. Catalysts. 2023; 13(6):939. https://doi.org/10.3390/catal13060939
Chicago/Turabian StyleGrondin, Joseph, Christian Aupetit, Jean-Marc Vincent, and Thierry Tassaing. 2023. "Cyclic Carbonates through the Photo-Induced Carboxylative Cyclization of Allylic Alcohol with CO2: A Comprehensive Kinetic Study of the Reaction Mechanism by In Situ ATR-IR Spectroscopy" Catalysts 13, no. 6: 939. https://doi.org/10.3390/catal13060939