
A Mild and Sustainable Procedure for the Functionalization of Morpholin-2-Ones by Oxidative Imidation Reactions

1
Coordination Chemistry and Catalysis Group, Centro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal
2
Research Institute of Chemistry, Peoples’ Friendship University of Russia (RUDN University), 117198 Moscow, Russia
*
Author to whom correspondence should be addressed.
Catalysts 2023, 13(7), 1072; https://doi.org/10.3390/catal13071072
Submission received: 9 June 2023
/
Revised: 30 June 2023
/
Accepted: 3 July 2023
/
Published: 5 July 2023
(This article belongs to the Special Issue Advances in Green Catalysis for Sustainable Organic Synthesis, 2nd Edition)
Abstract
:Nitrogen-containing heterocycles such as morpholin-2-ones are structural elements of many biologically active substances, as well as useful synthetic intermediates. To be able to functionalize them regioselectively in an easy, atom-efficient, and environmentally friendly manner is highly desirable. A procedure for cross-dehydrogenative coupling between morpholinones and cyclic imides was developed addressing these requirements. An earth-abundant metal catalyst, copper(I) chloride, in the presence of acetic acid, and with molecular oxygen as the sole oxidant, operating under mild conditions, afforded the desired C–N coupled products in high yields. Besides being potentially biologically active, as many members of both families of compounds are, the products themselves may be suitable substrates for functionalized polymers, e.g., poly(β-aminoesters) or even for PROTACs.
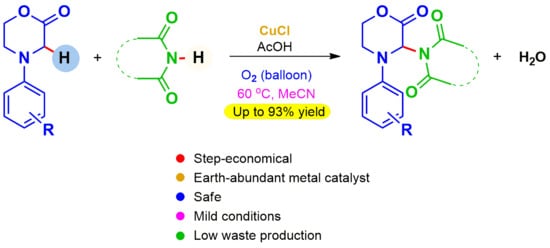
1. Introduction
Cross-dehydrogenative coupling (CDC) involving C(sp3)–H bonds has evolved in recent years as a mild, atom- and step-economical, cost-efficient, environmentally friendly way of undergoing synthesis [1,2,3,4,5,6,7,8,9]. Direct C–H/C–H coupling is possible, as well as C–X coupling (X = heteroatom), without the requirement for any pre-functionalization of the reagents. The possibility of using earth-abundant metal salts like copper or iron as catalysts has made this type of chemistry even more attractive. A variety of oxidants have been used, which act as the terminal acceptors of the two hydrogen atoms lost.
Amongst the substrates explored successfully for CDC reactions have been α-aminocarbonyl compounds, of which amino acids and peptides/peptidomimetics are the best representatives [10,11,12,13]. α-Alkylation or α-heterofunctionalization of these substances leads to the formation of modified amino acids and peptides which are of prime interest for the pharmaceutical industry [14,15,16,17]. For example, simple aryl glycines, which are nonproteogenic amino acids, are abundant in a large range of glycopeptide antibiotics and are also common key intermediates in the production of β-lactam antibiotics [18]. Until recently, classical approaches were utilized for α-functionalization to introduce various sidechains, relying mostly on carbanion chemistry [19,20,21,22], on the use of enzymes immobilized to a solid support [23,24,25,26], or on transition metal-catalyzed processes [23,24,25,26].
Problems related to side reactions or racemization with sensitive substrates may occur and the processes may be either more complex or less general. Several synthetic steps may also be involved. CDC by-passes these problems and also the possibility of racemization of chiral centres. We were interested in the prospect of extending this chemistry to conformationally constrained amino acids. Incorporating one or more amino acids into a ring is one of the ways to restrict conformational freedom, which is of great interest in medicinal chemistry for the development of new drugs, to include in peptidomimetics to probe protein–protein interactions, and even for synthetic intermediates [14,27,28,29,30,31]. Protein–protein interactions mediate processes in biological systems and play important roles in disease development [29]. Constraining by cyclization is a technique often used to improve stability and biological activity in peptides, since the molecules become more resistant to hydrolysis by peptidases either due to the conformational constraint or to the lack of amino and carboxyl termini [27,28,29,30,31]. In this context, morpholinones, which can subsequently be incorporated into peptides, seemed a good subject for research. CDC methodologies have been so far utilized for the α-functionalization of α-amino esters and α-amino amides or small peptides in some instances with alkynes, alkenes, indoles and other heteroarenes, ketones, β-ketoesters, nitroalkanes, ethers, diphenyl phosphine oxide and H-phosphonates, α-diazocarbonyl compounds, aziridines, as well as alcohols, thiols, naphthols, pyrroles [10,11,32] and amides and imides (Figure 1a) [33,34,35]. The α-functionalization of 3,4-dihydro-1,4-benzoxazin-2-ones with indoles (Figure 1b) [36] and with malonates (Figure 1c) [37] have been described, but to the best of our knowledge, morpholinones have not been employed in CDC reactions so far and became the subject of our studies (Figure 1d). The morpholinones can be obtained from amino acid esters, for example, by reaction with an ethylene equivalent [38,39].
Morpholinones are very interesting compounds in their own right [40,41,42,43,44]. They are useful intermediates for the pharmaceutical industry, an example being the synthesis of Aprepitant (A, Figure 2), a drug utilized to prevent chemotherapy-induced nausea or vomiting [40]. However, they also have important properties, including T-type Ca2+ channel activity blocking [41], anti-cancer [42], antifungal [43], amongst others. Some examples are shown in Figure 2 (structures B–D, J). In addition, morpholinones are good substrates for ring-opening reactions, including polymerization. For example, ring-opening polymerization of N-acyl morpholin-2-ones with N-acylated amines yielded functionalized poly(aminoesters) (PβAE) [45], and alcoholysis of optically active morpholin-2-ones yielded hydroxyl amides in a stereoselective fashion [46]. Substituted PβAEs have numerous applications due to their excellent properties. They are considered to be the most potent alternative to viral vectors for gene delivery, destined to treat or to eliminate diseases involving genetic factors, which are used instead of medicines to treat their symptoms [47,48,49]. However, PβAE are also important in other areas, which range from cancer therapy to tissue engineering [47,48,49]. New ways to functionalize morpholinones may find applications in the synthesis of novel PβAEs with different substitution patterns and different properties.
Inspired by recent developments in this field, we thought of using cyclic imides as reaction partners. CDC reactions of α-amino esters with imides have been reported but not with morpholinones [33,34,35]. CDC could yield new C–N bonded structures with various properties or for further synthetic manipulations, e.g., polymerization reactions for substituted PβAEs [50,51,52]. Cyclic imides are on their own an important group of compounds with many applications in medicine: as anticancer agents, antiepileptics, sedatives, hypnotics, anticonvulsants, hypotensive agents and antitubercular agents [53,54,55,56]. They are also valuable synthetic intermediates and of interest for the preparation of advanced polymers. An application in medicine is the glutarimide derivative Lenalidomide (E) (trade name Revlimid) the first oral medicine developed for the treatment of multiple myeloma and other hematological malignancies, which was the sixth most sold pharmaceutical in the USA in 2021 [57] and the second blockbuster drug in terms of retail sales [58]. This and other examples, some also sales blockbusters in 2021, are shown in Figure 2 (structures F–I) [53,54,55,56].
In recent years, certain cyclic imides, e.g., phthalimide, have also been identified as key structural units for PROTACs (Proteolysis Targeting Chimerics), small molecules used to bind to a target pathogenic protein as well as to an E3 ubiquitin ligase which triggers ubiquitination and induces degradation of the pathogenic protein by the ubiquitin-proteasome system [59,60,61,62,63,64]. Different from protein inhibition, this is a highly selective novel approach to disease control, for example, to destroy cancer cells. PROTACs are regarded as the next generation drugs and some are even undergoing clinical trials, e.g., K (Figure 2) [59]. Considering the importance and many applications of cyclic amides, sustainable and environmentally friendly approaches to synthesize new derivatives, which may display novel properties, are highly desirable. Although the imide proton is weakly acidic and many reactions of imides involve treatment with a base to form an anion to increase its nucleophilic reactivity [65], the Gabriel synthesis of primary amines being an example, recent publications show its usefulness in CDC reactions, under oxidative conditions, playing the role of nucleophilic partner [33,34,35]. We have developed a novel regioselective and environmentally friendly method to couple 2-morpholinones to cyclic imides by cross-dehydrogenative C–N coupling (Figure 1d), which works under mild conditions, using an earth abundant copper salt as catalyst and molecular oxygen as oxidant, thus avoiding by-product formation. Our results are described next.
2. Results
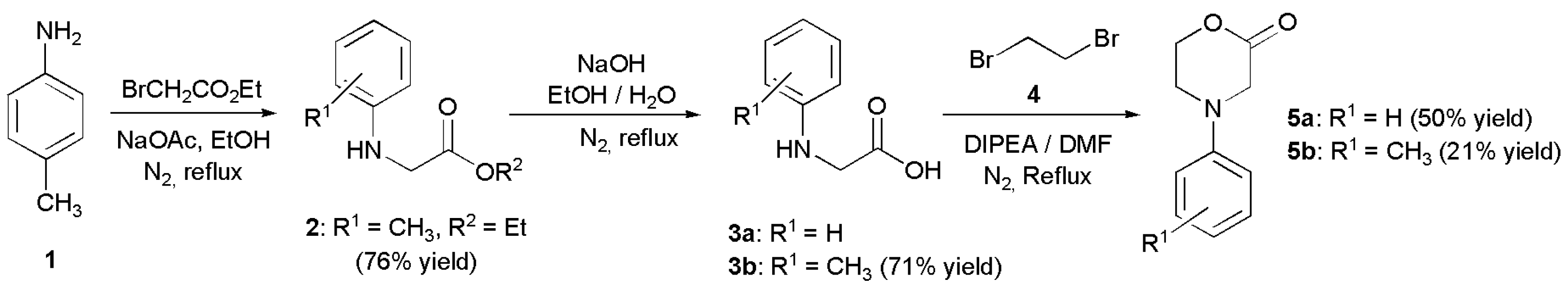
To develop our method, we chose known morpholinone, N-phenyl morpholin-2-one (5a), as the starting material, which was synthesized by an adaptation of a known methods from the corresponding commercially available protected amino acid 3a. The morpholinones may also be obtained from aryl amines as shown in Figure 3 [38,66,67]. The initial product of 1,2-dibromoethane addition is an intermediate bromoester, which upon prolonged heating cyclizes to the desired product 5 [38]. Morpholinone 5a was stable for prolonged periods at 4 °C (in a refrigerator), but if stored at room temperature for a few weeks it tended to decompose.
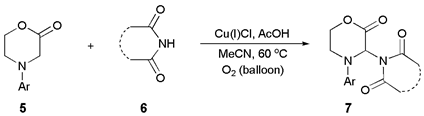
The cross-dehydrogenative coupling of morpholinone 5a with phthalimide (Phth, 6a) was selected as the model reaction to study. As starting reaction conditions, those used by Raman and Chandrasekharam to couple N-arylglycine ester derivatives with phthalimides were used, namely 1,2-dichloroethane as solvent, at room temperature, with atmospheric oxygen (air) as oxidant, and Cu(I)Cl as catalyst [33]. However, under these conditions, only traces of product 6a were observed. When the temperature was raised to 60 °C, and the reaction was performed under molecular O2 (in a balloon), the desired CDC product 6a was obtained with a 23% yield after 23 h (Table 1, entry 1). Coupling took place at C-3 exclusively, bearing the most activated C–H bond, since it is doubly activated by the nitrogen atom and the carbonyl function. No coupling was observed with C(6)–H. These results were not surprising in view of the fact that Raman and Chandrasekharam had found that under similar conditions, a substrate containing a tertiary nitrogen atom, i.e., N-methyl-N-phenylglycine ester, did not react, and that the presence of a free NH group was a requirement for coupling. When morpholinone 5a was reacted in DMSO no product was obtained either (entry 2); however, in MeCN, at a higher temperature (60 °C), with Cu(I)Cl as catalyst and under air, the desired C-3 imidated morpholinone was obtained in 37% yield (in a 0.2 M solution) during a similar period of time (cf. entries 2 and 3) although under an atmosphere of molecular oxygen, not air, a further increase was observed (entries 3 and 4). Since a large amount of starting material remained unreacted, as evidenced by 1H NMR spectroscopy, the reaction time was increased (entry 5) with only a small increase in yield. Preliminary observations had already shown that the reaction tends to stall after some time, with the conversion hardly increasing with time. Lowering the concentration while raising the catalyst loading had a beneficial effect on the yield (cf. entries 4 and 6), but a similar result was obtained when the reaction was performed at higher temperature with a larger excess of morpholinone (cf. entries 5, 6 and 7). Increasing the concentration even more made the results worse, and by-product formation was higher (cf. entries 6 and 8).
Higher catalyst loadings were also no better (entry 9). A concentration of 0.1 M with respect to the imide still gave the best result (entry 6), and this was preferable to running the reaction at a higher temperature, with a lower catalyst loading but a larger excess of morpholinone (entry 7). It thus seemed that this CDC reaction is highly susceptible to the solvent used, as well as to the concentration of the terminal oxidant, molecular oxygen, and that of the imide.
We also tried to use other metal salts as catalysts (entries 10–13), but Cu(I)Cl remained the best catalyst. A search for oxidants showed, in preliminary experiments, that the reaction did proceed in the presence of t-BuOOH in decane, but much more product was obtained with DTBP (di-tert-butyl peroxide) during the same period of time. Increasing the concentration of DTBP from 2.0 to 3.0 equiv caused a small increase in yield (cf. entries 14 and 15), but less product was obtained than when the reaction was performed under O2 (entry 9). However, with 1.0 equiv of DTBP, the product could be obtained in 80% yield (entry 16).
The use of additives was also explored in an attempt to improve results. The presence of triethylamine or molecular sieves practically suppressed the reaction completely, but with addition of pyridine a 30% yield could be obtained. Surprisingly, when acetic acid was added, the yield jumped to 87% and increasing its concentration caused an even higher increase in yield to 93% (entries 20 and 21). In the presence of this weak acid a clear reaction mixture was obtained, whereas in its absence, a small amount of precipitate was observed as the reaction proceeded. In recent times several reports have documented the beneficial effect of acetic acid in accelerating CDC reactions of nitrogen heterocycles and other amines, although the true nature of its role is not clear [68,69,70,71,72]. Finally, surprisingly, we also observed that acetic acid alone, under an atmosphere of air, could promote this CDC reaction affording the product in high yield (entry 22). If the acetic acid reaction was performed under nitrogen, i.e., excluding all oxygen, the yield obtained was substantially lower (entry 23). In the absence of both the copper salt and acetic acid, a small amount of background reaction was also observed. However, in all cases, even at very high conversions, there was always a small amount of unreacted imide present (less than 10%). Continuing the reaction for long periods did not help. The best reaction conditions overall, as shown in entry 21, were selected for further work, to explore the scope of the reaction.
It was found that cyclic imides with a variety of substitution patterns were compatible with the reaction conditions, and the corresponding products were obtained in good to high yields (Table 2). Unlike the starting morpholinones, the products were stable at room temperature for a long period of time (months). In some cases, it was difficult to separate by chromatography the product from the starting morpholinone. This led to lower yields of product being obtained in some cases. An excess of morpholinone over the imine helped to raise the yield, as observed in preliminary experiments. The reactions were regioselective, with the 3-substituted morpholinone being produced exclusively. This is to be expected, in view of the fact that a NC(3)HCOO radical is more stable than a NC(5)HCH2 radical, due to the additional resonance stabilization provided by the CO group. Hence, it is easier to form a C(3) radical than a C(5) radical, and loss of hydrogen occurs at this carbon atom preferentially. A similar behavior is observed in reactions of β-keto esters [73,74].
Cyclic imides with a five-membered ring reacted smoothly at 60 °C, affording the products in in up to 93% yield. The meso phthalimide cis-1,2,3,6-tetrahydrophthalimide produced 7b with an excellent yield of 93%. The linear imide N-acetylacetamide failed to react to yield the desired product 7g. The reaction conditions were, however, compatible with 6-membered cyclic imides and glutarimide afforded the product 7h, although it was slower to react than the five-membered ring imides. It was found necessary to perform the reaction at a higher temperature of 80 °C for a good yield of product to be obtained. In this case, the absence of acid was also found to favor the reaction. The presence of double bonds in the substrates, i.e., in 7b and 7d, did not influence the results and the reactions were also regioselective. The presence of additional electron withdrawing groups was counter-productive, and lower yields of product were obtained, e.g., the reaction of 4-nitrophthalimide to afford 7c. Presumably for a similar reason, barbituric acid did not react to produce the corresponding functionalized morpholinone (7i), neither did saccharin acid yield 7j in a catalytic CDC reaction either at 60 or at 80 °C, in the presence or absence of acid.
The enantiomerically pure drug (R)-ethosuximide (3-ethyl-3-methyl-pyrrolidine-2,5-dione) also reacted smoothly, providing morpholinone adduct 7f in high yield as a 1:1 mixture of two diastereoisomers. Ethosuximide is a medication used to treat absence seizures, sold under the brand name Zarontin amongst others. It is on the World Health Organization’s List of Essential Medicines [75]. In addition, when the phenyl nitrogen protecting group was replaced by tolyl, the desired CDC reaction proceeded smoothly to afford the desired product 7k in 75% yield after chromatography. The structures of all the new compounds were confirmed by NMR and infrared spectroscopy and by elemental analysis [also confirmed by mass spectrometry (ESI-MS)]. Copies of the spectra are available in the Supplementary information file.
Although in recent years, molecular dioxygen has emerged as an oxidant for oxidative coupling reactions, the actual reaction mechanisms by which it operates are still the subject of debate [76,77,78]. O2 is both thermodynamically and kinetically stable, with a triplet ground state, and, hence, its activation is challenging. First row transition metals like copper tend to work, probably because they are prone to function via single electron steps.
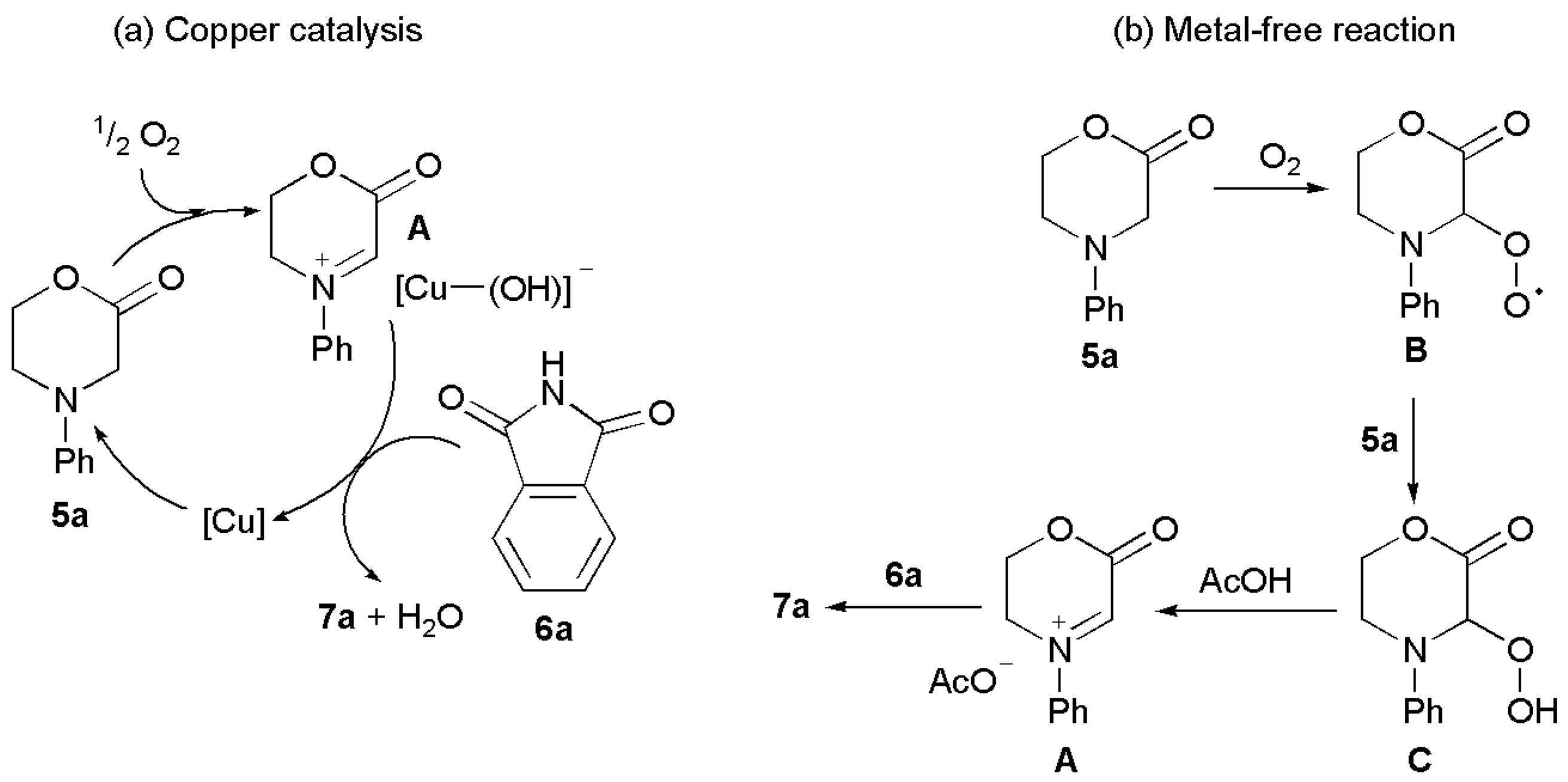
The exact mechanism of the present imidation reaction is not known. We performed an experiment in which 1.0 mol equiv of the radical trap 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) was added to the reaction mixture of morpholinone 5a and phthalimide, under the standard reaction conditions of Cu(I)Cl and AcOH catalysis, and some reaction suppression was observed, i.e., there was only ~70% conversion of the phthalimide into product 7a over the usual 28 h. When the reaction was performed under identical conditions with 1.0 equiv of TEMPO and Cu(I)Cl as catalyst, but without AcOH, a large suppression was observed, and there was only ~25% conversion of the Phth into product 6a. These results suggest that when Cu(I)Cl is used alone as a catalyst the reaction follows a radical pathway. However, when AcOH is added, the reaction mechanism changes, although some radical-mediated reaction may still take place to a smaller extent. Based on these results and previous findings by others on oxidative couplings of other benzylic species [76,77,78], we tentatively propose that a mechanism similar to that shown in Figure 4a may be operating, with copper-catalyzed oxidation of the tertiary amine giving rise to an iminium ion A, which is subsequently trapped by the weakly nucleophilic imides. The species [HOOCuBr]− has also been proposed as a very probable iminium counterion by Zhang, Wiest, Wu, and coworkers in a recent computational study on the mechanism of copper-catalyzed sp3-CH CDC reactions [79]. The role of the acetic acid in the present study may be to protonate nitrogen and hence accelerate iminium ion formation, which would lead to the rate acceleration observed.
In the case of the metal-free reaction (Figure 4b), in the presence of AcOH and molecular oxygen, a mechanism similar to that proposed by Tokuyama and co-workers for oxidative coupling reactions at the α-position of tertiary amines is a possibility. It involves the formation of an intermediate radical B by auto-oxidation, which reacts further with molecular oxygen to generate peroxide C. Radical C can abstract a hydrogen atom from another substrate molecule 5a, producing D and more B. In the presence of acetic acid, the iminium ion A is formed, which reacts with the nucleophilic imide to giving rise to the final product 7a [70]. The two mechanisms could also act simultaneously, through the common intermediate A, which would increase the reaction rate, in the metal/AcOH catalyzed reaction.
To the best of our knowledge, the functionalized morpholinones 7 obtained in this work are all new compounds, so far undocumented.
3. Materials and Methods
3.1. General Methods
All the reagents were of analytical grade, acquired from commercial suppliers [Acros Organics (Geel, Belgium), Alfa Aesar (Kandel, Germany), Fluka (Buchs, Switzerland),TCI (Zwijndrecht, Belgium), Fisher Chemical (Porto Salvo, Portugal), or Sigma-Aldrich (Merck, Darmstadt, Germany)]. unless otherwise stated below, and were used without further purification. Compressed oxygen gas (purity 99.999%) was supplied by Air Liquide (Portugal). One-dimensional (1H, 13C, and DEPT) and two-dimensional (COSY, HSQC and HMBC) NMR spectra were recorded on a Bruker Avance (300 MHz) spectrometer or on a Bruker Avance (400 MHz) spectrometer (Brucker, Bremen, Germany) in deuterated chloroform (CDCl3) as solvent. Chemical shifts are reported in ppm relative to the residual CHCl3 solvent peaks and coupling constants (J) are reported in Hertz. Resonance and structural assignments were based on the analysis of coupling patterns, including the 13C−1H coupling profiles obtained in bidimensional heteronuclear multiple bond correlation (HMBC) and heteronuclear single quantum coherence (HSQC) experiments, performed with standard pulse programs. Multiplicities in 13C were determined by DEPT experiments. Infrared spectra were recorded neat on an Agilent Cary 630 FTIR spectrophotometer (Agilent, Santa Clara, CA, USA). Elemental analyses (C, H and N) were performed by the microanalytical services of the Laboratório de Análises do Instituto Superior Técnico (LAIST-IST, Universidade de Lisboa, Portugal). High resolution mass spectra were performed by the Mass Spectrometry Laboratory of IST (Universidade de Lisboa, Portugal). They were obtained on a QqTOF Impact IITM mass spectrometer (Bruker Daltonics, Bremen, Germany) interfaced with an ESI source operating in the positive mode. Samples were analyzed by direct infusion (DI), and the mass analyser was calibrated with a solution of ammonium formate (10 mM) at the beginning of each analysis. The full scan mass spectra were acquired over a mass range of 100–1000 m/z at a spectra rate of 1 Hz. Data were acquired and processed using Bruker Compass Data Analysis ver 5.1 software of LAIST (Bruker Daltonics, Bremen, Germany).
The starting morpholinones are not commercially available and were synthesized by known methods [38,66,67], and so was 4-nitrophthalimide (5-nitroisoindole-1,3-dione) [80]. The substances synthesized produced NMR spectra (1H and 13C) identical to those described in the literature as follows: N-phenyl morpholine-2-one [45]; 4-nitrophthalimide [81]. Copies are available below. Melting points were determined either by a Büchi melting point apparatus B-540 (Büchi, Flawil, Switzerland) or by a Leica Galen III melting point apparatus (Aigner-Unilab Laborfachhandel, Vienna, Austria), and they are uncorrected.
3.2. General Procedure for the CDC Reaction between Morpholinones and Imides
The morpholinone (2.0 mmol), the imide (1.0 mmol) and Cu(I)Cl (0.15 mmol) were dissolved in acetonitrile (10 mL). Acetic acid (1.5 equiv., 1.5 mmol) was added dropwise with a syringe. The flask was topped-up with an oxygen balloon. The reaction mixture was heated to 60 or to 80 °C as required and stirred for the appropriate time at that temperature. After cooling, the reaction mixture was filtered through a small plug of silica gel with the aid of more acetonitrile. Subsequently, the solvent was evaporated off in a rotary evaporator to afford the crude dry product, which was purified by silica gel chromatography. The purified derivatized morpholinones 7 were stable at room temperature for long periods of time (months). They were slightly hygroscopic.
3.3. Characterization Data of Products and Starting Materials
- 3-(1,3-Dioxoindolin-2-yl)-4-phenylmorpholin-2-one (7a): Prepared from N-phenyl morpholine-2-one and phthalimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate (8:1) as eluent. Obtained as buff-colored crystals, 57.8 mg, 68% yield, m.p. 166 °C. 1H NMR (CDCl3): δ 3.758 (dt, 1 H, J 2.5, 13.4 Hz, C(5)HH), 4.249 (overlapp ddd, 1 H, J 1.3, 10.8, 13.6 Hz, C(5)HH), 4.62–4.79 (m, 2 H, 2 × H-6), 6.516 (s, 1 H, H-3), 6.911 (t, 1 H, J 7.3 Hz, p-Ph-H), 7.020 (d, 2 H, J 8.5 Hz, 2 × o-Ph-H), 7.279 (overlapp. t, 1 H, J 6.9 Hz, m-Ph-H), 7.282 (overlapp. t, 1 H, J 7.4 Hz, m-Ph-H), 7.734 (dd, 2 H, J 3.1 Hz, 5.5 Hz, H-6′ and H-7′), 7.835 (dd, 2 H, J 3.0, 5.5 Hz, H-5′, H-8′) ppm. 13C NMR (CDCl3): δ 42.27 (CH2, C-5), 61.72 (CH, C-3), 69.32 (CH2, C-6), 115.78 (CH, 2 × o-Ph-H), 121.13 (CH, p-C, Ph), 123.78 (CH, C-5′ and C-8′), 129.41 (CH, 2 × m-Ph-H), 131.55 (Cq, i-C, Ph), 134.42 (CH, C-6′ and C-7′), 144.83 (Cq, C-4′ and C-9′), 164.41 (Cq, CO, C-2), 167.43 (Cq, 2 × Phth-CO) ppm. IR (neat): ṽ 2957, 1742, 1714, 1600, 1500, 1380, 1221, 1186, 1080, 1000, 979, 886, 765, 714, 693, 656, 521 cm−1. Elemental analysis: Calcd for C18H14N2O4·¼H2O: C, 66.15; H, 4.47; N, 8.57. Found C, 66.19; H, 4.12; N, 8.49. HRMS (ESI): Calcd for C18H14N2NaO4: 345.0847. Found: 345.0846. Calcd for C18H14KN2O4: 361.0589. Found: 361.0585.
- 2-(2-Oxo-4-phenylmorpholin-3-yl)-3a,4,7,7a-tetrahydroisoindole-1,3-dione (7b): Prepared from N-phenyl morpholine-2-one and cis-1,2,3,6-tetrahydrophthalimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate (8:1) as eluent. Obtained as buff-colored crystals, 38.2 mg, 93% yield, m.p. 131–133 °C. 1H NMR (CDCl3): δ 2.129 (dd, 2 H, J 6.6, 15.0 Hz, C(5′)HH and C(8′)HH), 2.363 (d, 2 H, J 13.9 Hz, C(5′)HH and C(8′)HH), 2.94–3.11 (m, 2 H, H-4′ and H-9′), 3.584 (apparent. d, 1 H, 13.0 Hz, C(5)HH), 4.136 (appar. t, 1 H, J 12.7 Hz, C(5)HH), 4.53–4.71 (m, 2 H, 2 × H-6), 5.536 (apparent. s, 2 H, 6′ and 7′), 6.327 (s, 1 H, H-3), 6.880 (d, 2 H, J 7.8 Hz, 2 × o-Ph-H), 6.934 (t, 1 H, J 7.0 Hz, p-Ph-H), 7.258 (t, 2 H, J 7.4 Hz, 2 × m-Ph-H) ppm. 13C NMR (CDCl3): δ 23.15/23.27 (2 × CH2, C-5′ and C-8′), 38.84/39.23 (2 × CH, C-4′ and C-9′), 42.61 (CH2, C-5), 61.88 (CH, C-3), 69.28 (CH2, C-6), 116.46 (CH, 2 × o-Ph-C), 121.28 (CH, p-Ph-C), 126.73/127.06 (2 × CH, C-6 and C-7), 129.19 (CH, 2 × m-Ph-C), 144.70 (Cq, i-Ph-C), 163.99 (Cq, C-2), 179.12/179.19 (2 × Cq, C-1′ and C-3′) ppm. IR (neat): ṽ 3063, 3042, 2971, 2945, 2910, 2853, 1735, 1707, 1600, 1496, 1347, 1274, 1234, 1194, 1165, 1087, 1029, 995, 981, 894, 753, 684 cm−1. Elemental analysis: Calcd for C18H18N2O4·½H2O: C, 64.47; H, 5.71; N, 8.36. Found C, 64.88; H, 5.67; N, 8.49.
- 3-(6-Nitro-1,3-dioxo-indolin-2-yl)-4-phenylmorpholin-2-one (7c): Prepared from N-phenyl morpholine-2-one and 4-nitrophthalimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with ethyl acetate/hexane (1:3) as eluent, followed by an elution with dichloromethane/ethyl acetate (8:1). Obtained as pale orange crystals, 12.5 mg, 27% yield, m.p. 192–193 °C. 1H NMR (CDCl3): δ 3.768 (d, 1 H, J 13.4 Hz, C(5)HH), 4.213 (t, 1 H, 11.8 Hz, C(5)HH), 4.65–4.80 (m, 2 H, 6-H), 6.549 (s, H-3), 6.923 (t, 1 H, J 7.1 Hz, p-Ph-H), 6.994 (d, 2 H, 2 × o-Ph-H), 7.23–7.34 (m, 2 H, 2 × m-Ph-H), 8.028 (d, 1 H, J 8.0 Hz, H-8′), 8.597 (d, 1 H, J 8.1 Hz, H-7′), 8.635 (s, 1 H, H-5′) ppm. 13C NMR (CDCl3): δ 42.550 (CH2, C-5), 62.272 (CH, C-3), 69.444 (CH2, C-6), 115.93 (CH, 2 × o-Ph-C), 119.20 (CH, C-5′), 121.58 (CH, p-Ph-C), 125.06 (CH, C-7′), 129.54/129.58 (CH, 2 × m-Ph-C), 132.92 (Cq, C-9′), 135.84 (Cq, C-4′), 144.54 (Cq, i-Ph-C), 151.89 (Cq, C-6′), 163.92 (Cq, C(O)-2), 165.10/165.38 (2 × Cq, C(O)-1′ and C(O)-3′) ppm. IR (neat): ṽ 3100, 2900, 1776, 1722, 1600, 1500, 1338, 1250, 1225, 1200, 1100, 1056, 1013, 962, 932, 856, 744, 716, 687, 641, 524, 500 cm−1. Elemental analysis: Calcd for C18H13N3O6·¼H2O: C, 58.15; H, 3.65; N, 11.30. Found C, 58.01; H, 3.57; N, 11.00.
- 3-(2,5-Dioxopyrrol-1-yl)-4-phenylmorpholin-2-one (7d): Prepared from N-phenyl morpholine-2-one and maleimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with ethyl acetate/hexane (1:2) as eluent, followed by an elution with dichloromethane/ethyl acetate (8:1). Obtained as pale yellow crystals, 15.6 mg, 36% yield, m.p. 121 °C. 1H NMR (CDCl3): δ 3.697 (td, 1 H, J 2.6, 13.4 Hz, C(5)HH), 4.104 (overlapp ddd, 1 H, J 3.4, 7.44, 13.7 Hz, C(5)HH), 4.59–4.73 (m, 2 H, 2 × H-6), 6.299 (s, 1 H, H-3), 6.605 (s, 2 H, H-3′ and H-4′), 6.92–6.99 (m, 3 H, o-Ph-H and p-Ph-H), 7.294 (t, 2 H, J 7.8 Hz, 2 × m-Ph-H) ppm. 13C NMR (CDCl3): δ 12.159 (CH2, C-5), 61.651 (CH, C-3), 69.272 (CH2, C-6), 115.92 (2 × CH, o-Ph-C), 121.35 (CH, p-Ph-C), 129.41 (2 × CH, C-3′ and C-4′), 134.41 (2 × CH, m-Ph-C), 144.71 (CH, i-Ph-C), 164.22 (Cq, C-2), 169.71 (2 × Cq, C-2′ and C-5′) ppm. IR (neat): ṽ 3102, 2968, 2909, 2867, 1738, 1704, 1603, 1505, 1462, 1372, 1361, 1341, 1272, 1210, 1145, 1082, 1033, 983, 851, 828, 796, 750, 693, 649, 439 cm−1. Elemental analysis: Calcd for C14H12N2O4·½H2O: C, 59.78; H, 4.66; N, 9.96. Found C, 59.46; H, 4.26; N, 9.39. HRMS (ESI): Calcd for C14H12N2NaO4: 295.0689. Found: 295.08687. Calcd for C14H12KN2O4: 331.0430. Found: 311.0429.
- 3-(2,5-Dioxopyrrolidin-1-yl)-4-phenylmorpholin-2-one (7e): Prepared from N-phenyl morpholine-2-one and succinimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate (8:1) as eluent. Obtained as buff-colored crystals, 30.6 mg, 83% yield, m.p. 157 °C. 1H NMR (CDCl3): δ 2.663 (s, 4 H, 2 × H-3′ and 2 × H-4′), 3.700 (d, 1 H, J 13.4 Hz, C(5)HH), 4.15 (t, 1 H, J 13.5 Hz, C(5)HH), 4.55–4.71 (m, 2 H, 2 × H-6), 6.340 (s, 1 H, H-3), 6.89–6.98 (m, 3 H, 2 × o-Ph-H + p-Ph-H), 7.290 (t, 1 H, J 7.7 Hz, 2 × m-Ph-H) ppm. 13C NMR (CDCl3): δ 27.98 (2 × CH2, C-3′ and C-4′), 42.55 (CH2, C-5), 61.873 (CH2, C-3), 69.30 (CH2, C-6), 115.59 (2 × CH, o-Ph-C), 121.10 (CH, p-Ph-C), 129.43 (2 × CH, m-Ph-C), 144.64 (CH, i-Ph-C), 164.03 (Cq, C-2), 176.19 (2 × Cq, C-2′ and C-5′) ppm. IR (neat): ṽ 2988, 2937, 2906, 2865, 1741, 1706, 1597, 1496, 1375, 1269, 1208, 1177, 1084, 984, 753, 698 cm−1. Elemental analysis: Calcd for C14H14N2O4·¼H2O: C, 60.31; H, 5.24; N, 10.04. Found C, 60.26; H, 5.22; N, 9.64.
- 3-(3-Ethyl-3-methyl-2,5-dioxopyrrolidin-1-yl)-4-phenylmorpholin-2-one (7f): Prepared N-morpholine-2-one and (R)-ethosuximide according to the general procedure in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate (8:1) as eluent to afford a 1:1 mixture of two inseparable diastereoisomers. Obtained as buff-colored crystals, 29.1 mg, 71% yield, m.p. 78 °C. 1H NMR (CDCl3): δ (mixture of two diastereoisomers) 0.629 and 0.682 (t, 3 H, J 7.4 Hz, CH3-8′), 1.121 (s, 3 H, CH3-6′), 1.36–1.49 (m, 1 H, CHH-7′), 1.52–1.66 (m, 1 H, CHH-7′), 2.319 (2 × overlapp. d, 1 H, CHH-4′), 2.542 (2 × overlapp. d, 1 H, CHH-4′), 3.56–3.66 (m, 1 H, CHH-5), 4.160 (appar. t, 1 H, CHH-5), 4.57–4.71 (m, 2 H, H-6), 6.334 and 6.348 (overlapp. s, 1 H, H-3), 6.87–6.97 (m, 3 H, 2 × o-Ph-H and p-Ph-H), 7.22–7.33 (m, 2 × m-Ph-H) ppm. 13C NMR (CDCl3): δ (mixture of two diastereoisomers) 8.142 and 8.244 (CH3, C-8′), 23.638 and 23.844 (CH3, C-6′), 30.483 and 30.720 (CH2, C-7′), 39.985 and 40.045 (CH2, C-4′), 42.672 and 42.713 (CH2, C-5), 44.166 and 44.336 (Cq, C-3), 61.762 and 61.939 (CH, C-3), 69.234 and 69.293 (CH2, C-6), 116.11 and 116.54 (CH, 2 × o-Ph-C), 121.26 and 121.45 (CH, p-Ph-C), 129.23 (CH, 2 × m-Ph-C), 144.72 and 144.79 (Cq, i-Ph-C), 164.06 (Cq, C(O)-2), 175.12 and 175.22 (Cq, C(O)-2′ and C(O)-5′) ppm. IR (neat): ṽ 3012, 2940, 2900, 2858, 1760, 1712, 1600, 1500, 1383, 1267, 1208, 1146, 1092, 983, 742, 692, 667 cm−1. Elemental analysis: Calcd for C17H20N2O4·½H2O: C, 62.76; H, 6.50; N, 8.61. Found C, 62.84; H, 6.70; N, 8.48.
- 3-(2,6-Dioxopiperidin-1-yl)-4-phenylmorpholin-2-one (7h): Prepared from N-phenyl morpholine-2-one and glutarimide according to the general procedure, but omitting the addition of acetic acid, in a reaction performed at 80 °C. Purified by plate chromatography (silica gel) with hexane/ethyl acetate (2:1) as eluent. Obtained as buff-colored crystals, 16.0 mg, 36% yield, m.p. 165 °C. 1H NMR (CDCl3): δ 1.723 (br s, 2 H, 2 × H-4′), 2.574 (t, J 11.1 Hz, 4 H, 2 × H-3′ and 2 × H-5′), 3.568 (dd, 1 H, J 1.8, 12.7 Hz, C(5)HH), 4.104 (m, 1 H, C(5)HH), 4.50–4.69 (m, 2 H, 2 × H-6), 6.866 (d, 2 H, J 7.5 Hz, 2 × o-Ph-H), 6.923 (t, 1 H, J 7.3 Hz, p-Ph-H), 6.905 (s, 1 H, H-3), 7.261 (t, 2 H, J 7.3. Hz, m-Ph-H) ppm. 13C NMR (CDCl3): δ 16.416 (CH2, C-4′), 32.456 (2 × CH2, C-3′ and C-5′), 43.367 (CH2, C-5), 61.424 (CH2, C-3), 68.907 (CH2, C-6), 116.48 (CH, 2 × o-Ph-C), 120.96 (CH, p-Ph-C), 129.14 (CH, 2 × m-Ph-C), 145.16 (Cq, i-Ph-C), 165.25 (Cq, C(O)-2), 172.23 (Cq, 2 × C(O), C-2′ and C-6′) ppm. IR (neat): ṽ 2967, 2932, 2906, 2862, 1749, 1722, 1671, 1595, 1494, 1368, 1344,1310, 1271, 1247, 1213, 1171, 1134, 1086, 1012, 988, 750, 701, 438 cm−1. Elemental analysis: Calcd for C15H16N2O4·¼H2O: C, 61.68; H, 5.84; N, 9.25. Found C, 61.30; H, 5.76; N, 9.74.
- 3-(1,3-Dioxoindolin-2-yl)-4-(p-tolyl)-morpholin-2-one (7k): Prepared from N-tolylmorpholine-2-one and phthalimide according to the general procedure in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate/hexane (8:1:3) as eluent. Obtained as buff-colored crystals, 33.4 mg, 75% yield. 1H NMR (CDCl3): δ 2.240 (s, 3 H, CH3), 3.669 (d, J 13.3 Hz, C(5)HH), 4.179 (overlapp. ddd, J 7.6, 13.5 Hz, C(5)HH), 4.61–4.79 (m, 2 × H-6), 6.457 (s, 1 H, H-3), 6.924 (d, 2 H, J 8.5 Hz, 2 × o-Ph-H), 7.071 (d, 2 H, J 8.5 Hz, 2 × m-Ph-H), 7.719 (dd, J 3.1, 5.4 Hz, C-6′ and C-7′), 7.824 (dd, J 3.1, 5.5 Hz, H-5′ and H-8′) ppm. 13C NMR (CDCl3): δ 20.402 (CH3), 42.688 (CH2, C-5), 62.113 (CH, C-3), 69.359 (CH2, C-6), 116.46 (CH, 2 × o-Ph-C), 123.76 (CH, C-5′ and C-8′), 129.90 (CH, 2 × m-Ph-C), 130.94 (Cq, p-Ph-C), 131.55 (Cq, C-4′ and C-9′), 134.37 (CH, C-6′ and C-7′), 142.62 (Cq, i-Ph-C), 164.53 (Cq, C(O)-2), 167.44 (Cq, C(O)-1′ and C(O)-3′) ppm. IR (neat): ṽ 3037, 2960, 2922, 2861, 1756, 1712, 1617, 1519, 1470, 1379, 1269, 1204, 1084, 986, 896, 808, 716, 644, 516 cm−1. Elemental analysis: Calcd for C19H16N2O4·¼H2O: C, 63.12; H, 5.24; N, 7.74. Found C, 63.33; H, 4.74; N, 7.37.
- 4-(p-Tolyl)-morpholin-2-one (5b): Prepared as 5a. Purified by column chromatography (silica gel) with ethyl acetate/hexane (1:2) as eluent. Obtained as buff-coloured crystals, 0.188 mg, 21% yield, m.p. 72 °C. 1H NMR (CDCl3): 2.313 (s, 3 H, CH3), 3.469 (m, 2 × H-5), 4.089 (s, 2 × H-3), 4.575 (m, 2 × H-6),6.76 (d, 2 H, J 7.4 Hz, 2 × o-CH ), 7.15 (d, 2 H, J 7.4 Hz, 2 × m-CH) ppm. 13C NMR (CDCl3): δ 20.355 (CH3), 44.757, (CH2, C-5), 50.868, (CH2, C-3), 67.793, (CH2, C-6), 114, 39 (CH, 2 × o-Ph-C), 129.73 (Cq, p-C), 130.03 (CH, 2 × m-Ph-C), 145.80, (Cq, i-Ph-C), 167.49 (Cq, C(O)-2) ppm. IR (neat): ṽ 3037, 2999, 2960, 2916, 2852, 1718, 1617, 1514, 1462, 1381, 1275, 1234, 1079, 978, 938, 814, 796, 520 cm−1. Elemental analysis: Calcd for C11H13NO2·¼H2O: C, 67.50; H, 6.95; N, 7.16. Found C, 67.12; H, 6.83; N, 7.12.
4. Conclusions
We have developed an environmentally friendly and safe method to couple morpholinones to imides via C–N cross-dehydrogenative coupling, which affords products in good to high yields. This method uses an earth abundant, easily accessible metal, copper, as catalyst, and it relies solely on molecular dioxygen as oxidant and terminal hydrogen acceptor, thus avoiding the use of more expensive reagents and the formation of waste by-products, since in this case only water is also produced. Air may replace oxygen, but the product yields are lower. No bases are required, avoiding the racemization of sensitive substrates. The reaction shows good functional group compatibility, and the products may be useful intermediates for the synthesis of even higher value products, such as advanced polymers like poly(β-aminoesters) or even for PROTACs, besides the potential biological activity.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13071072/s1, Copies of NMR spectra of the new compounds and of the starting materials prepared in the course of this work: 1H, 13C, DEPT, HSQC.
Author Contributions
A.M.F.P.: Conceptualization, investigation, methodology, formal analysis, writing; A.J.L.P.: Discussions, supervision, resources. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Fundação para a Ciência e a Tecnologia (FCT), Portugal, in the form of projects UIDB/00100/2020 and UIDP/00100/2020 of Centro de Química Estrutural and project LA/P/0056/2020 of the Institute of Molecular Sciences. It was also supported by the RUDN University Strategic Academic Leadership Program (recipient A.J.L.P.).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tian, T.; Li, Z.; Li, C.-J. Cross-dehydrogenative coupling: A sustainable reaction for C–C bond formations. Green Chem. 2021, 23, 6789–6862. [Google Scholar] [CrossRef]
- Faisca Phillips, A.M.; Guedes da Silva, M.d.F.C.; Pombeiro, A.J.L. New trends in enantioselective cross-dehydrogenative coupling. Catalysts 2020, 10, 529. [Google Scholar] [CrossRef]
- Huang, C.Y.; Kang, H.; Li, J.; Li, C.J. En route to intermolecular cross-dehydrogenative coupling Reactions. J. Org. Chem. 2019, 84, 12705–12721. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S. Catalytic enantioselective cross dehydrogenative coupling of sp3 C–H of heterocycles. Org. Biomol. Chem. 2019, 17, 9683–9692. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Pombeiro, A.J.L. Recent developments in transition metal-catalyzed cross-dehydrogenative coupling reactions of ethers and thioethers. ChemCatChem 2018, 10, 3354–3383. [Google Scholar] [CrossRef]
- Varun, B.V.; Dhineshkumar, J.; Bettadapur, K.R.; Siddaraju, Y.; Alagiri, K.; Prabhu, K.R. Recent advancements in dehydrogenative cross coupling reactions for C–C bond formation. Tetrahedron Lett. 2017, 58, 803–824. [Google Scholar] [CrossRef]
- Kozlowski, M.C. Oxidative coupling in complexity building transforms. Acc. Chem. Res. 2017, 50, 638–643. [Google Scholar] [CrossRef]
- Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Oxidative coupling between two hydrocarbons: An update of recent C-H functionalizations. Chem. Rev. 2015, 115, 12138–12204. [Google Scholar] [CrossRef]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Pombeiro, A.J.L. The functionalization of amino acids, peptides, and derivatives by cross-dehydrogenative coupling. In Handbook of CH-Functionalization; Maiti, D., Ed.; Wiley-VCH: Weinheim, Germany, 2022. [Google Scholar] [CrossRef]
- San Segundo, M.; Correa, A. Cross-dehydrogenative coupling reactions for the functionalization of α-amino acid derivatives and peptides. Synthesis 2018, 50, 2853–2866. [Google Scholar] [CrossRef]
- Noisier, A.F.M.; Brimble, M.A. C–H functionalization in the synthesis of amino acids and peptides. Chem. Rev. 2014, 114, 8775–8806. [Google Scholar] [CrossRef] [PubMed]
- San Segundo, M.; Guerrero, I.; Correa, A. Co-catalyzed C(sp3)–H oxidative coupling of glycine and peptide derivatives. Org. Lett. 2017, 19, 5288–5291. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; Bernardes, G.J.L. Advances in chemical protein modification. Chem. Rev. 2015, 115, 2174–2195. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.-Y.; Berti, F.; Adamo, R. Towards the next generation of biomedicines by site-selective conjugation. Chem. Soc. Rev. 2016, 45, 1691–1719. [Google Scholar] [CrossRef]
- Qvit, N.; Rubin, S.J.S.; Urban, T.J.; Mochly-Rosen, D.; Gross, E.C.R. Peptidomimetic therapeutics: Scientific approaches and opportunities. Drug Discov. Today 2017, 22, 454–462. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T. Unusual amino acids in medicinal chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef]
- Elander, R.P. Industrial production of β-lactam antibiotics. Appl. Microbiol. Biotechnol. 2003, 61, 385–392. [Google Scholar] [CrossRef]
- Ballard, A.; Narduolo, S.; Ahmad, H.O.; Keymer, N.I.; Asaad, N.; Cosgrove, D.A.; Buurma, N.J.; Leach, A.G. Racemisation in Chemistry and Biology. Chem. Eur. J. 2020, 26, 3661–3687. [Google Scholar] [CrossRef]
- Shirakawa, S.; Maruoka, K. Recent developments in asymmetric phase-transfer reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent development and application of chiral phase-transfer catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef]
- Bada, J.L. Racemization of Amino Acids. In Chemistry and Biochemistry of the Amino Acids; Barrett, G.C., Ed.; Springer: Dordrecht, Germany, 1985; pp. 399–414. [Google Scholar]
- King, T.A.; Mandrup Kandemir, J.; Walsh, S.J.; Spring, D.R. Photocatalytic methods for amino acid modification. Chem. Soc. Rev. 2021, 50, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Ohata, J.; Martin, S.C.; Ball, Z.T. Metal-Mediated Functionalization of Natural peptides and proteins: Panning for bioconjugation gold. Angew. Chem. Int. Ed. 2019, 58, 6176–6199. [Google Scholar] [CrossRef] [PubMed]
- de Gruyter, J.N.; Malins, L.R.; Baran, P.S. Residue-specific peptide modification: A chemist’s guide. Biochemistry 2017, 56, 3863–3873. [Google Scholar] [CrossRef] [PubMed]
- Faraggi, T.M.; Rouget-Virbel, C.; Rincón, J.A.; Barberis, M.; Mateos, C.; García-Cerrada, S.; Agejas, J.; de Frutos, O.; MacMillan, D.W.C. Synthesis of enantiopure unnatural amino acids by metallaphotoredox catalysis. Org. Process Res. Dev. 2021, 25, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Mani, V.S.; Narayanasamy, R. Conformationally constrained amino acids in peptide design. SSRN 2016. [Google Scholar] [CrossRef]
- Perez, J.J. Designing peptidomimetics. Curr. Top. Med. Chem. 2018, 18, 566–590. [Google Scholar] [CrossRef]
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef]
- Robertson, N.S.; Spring, D.R. Using peptidomimetics and constrained peptides as valuable tools for inhibiting protein–protein interactions. Molecules 2018, 23, 959. [Google Scholar] [CrossRef]
- Morrison, C. Constrained peptides’ time to shine? Nat. Rev. Drug Discov. 2018, 17, 531–533. [Google Scholar] [CrossRef]
- Chowdhury, R. Eosin-Y/Cu(OAc)2-catalyzed aerobic oxidative coupling reactions of glycine esters in the dark. Org. Biomol. Chem. 2022, 20, 5387–5392. [Google Scholar] [CrossRef]
- Daggupati, R.V.; Malapaka, C. Cu(i)-Catalyzed amidation/imidation of N-arylglycine ester derivatives via C–N coupling under mild conditions. Org. Chem. Front. 2018, 5, 788–792. [Google Scholar] [CrossRef]
- Sun, B.; Wang, Y.; Li, D.; Jin, C.; Su, W. A copper/O2-mediated direct sp3 C–H/N–H cross-dehydrogen coupling reaction of acylated amines and N-aryl glycine esters. Org. Biomol. Chem. 2018, 16, 2902–2909. [Google Scholar] [CrossRef]
- Xiao, L.-J.; Zhu, Z.-Q.; Guo, D.; Xie, Z.-B.; Lu, Y.; Le, Z.-G. Copper-catalyzed cross-dehydrogenative-coupling reaction of N-arylglycine esters with imides or amides for synthesis of α-substituted α-amino acid esters. Synlett 2018, 29, 1659–1663. [Google Scholar] [CrossRef]
- Huo, C.; Dong, J.; Su, Y.; Tang, J.; Chen, F. Iron-catalyzed oxidative sp3 carbon-hydrogen bond functionalization of 3,4-dihydro-1,4-benzoxazin-2-ones. Chem. Commun. 2016, 52, 13341–13344. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, A.; Moazami, M.; Abaee, M.S.; Mirzaei, M. Ionic liquid-catalyzed synthesis of (1,4-benzoxazin-3-yl) malonate derivatives via cross-dehydrogenative-coupling reactions. Heterocycl. Commun. 2022, 28, 51–57. [Google Scholar] [CrossRef]
- Ashwood, M.S.; Cottrell, I.F.; Davies, A.J. Stereoselective synthesis of 2-(S)-(3,5-bis(trifluoromethyl)benzyloxy)-3-(S)-phenyl-1,4-oxazine. Tetrahedron Asymmetry 1997, 8, 957–963. [Google Scholar] [CrossRef]
- Trstenjak, U.; Ilaš, J.; Kikelj, D. Advances in the synthesis of morpholin-3-ones and morpholin-2-ones. Synthesis 2012, 44, 3551–3578. [Google Scholar] [CrossRef]
- Nelson, T.D. Synthesis of aprepitant. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Academic Press: Amsterdam, The Netherlands, 2005; Volume 6, pp. 321–351. [Google Scholar]
- Ku, I.W.; Cho, S.; Doddareddy, M.R.; Jang, M.S.; Keum, G.; Lee, J.H.; Chung, B.Y.; Kim, Y.; Rhim, H.; Kang, S.B. Morpholin-2-one derivatives as novel selective T-type Ca2+ channel blockers. Bioorg. Med. Chem. Lett. 2006, 16, 5244–5248. [Google Scholar] [CrossRef]
- Gonzalez, A.Z.; Eksterowicz, J.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Fox, B.M.; Fu, J.; et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J. Med. Chem. 2014, 57, 2472–2488. [Google Scholar] [CrossRef]
- Mock, J.N.; Taliaferro, J.P.; Lu, X.; Patel, S.K.; Cummings, B.S.; Long, T.E. Haloenol pyranones and morpholinones as antineoplastic agents of prostate cancer. Bioorg. Med. Chem. Lett. 2012, 22, 4854–4858. [Google Scholar] [CrossRef]
- Bardiot, D.; Thevissen, K.; De Brucker, K.; Peeters, A.; Cos, P.; Taborda, C.P.; McNaughton, M.; Maes, L.; Chaltin, P.; Cammue, B.P.; et al. 2-(2-oxo-morpholin-3-yl)-acetamide derivatives as broad-spectrum antifungal agents. J. Med. Chem. 2015, 58, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Blake, T.R.; Waymouth, R.M. Organocatalytic ring-opening polymerization of morpholinones: New strategies to functionalized polyesters. J. Am. Chem. Soc. 2014, 136, 9252–9255. [Google Scholar] [CrossRef] [PubMed]
- Kashima, C.; Harada, K. Nucleophilic ring-opening reactions of morpholin-2-ones. A resolution of dl-(secondary-alkyl)amines. J. Org. Chem. 1989, 54, 789–792. [Google Scholar] [CrossRef]
- Basu, A.; Kunduru, K.R.; Katzhendler, J.; Domb, A.J. Poly(α-hydroxy acid)s and poly(α-hydroxy acid-co-α-amino acid)s derived from amino acid. Adv. Drug Deliv. Rev. 2016, 107, 82–96. [Google Scholar] [CrossRef]
- Cordeiro, R.A.; Serra, A.; Coelho, J.F.J.; Faneca, H. Poly(β-amino ester)-based gene delivery systems: From discovery to therapeutic applications. J. Control. Release 2019, 310, 155–187. [Google Scholar] [CrossRef]
- Iqbal, S.; Qu, Y.; Dong, Z.; Zhao, J.; Khan, A.R.; Rehman, S.; Zhao, Z. Poly(β-amino esters) based potential drug delivery and targeting polymer; an overview and perspectives (review). Eur. Polym. J. 2020, 141, 110097. [Google Scholar] [CrossRef]
- Zheng, Y.-N.; Zheng, H.; Li, T.; Wei, W.-T. Recent advances in copper-catalyzed C–N bond formation involving N-centered radicals. ChemSusChem 2021, 14, 5340–5358. [Google Scholar] [CrossRef]
- Park, Y.; Kim, Y.; Chang, S. Transition metal-catalyzed C–H amination: Scope, mechanism, and applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef]
- Bariwal, J.; Van der Eycken, E. C–N bond forming cross-coupling reactions: An overview. Chem. Soc. Rev. 2013, 42, 9283–9303. [Google Scholar] [CrossRef]
- Lamberth, C. Synthesis and applications of cyclic imides in agrochemistry. In Imides: Medicinal, Agricultural, Synthetic Applications and Natural Products Chemistry; Luzzio, F.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 335–352. [Google Scholar] [CrossRef]
- Li, J.J. Imide-containing synthetic drugs. In Imides: Medicinal, Agricultural, Synthetic Applications and Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 353–366. [Google Scholar] [CrossRef]
- Luzzio, F.A. Thalidomide and analogues. In Imides: Medicinal, Agricultural, Synthetic Applications and Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 367–429. [Google Scholar] [CrossRef]
- Zerilli, T.; Ocheretyaner, E. Apremilast (Otezla): A new oral treatment for adults with psoriasis and psoriatic arthritis. Pharm. Ther. 2015, 40, 495–500. [Google Scholar]
- In Top 200 Pharmaceuticals by Retail Sales in 2021, compiled and produced by M. H. Qureshi from the Njarðarson group (University of Arizona) as described by McGrath, N.A.; Brichacek, M.; Njardarson, J.T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Ed. 2010, 87, 1348–1349. Available online: https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top%20200%20Pharmaceuticals%202021V2.pdf (accessed on 8 June 2023). [CrossRef]
- In Top 200 Small Molecule Pharmaceuticals by Retail Sales in 2021, compiled and produced by M. H. Qureshi from the Njarðarson group (University of Arizona) as described by McGrath, N.A.; Brichacek, M.; Njardarson, J.T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Ed. 2010, 87, 1348–1349. Available online: https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top%20200%20Small%20Molecules%202021V3.pdf (accessed on 8 June 2023). [CrossRef]
- Mullard, A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Ishoey, M.; Chorn, S.; Singh, N.; Jaeger, M.G.; Brand, M.; Paulk, J.; Bauer, S.; Erb, M.A.; Parapatics, K.; Müller, A.C.; et al. Translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem. Biol. 2018, 13, 553–560. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Breugst, M.; Tokuyasu, T.; Mayr, H. Nucleophilic reactivities of imide and amide anions. J. Org. Chem. 2010, 75, 5250–5258. [Google Scholar] [CrossRef]
- Rohlmann, R.; Stopka, T.; Richter, H.; García Mancheño, O. Iron-catalyzed oxidative tandem reactions with TEMPO oxoammonium salts: Synthesis of dihydroquinazolines and quinolines. J. Org. Chem. 2013, 78, 6050–6064. [Google Scholar] [CrossRef]
- Johnston, A.D.; Asmussen, E.; Bowen, R.L. Substitutes for N-phenylglycine in adhesive bonding to dentin. J. Dent. Res. 1989, 68, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Behbehani, H.; Ibrahim, H.M. Synthetic strategy for pyrazolo[1,5-a]pyridine and pyrido[1,2-b]indazole derivatives through AcOH and O2-promoted cross-dehydrogenative coupling reactions between 1,3-dicarbonyl compounds and N-amino-2-iminopyridines. ACS Omega 2019, 4, 15289–15303. [Google Scholar] [CrossRef] [PubMed]
- Forkosh, H.; Vershinin, V.; Reiss, H.; Pappo, D. Stereoselective synthesis of optically pure 2-amino-2′-hydroxy-1,1′-binaphthyls. Org. Lett. 2018, 20, 2459–2463. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Yoshida, K.; Tokuyama, H. Acetic acid promoted metal-free aerobic carbon-carbon bond forming reactions at α-position of tertiary amines. Org. Lett. 2014, 16, 4194–4197. [Google Scholar] [CrossRef]
- Froehr, T.; Sindlinger, C.P.; Kloeckner, U.; Finkbeiner, P.; Nachtsheim, B.J. A metal-free amination of benzoxazoles–The first example of an iodide-catalyzed oxidative amination of heteroarenes. Org. Lett. 2011, 13, 3754–3757. [Google Scholar] [CrossRef]
- Ma, S.; Wu, L.; Liu, M.; Xu, X.; Huang, Y.; Wang, Y. Highly enantioselective aza-Michael addition reactions of 4-nitrophthalimide with α,β-unsaturated ketones. RSC Adv. 2013, 3, 11498–11501. [Google Scholar] [CrossRef]
- Narute, S.; Pappo, D. Iron phosphate catalyzed asymmetric cross-dehydrogenative coupling of 2-naphthols with β-ketoesters. Org. Lett. 2017, 19, 2917–2920. [Google Scholar] [CrossRef]
- Lee, A.; Betori, R.C.; Crane, E.A.; Scheidt, K.A. An enantioselective cross-dehydrogenative coupling catalysis approach to substituted tetrahydropyrans. J. Am. Chem. Soc. 2018, 140, 6212–6216. [Google Scholar] [CrossRef]
- World Health Organization. World Health Organization Model List of Essential Medicines: 22nd List 2019; World Health Organization: Geneva, Switzerland, 2019.
- Funes-Ardoiz, I.; Maseras, F. Oxidative coupling mechanisms: Current state of understanding. ACS Catal. 2018, 8, 1161–1172. [Google Scholar] [CrossRef]
- Allen, S.E.; Walvoord, R.R.; Padilla-Salinas, R.; Kozlowski, M.C. Aerobic copper-catalyzed organic reactions. Chem. Rev. 2013, 113, 6234–6458. [Google Scholar] [CrossRef]
- Wendlandt, A.E.; Suess, A.M.; Stahl, S.S. Copper-catalyzed aerobic oxidative C–H functionalizations: Trends and mechanistic insights. Angew. Chem. Int. Ed. 2011, 50, 11062–11087. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.-J.; Song, L.-J.; Yang, Y.-F.; Zhang, X.; Wiest, O.; Wu, Y.-D. Computational studies on the mechanism of the copper-catalyzed sp3-C–H cross-dehydrogenative coupling reaction. ChemPlusChem 2013, 78, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Young, J.G.; Onyebuagu, W. Synthesis and characterization of di-disubstituted phthalocyanines. J. Org. Chem. 1990, 55, 2155–2159. [Google Scholar] [CrossRef]
- Howell, B.A.; Dangalle, H.; Al-Omari, M. Thermal characteristics of precursors to a difunctional imide monomer. J. Therm. Anal. Calorim. 2011, 106, 129–137. [Google Scholar] [CrossRef]
Figure 1.
CDC reactions related to this work: (a) Cu(I)-catalyzed imidation of N-arylglycine ester derivatives via C–N coupling; (b) Oxidative sp3 C-H bond functionalization of 3,4-dihydro-1,4-benzoxazin-2-ones with indoles; (c) Synthesis of (1,4-benzoxazin-3-yl)malonate derivatives via CDC reactions; (d) The new work plan proposed.
Figure 1.
CDC reactions related to this work: (a) Cu(I)-catalyzed imidation of N-arylglycine ester derivatives via C–N coupling; (b) Oxidative sp3 C-H bond functionalization of 3,4-dihydro-1,4-benzoxazin-2-ones with indoles; (c) Synthesis of (1,4-benzoxazin-3-yl)malonate derivatives via CDC reactions; (d) The new work plan proposed.
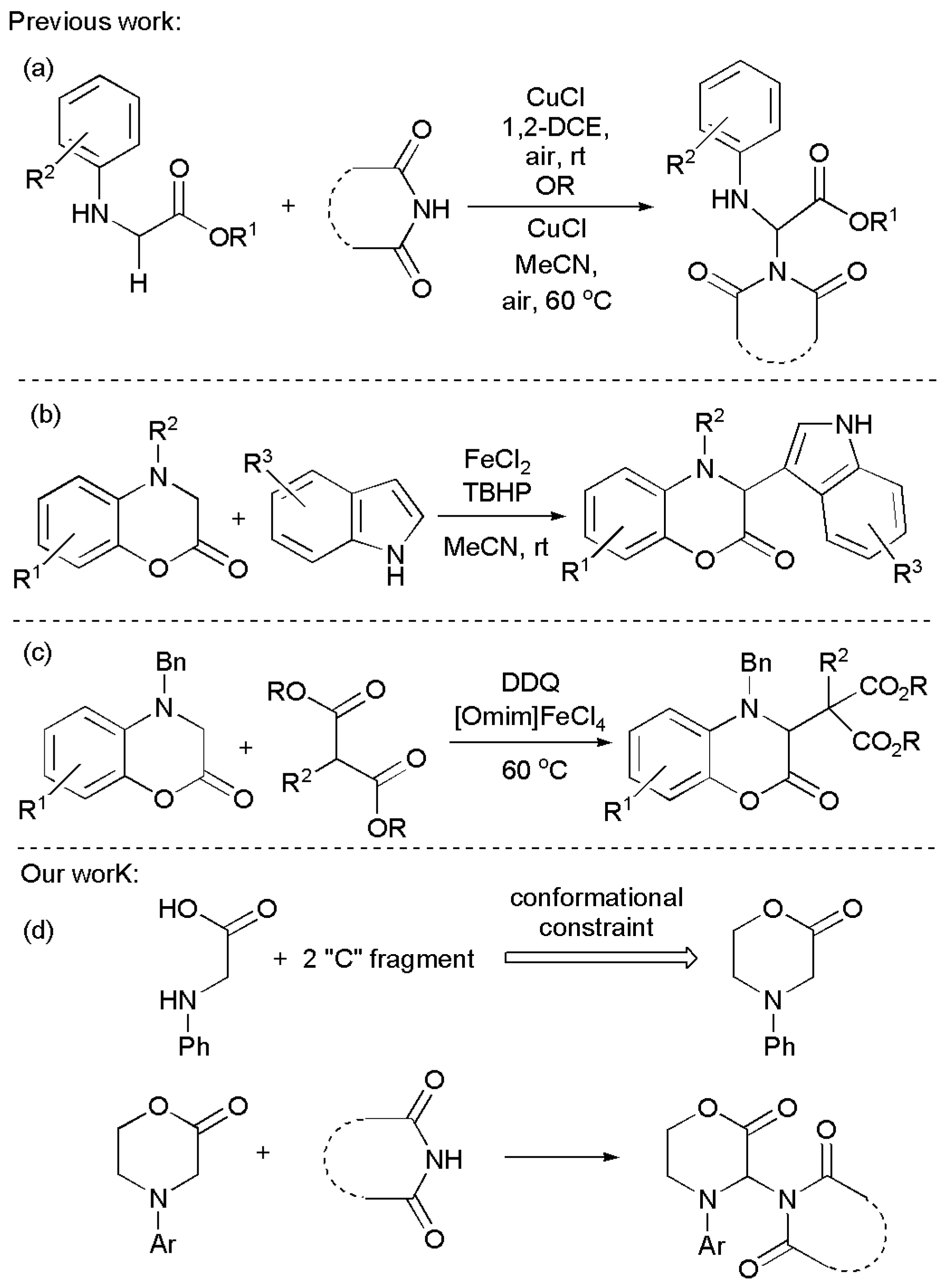
Figure 2.
Examples of biologically active morpholinones, imides and derivatives.
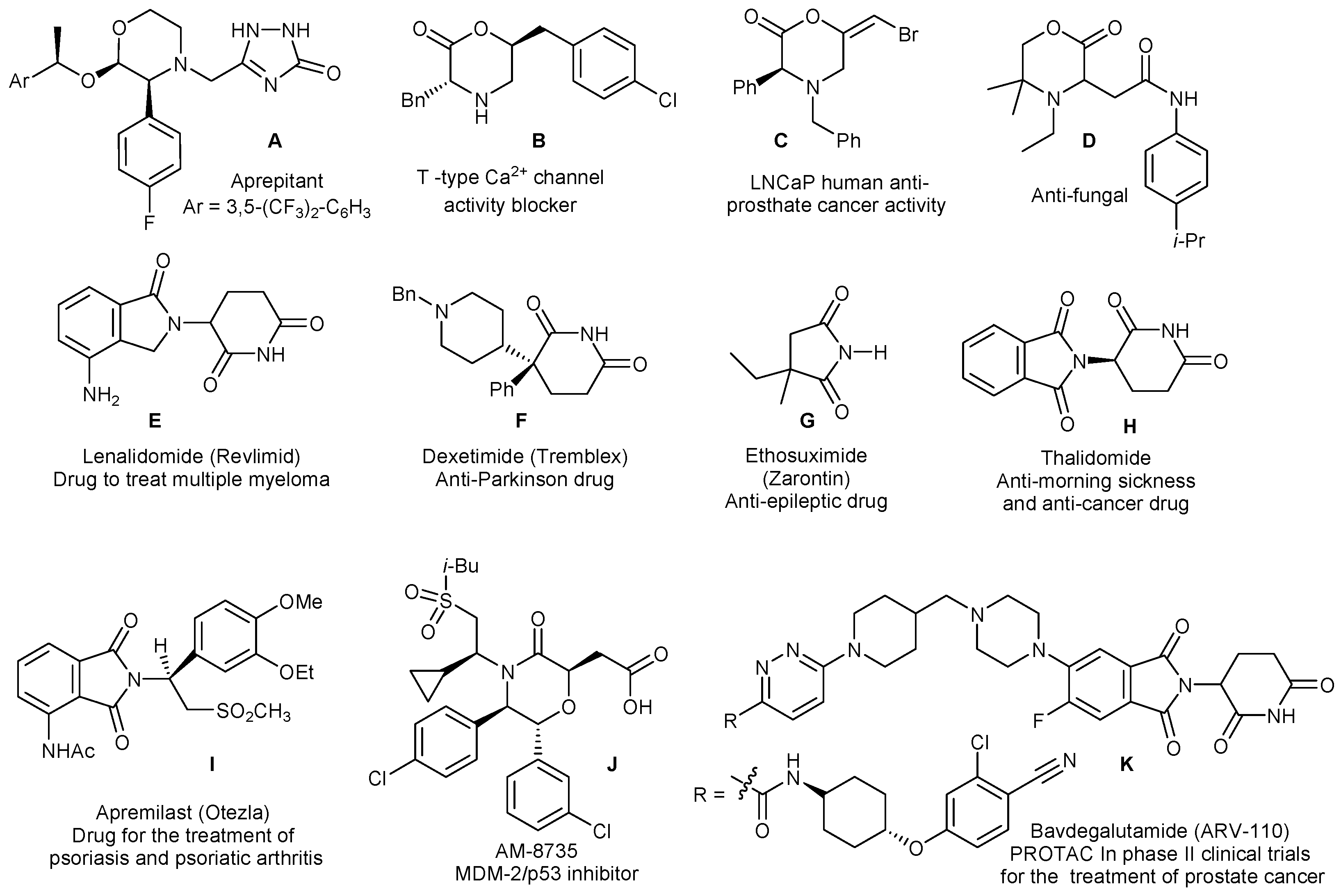
Figure 3.
Synthetic route towards the 2-morpholinones. For the preparation of compound 5a, commercially available 3a was used.
Figure 3.
Synthetic route towards the 2-morpholinones. For the preparation of compound 5a, commercially available 3a was used.

Figure 4.
(a) The mechanism proposed for the copper(I)-AcOH-catalyzed reaction and (b) for the metal-free reaction. [Cu] represents CuCl.
Figure 4.
(a) The mechanism proposed for the copper(I)-AcOH-catalyzed reaction and (b) for the metal-free reaction. [Cu] represents CuCl.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of the reaction conditions 1.

| Ent. | Catalyst (mol%) | Solvent | Oxidant | [Phth] (M) | Additive | Yield 2 (%) |
|---|---|---|---|---|---|---|
| 1 | Cu(I)Cl | DCE | O2 | 0.1 | None | 23 |
| 2 | Cu(I)Cl | DMSO | air | 0.1 | None | Traces |
| 3 | Cu(I)Cl (10) | MeCN | air | 0.2 | None | 37 |
| 4 | Cu(I)Cl (10) | MeCN | O2 | 0.2 | None | 54 |
| 5 3 | Cu(I)Cl (10) | MeCN | O2 | 0.2 | None | 61 |
| 6 | Cu(I)Cl | MeCN | O2 | 0.1 | None | 70 |
| 7 4 | Cu(I)Cl (10) | MeCN | O2 | 0.1 | None | 70 |
| 8 | Cu(I)Cl | MeCN | O2 | 0.3 | None | 60 |
| 9 | Cu(I)Cl (30) | MeCN | O2 | 0.1 | None | 65 |
| 10 | Cu(II)Cl2 | MeCN | O2 | 0.1 | None | Traces |
| 11 | Cu(I)Br | MeCN | O2 | 0.1 | None | 49 |
| 12 | Fe(II)Cl2 | MeCN | O2 | 0.1 | None | 3 |
| 13 | Cu(II)(OAc)2 | MeCN | O2 | 0.1 | None | 42 |
| 14 5 | Cu(I)Cl (10) | MeCN | DTBP (2 equiv)/N2 | 0.1 | None | 40 |
| 15 5 | Cu(I)Cl (10) | MeCN | DTBP (3 equiv)/N2 | 0.1 | None | 55 |
| 16 | Cu(I)Cl | MeCN | DTBP (1 equiv)/N2 | 0.1 | None | 80 |
| 17 | Cu(I)Cl | MeCN | O2 | 0.1 | Et3N (1.0 equiv) | 0 |
| 18 | Cu(I)Cl | MeCN | O2 | 0.1 | Mol. sieves | Traces |
| 19 | Cu(I)Cl | MeCN | O2 | 0.1 | Pyridine (1.0 equiv) | 31 |
| 20 | Cu(I)Cl | MeCN | O2 | 0.1 | AcOH (1.0 equiv) | 87 |
| 21 | Cu(I)Cl | MeCN | O2 | 0.1 | AcOH (1.5 equiv) | 93 |
| 22 | None | MeCN | air | 0.1 | AcOH (1.5 equiv) | 80 |
| 23 | None | MeCN | N2 | 0.1 | AcOH (1.5 equiv) | 53 |
| 24 | none | MeCN | air | 0.1 | None | 8 |
1 Conditions: morpholinone:imide:catalyst 2:1:0.15; [imide] = 0.1 M; AcOH: 1.5 equiv; reaction time 28 h. Reactions at 60 °C unless otherwise indicated. Add. = additive. 2 Determined by 1H NMR analysis with 4-phenylcyclohexanone as internal standard in relation to the total amount of imide reacted. 3 Morpholinone:imide 2.5:1; 80 °C. 4 Reaction time = 41 h. 5 [imide] = 0.15 M.
Table 2.
Scope of the CDC reaction between morpholin-2-ones and imides under the optimized reaction conditions 1.
Table 2.
Scope of the CDC reaction between morpholin-2-ones and imides under the optimized reaction conditions 1.
| Entry | Morpholinone | Imide | Product (M) | Yield (%) | |
|---|---|---|---|---|---|
| 1 | 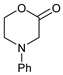 | 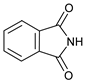 | 7a | 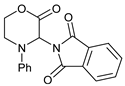 | 68 (93) |
| 2 | | 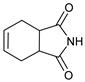 | 7b |  | 93 2 |
| 3 | | 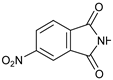 | 7c |  | 27 |
| 4 | | 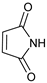 | 7d | 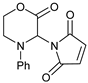 | 36 (85) |
| 5 | | 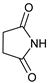 | 7e | 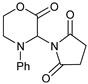 | 83 3 |
| 6 | |  | 7f | 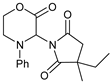 | 71 (dr = 1:1) |
| 7 | |  | 7g |  | ND |
| 8 | | 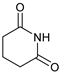 | 7h |  | 36 (75) (at 80 °C) 4 |
| 9 | | 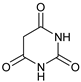 | 7i | 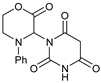 | ND |
| 10 | | 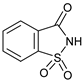 | 7j | 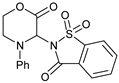 | ND |
| 11 |  | 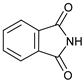 | 7k | 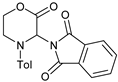 | 75 |
1 Reaction conditions: morpholinone:imide:catalyst 2:1:0.15; [imide] = 0.1 M; reaction time 28 h. The yields reported are of isolated products, after chromatographic purification. Numbers within brackets are yields determined by 1H NMR spectroscopy, relative to the internal standard 4-phenylcyclohexanone. 2 Reaction performed for 48 h; 3 Reaction time = 31 h; 4 No AcOH used; ND = Not detected.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Faisca Phillips, A.M.; Pombeiro, A.J.L. A Mild and Sustainable Procedure for the Functionalization of Morpholin-2-Ones by Oxidative Imidation Reactions. Catalysts 2023, 13, 1072. https://doi.org/10.3390/catal13071072
AMA Style
Faisca Phillips AM, Pombeiro AJL. A Mild and Sustainable Procedure for the Functionalization of Morpholin-2-Ones by Oxidative Imidation Reactions. Catalysts. 2023; 13(7):1072. https://doi.org/10.3390/catal13071072
Chicago/Turabian StyleFaisca Phillips, Ana Maria, and Armando J. L. Pombeiro. 2023. "A Mild and Sustainable Procedure for the Functionalization of Morpholin-2-Ones by Oxidative Imidation Reactions" Catalysts 13, no. 7: 1072. https://doi.org/10.3390/catal13071072
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.