
Optimizing Citrate Combustion Synthesis of A-Site-Deficient La,Mn-Based Perovskites: Application for Catalytic CH4 Combustion in Stoichiometric Conditions

Abstract
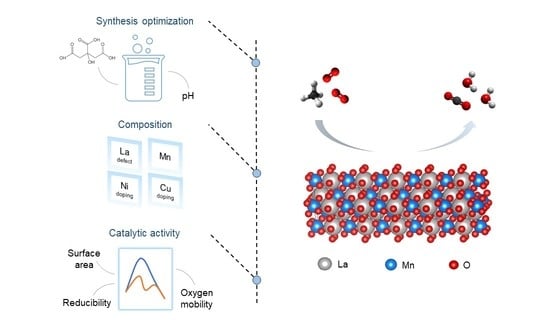
:
1. Introduction
2. Results and Discussion
2.1. Physicochemical Characterizations
2.1.1. Structural and Morphological Features
2.1.2. Precursor Decomposition
2.1.3. Bulk and Surface Composition
2.1.4. Bulk Reducibility and Oxygen Mobility
2.2. Catalytic Activity
3. Materials and Methods
3.1. Synthesis Protocol
3.2. Characterizations
3.3. Catalytic Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, J.; Li, H.; Zhong, L.; Xiao, P.; Xu, X.; Yang, X.; Zhao, Z.; Li, J. Perovskite Oxides: Preparation, Characterizations, and Applications in Heterogeneous Catalysis. ACS Catal. 2014, 4, 2917–2940. [Google Scholar] [CrossRef]
- Peña, M.A.; Fierro, J.L.G. Chemical Structures and Performance of Perovskite Oxides. Chem. Rev. 2001, 101, 1981–2017. [Google Scholar] [CrossRef] [PubMed]
- Najjar, H.; Batis, H. Development of Mn-Based Perovskite Materials: Chemical Structure and Applications. Catal. Rev. Sci. Eng. 2016, 58, 371–438. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Sun, Y.; Wang, W.; Shao, Z. Perovskite Oxides in Catalytic Combustion of Volatile Organic Compounds: Recent Advances and Future Prospects. Energ. Environ. Mater. 2022, 5, 751–776. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Y. Nanostructured Perovskite Oxides as Promising Substitutes of Noble Metals Catalysts for Catalytic Combustion of Methane. Chinese Chem. Lett. 2018, 29, 252–260. [Google Scholar] [CrossRef]
- Alifanti, M.; Kirchnerova, J.; Delmon, B.; Klvana, D. Methane and Propane Combustion over Lanthanum Transition-Metal Perovskites: Role of Oxygen Mobility. Appl. Catal. A Gen. 2004, 262, 167–176. [Google Scholar] [CrossRef]
- Spinicci, R.; Delmastro, A.; Ronchetti, S.; Tofanari, A. Catalytic Behaviour of Stoichiometric and Non-Stoichiometric LaMnO3 Perovskite towards Methane Combustion. Mater. Chem. Phys. 2002, 78, 393–399. [Google Scholar] [CrossRef]
- Royer, S.; Alamdari, H.; Duprez, D.; Kaliaguine, S. Oxygen Storage Capacity of La1−xA′xBO3 Perovskites (with A′=Sr, Ce; B=Co, Mn)—Relation with Catalytic Activity in the CH4 Oxidation Reaction. Appl. Catal. B Environ. 2005, 58, 273–288. [Google Scholar] [CrossRef]
- Chen, J.; Shen, M.; Wang, X.; Qi, G.; Wang, J.; Li, W. The Influence of Nonstoichiometry on LaMnO3 Perovskite for Catalytic NO Oxidation. Appl. Catal. B 2013, 134–135, 251–257. [Google Scholar] [CrossRef]
- Pinto, D.; Glisenti, A. Pulsed Reactivity on LaCoO3-Based Perovskites: A Comprehensive Approach to Elucidate the CO Oxidation Mechanism and the Effect of Dopants. Catal. Sci. Technol. 2019, 9, 2749–2757. [Google Scholar] [CrossRef]
- Ponce, S.; Peña, M.A.; Fierro, J.L.G. Surface Properties and Catalytic Performance in Methane Combustion of Sr-Substituted Lanthanum Manganites. Appl. Catal. B Environ. 2000, 24, 193–205. [Google Scholar] [CrossRef]
- Marcilly, C.; Courty, P.; Delmon, B. Preparation of Highly Dispersed Mixed Oxides and Oxide Solid Solutions by Pyrolysis of Amorphous Organic Precursors. J. Am. Ceram. Soc. 1970, 53, 56–57. [Google Scholar] [CrossRef]
- Taguchi, H.; Matsu-ura, S.; Nagao, M.; Choso, T.; Tabata, K. Synthesis of LaMnO3 by Firing Gels Using Citric Acid. J. Solid. State Chem. 1997, 129, 60–65. [Google Scholar] [CrossRef]
- Ghiasi, E.; Malekzadeh, A.; Ghiasi, M. Moderate Concentration of Citric Acid for the Formation of LaMnO3 and LaCoO3 Nano-Perovskites. J. Rare Earths 2013, 31, 997–1002. [Google Scholar] [CrossRef]
- Sihaib, Z.; Puleo, F.; Pantaleo, G.; La Parola, V.; Valverde, J.L.; Gil, S.; Liotta, L.F.; Fendler, A.G. The Effect of Citric Acid Concentration on the Properties of LaMnO3 as a Catalyst for Hydrocarbon Oxidation. Catalysts 2019, 9, 226. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xue, L.; Fan, L.; Yan, Y. The Effect of Citric Acid to Metal Nitrates Molar Ratio on Sol-Gel Combustion Synthesis of Nanocrystalline LaMnO3 Powders. J. Alloys Compd. 2009, 478, 493–497. [Google Scholar] [CrossRef]
- Nakamura, T.; Misono, M.; Yoneda, Y. Catalytic Properties of Perovskite-Type Mixed Oxides, La1−xSrxCoO3. Bull. Chem. Soc. Jpn. 1982, 55, 394–399. [Google Scholar] [CrossRef] [Green Version]
- Schön, A.; Dujardin, C.; Dacquin, J.-P.; Granger, P. Enhancing Catalytic Activity of Perovskite-Based Catalysts in Three-Way Catalysis by Surface Composition Optimisation. Catal. Today 2015, 258, 543–548. [Google Scholar] [CrossRef]
- Esmaeilnejad-Ahranjani, P.; Khodadadi, A.; Ziaei-Azad, H.; Mortazavi, Y. Effects of Excess Manganese in Lanthanum Manganite Perovskite on Lowering Oxidation Light-off Temperature for Automotive Exhaust Gas Pollutants. Chem. Eng. J. 2011, 169, 282–289. [Google Scholar] [CrossRef]
- Zhang, C.; Zeng, K.; Wang, C.; Liu, X.; Wu, G.; Wang, Z.; Wang, D. LaMnO3 Perovskites via a Facile Nickel Substitution Strategy for Boosting Propane Combustion Performance. Ceram. Int. 2020, 46, 6652–6662. [Google Scholar] [CrossRef]
- Esmaeilnejad-Ahranjani, P.; Khodadadi, A.A.; Mortazavi, Y. Self-Regenerative Function of Cu in LaMnCu0.1O3 Catalyst: Towards Noble Metal-Free Intelligent Perovskites for Automotive Exhaust Gas Treatment. Appl. Catal. A Gen. 2020, 602, 117702. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomie Distances in Halides and Chaleogenides. Acta Cryst. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Gholizadeh, A. X-Ray Peak Broadening Analysis in LaMnO3+δ Nano-Particles with Rhombohedral Crystal Structure. J. Adv. Mater. Process. 2015, 3, 71–83. [Google Scholar]
- Frozandeh-Mehr, E.; Malekzadeh, A.; Ghiasi, M.; Gholizadeh, A.; Mortazavi, Y.; Khodadadi, A. Effect of Partial Substitution of Lanthanum by Strontium or Bismuth on Structural Features of the Lanthanum Manganite Nanoparticles as a Catalyst for Carbon Monoxide Oxidation. Catal. Commun. 2012, 28, 32–37. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zheng, Y.; Dacquin, J.P.; Djelal, N.; Cordier, C.; Dujardin, C.; Granger, P. Impact of Dual Calcium and Manganese Substitution of La-Deficient Perovskites on Structural and Related Catalytic Properties: Future Opportunities in next Three-Way-Catalyst Generation? Appl. Catal. A Gen. 2021, 619, 118137. [Google Scholar] [CrossRef]
- Feick, G.; Hainer, R.M. On the Thermal Decomposition of Ammonium Nitrate. Steady-State Reaction Temperatures and Reaction Rate. J. Am. Chem. Soc. 1954, 76, 5860–5863. [Google Scholar] [CrossRef]
- Brusamarello, E.; Blonda, C.; Salazar-Castro, C.; Pascui, A.E.; Canu, P.; Glisenti, A. Industrially Produced Fe- And Mn-Based Perovskites: Effect of Synthesis on Reactivity in Three-Way Catalysis: Part 1. ACS Omega 2021, 6, 24325–24337. [Google Scholar] [CrossRef]
- Brusamarello, E.; Blonda, C.; Salazar-Castro, C.; Canu, P.; Glisenti, A. Industrially Produced Fe- And Mn-Based Perovskites: Effect of Synthesis on Reactivity in Three-Way Catalysis: Part 2. ACS Omega 2021, 6, 24316–24324. [Google Scholar] [CrossRef]
- Giordano, F.; Trovarelli, A.; De Leitenburg, C.; Giona, M. A Model for the Temperature-Programmed Reduction of Low and High Surface Area Ceria. J. Catal. 2000, 193, 273–282. [Google Scholar] [CrossRef]
- Wei, T.; Jia, L.; Zheng, H.; Chi, B.; Pu, J.; Li, J. LaMnO3-Based Perovskite with in-Situ Exsolved Ni Nanoparticles: A Highly Active, Performance Stable and Coking Resistant Catalyst for CO2 Dry Reforming of CH4. Appl. Catal. A Gen. 2018, 564, 199–207. [Google Scholar] [CrossRef]
- Patcas, F.; Buciuman, F.C.; Zsako, J. Oxygen Non-Stoichiometry and Reducibility of B-Site Substituted Lanthanum Manganites. Termochimica Acta 2000, 360, 71–76. [Google Scholar] [CrossRef]
- Huang, C.; Shan, W.; Lian, Z.; Zhang, Y.; He, H. Recent Advances in Three-Way Catalysts of Natural Gas Vehicles. Catal. Sci. Technol. 2020, 10, 6407–6419. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations Carried out by Means of Vanadium Oxide Catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Hammami, R.; Aïssa, S.B.; Batis, H. Effects of Thermal Treatment on Physico-Chemical and Catalytic Properties of Lanthanum Manganite LaMnO3+y. Appl. Catal. A Gen. 2009, 353, 145–153. [Google Scholar] [CrossRef]
- Wu, J.; Dacquin, J.P.; Djelal, N.; Cordier, C.; Dujardin, C.; Granger, P. Calcium and Copper Substitution in Stoichiometric and La-Deficient LaFeO3 Compositions: A Starting Point in next Generation of Three-Way-Catalysts for Gasoline Engines. Appl. Catal. B 2021, 282, 119621. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CA/M* Molar Ratio | pH of Synthesis | Ignition T (°C) | Calcination T (°C) |
|---|---|---|---|---|
| La0.8MnO3-NH3-CA1.1 | 1.1 | 7 | 350 | 750 |
| La0.8MnO3-NH3-CA1.5 | 1.5 | 7 | 350 | 750 |
| La0.8MnO3-H+-CA1.1 | 1.1 | <1 | 200–230 | 750 |
| La0.8MnO3-H+-CA1.5 | 1.5 | <1 | 200–230 | 750 |
| La0.8Mn0.9Ni0.1O3-H+-CA1.1 | 1.1 | <1 | 200–230 | 750 |
| La0.88Mn0.9Ni0.1O3-H+-CA1.5 | 1.5 | <1 | 200–230 | 750 |
| La0.88Mn0.9Cu0.1O3-H+-CA1.1 | 1.1 | <1 | 200–230 | 750 |
| La0.88Mn0.9Cu0.1O3-H+-CA1.5 | 1.5 | <1 | 200–230 | 750 |
| Sample | SSA (m2/g) 1 | Pore Vol. (cm3/g) 2 | dXRD (nm) 3 |
|---|---|---|---|
| La0.8MnO3-NH3-CA1.1 | 4.9 | 0.013 | 69.3 |
| La0.8MnO3-NH3-CA1.5 | 4.0 | 0.0070 | 81.5 |
| La0.8MnO3-H+-CA1.1 | 25.0 | 0.081 | 33.1 |
| La0.8MnO3-H+-CA1.5 | 4.2 | 0.016 | 64.8 |
| La0.8Mn0.9Ni0.1O3-H+-CA1.1 | 43.9 | 0.152 | 22.7 |
| La0.88Mn0.9Ni0.1O3-H+-CA1.5 | 7.6 | 0.039 | 46.8 |
| La0.88Mn0.9Cu0.1O3-H+-CA1.1 | 37.1 | 0.145 | 33.0 |
| La0.88Mn0.9Cu0.1O3-H+-CA1.5 | 11.7 | 0.037 | 45.1 |
| Sample | La (at%) * | Mn (at%) * | Ni or Cu (at%) * | |
|---|---|---|---|---|
| La0.8MnO3-NH3-CA1.1 | EDX | 48.1 | 51.9 | - |
| XPS | 45.0 | 55.0 | - | |
| (nominal) | (44.5) | (55.5) | - | |
| La0.8MnO3-NH3-CA1.5 | EDX | 46.2 | 53.8 | - |
| XPS | 42.9 | 57.1 | - | |
| (nominal) | (44.5) | (55.5) | - | |
| La0.8MnO3-H+-CA1.1 | EDX | 50.1 | 49.9 | - |
| XPS | 41.7 | 58.3 | - | |
| (nominal) | (44.5) | (55.5) | - | |
| La0.8MnO3-H+-CA1.5 | EDX | 46.5 | 53.5 | - |
| XPS | 41.9 | 58.1 | - | |
| (nominal) | (44.5) | (55.5) | - | |
| EDX | 48.7 | 46.3 | 5.0 | |
| La0.8Mn0.9Ni0.1O3-H+-CA1.1 | XPS | 37.9 | 60.1 | 2.0 |
| (nominal) | (44.5) | (50.0) | (5.5) | |
| EDX | 48.3 | 45.7 | 6.0 | |
| La0.8Mn0.9Ni0.1O3-H+-CA1.5 | XPS | 39.7 | 56.2 | 4.1 |
| (nominal) | (44.5) | (50.0) | (5.5) | |
| EDX | 48.1 | 48.3 | 3.6 | |
| La0.8Mn0.9Cu0.1O3-H+-CA1.1 | XPS | 37.2 | 51.5 | 11.3 |
| (nominal) | (44.5) | (50.0) | (5.5) | |
| EDX | 48.2 | 45.7 | 6.1 | |
| La0.8Mn0.9Cu0.1O3-H+-CA1.5 | XPS | 35.7 | 53.7 | 10.6 |
| (nominal) | (44.5) | (50.0) | (5.5) |
| Sample | Low Temperature Range 1 | High Temperature Range 2 | ||||
|---|---|---|---|---|---|---|
| Tmax (°C) | H2 Uptake (mmol/g) | H/Mn + Cu | Tmax (°C) | H2 Uptake (mmol/g) | H/Mn + Ni | |
| La0.8MnO3-NH3-CA1.1 | 387 | 1.18 | 0.50 | 785 | 1.66 | 0.71 |
| La0.8MnO3-NH3-CA1.5 | 406 | 1.20 | 0.51 | 827 | 1.69 | 0.72 |
| La0.8MnO3-H+-CA1.1 | 346 | 1.85 | 0.79 | 794 | 1.32 | 0.56 |
| La0.8MnO3-H+-CA1.5 | 403 | 1.16 | 0.49 | 777 | 1.83 | 0.78 |
| La0.8Mn0.9Ni0.1O3-H+-CA1.1 | 315 | 1.35 | 0.58 | 740 | 1.73 | 0.74 |
| La0.8Mn0.9Ni0.1O3-H+-CA1.5 | 378 | 1.02 | 0.44 | 785 | 1.96 | 0.84 |
| La0.8Mn0.9Cu0.1O3-H+-CA1.1 | 239 | 1.98 | 0.85 | 745 | 1.45 | 0.62 |
| La0.8Mn0.9Cu0.1O3-H+-CA1.5 | 265 | 1.59 | 0.68 | 793 | 1.68 | 0.72 |
| Sample | O2 Desorption (μmol/g) | |
|---|---|---|
| Low T Range (<500 °C) | High T Range (>500 °C) | |
| La0.8MnO3-NH3-CA1.1 | - | 86 |
| La0.8MnO3-H+-CA1.1 | 28 | 296 |
| La0.8MnO3-H+-CA1.5 | - | 149 |
| La0.8Mn0.9Ni0.1O3-H+-CA1.1 | 46 | 145 |
| La0.8Mn0.9Cu0.1O3-H+-CA1.5 | 8.3 | 208 |
| Sample | T10 (°C) | T50 (°C) | T80 (°C) | rspecific (mol/s/g) | rintrinsic (mol/s/m2) |
|---|---|---|---|---|---|
| La0.8MnO3-NH3-CA1.1 | 583 | 696 | 783 | 3.7 × 10−6 | 7.5 × 10−7 |
| La0.8MnO3-NH3-CA1.5 | 570 | 732 | >800 | 3.5 × 10−6 | 8.8 × 10−7 |
| La0.8MnO3-H+-CA1.1 | 489 | 581 | 639 | 9.9 × 10−6 | 4.0 × 10−7 |
| La0.8MnO3-H+-CA1.5 | 569 | 750 | >800 | 3.6 × 10−6 | 8.7 × 10−7 |
| La0.8Mn0.9Ni0.1O3-H+-CA1.1 | 455 | 568 | 637 | 1.0 × 10−5 | 2.3 × 10−7 |
| La0.88Mn0.9Ni0.1O3-H+-CA1.5 | 528 | 681 | 790 | 5.6 × 10−6 | 7.4 × 10−7 |
| La0.88Mn0.9Cu0.1O3-H+-CA1.1 | 455 | 580 | 644 | 1.0 × 10−5 | 2.7 × 10−7 |
| La0.88Mn0.9Cu0.1O3-H+-CA1.5 | 524 | 664 | 736 | 6.2 × 10−6 | 5.3 × 10−7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osti, A.; Rizzato, L.; Cavazzani, J.; Glisenti, A. Optimizing Citrate Combustion Synthesis of A-Site-Deficient La,Mn-Based Perovskites: Application for Catalytic CH4 Combustion in Stoichiometric Conditions. Catalysts 2023, 13, 1177. https://doi.org/10.3390/catal13081177
Osti A, Rizzato L, Cavazzani J, Glisenti A. Optimizing Citrate Combustion Synthesis of A-Site-Deficient La,Mn-Based Perovskites: Application for Catalytic CH4 Combustion in Stoichiometric Conditions. Catalysts. 2023; 13(8):1177. https://doi.org/10.3390/catal13081177
Chicago/Turabian StyleOsti, Andrea, Lorenzo Rizzato, Jonathan Cavazzani, and Antonella Glisenti. 2023. "Optimizing Citrate Combustion Synthesis of A-Site-Deficient La,Mn-Based Perovskites: Application for Catalytic CH4 Combustion in Stoichiometric Conditions" Catalysts 13, no. 8: 1177. https://doi.org/10.3390/catal13081177