1. Introduction
The demand for highly efficient and stable supported catalysts has been increasing due to the growing need to address various energy and environmental concerns. Among different forms of catalysts, those applied as coatings of structured packings such as meshes, open-cell foams, and honeycomb monoliths deserve special attention due to their versatility and potential for improving process efficiency when applied in heterogeneous reactors [
1,
2]. Catalytic structured packings can be successfully employed in various processes, such as automotive gas treatment, the combustion of volatile organic compounds, CO
2 methanation, methane/methanol steam reforming, and many others [
3]. Compared to the conventional packed beds of particle catalysts, structured catalysts offer lower pressure drop, better mass and heat transfer, and vast possibilities for tailoring their design to specific applications, providing great potential for process intensification in demanding applications, such as CO
2 valorization [
4].
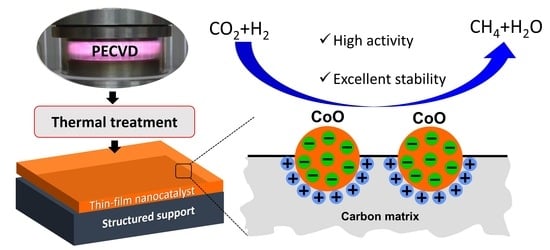
Recently, we developed an effective method for fabricating a novel and sophisticated CoO-based nanocatalyst. This catalyst is in the form of a thin film deposited on a wire-gauze support, with the aim of CO
2 hydrogenation to methane [
5]. Using the plasma-enhanced chemical vapor deposition (PECVD) technique and volatile organometallic complex precursors enabled the creation of thin-film catalytic coatings that did not alter the geometry of the fine support, which is a critical factor for future applications. Thin-film catalysts prepared in this manner differ significantly from analogous conventional catalytic systems, primarily in that they cover the entire surface of the support, thus making it inert to the ongoing catalytic process. Moreover, the material deposited from the organometallic precursor in the plasma process always contains carbon, which, for example, in the case of the above-mentioned CoO-based nanocatalysts, allows one to obtain a unique nanostructure of cobalt oxide embedded in a carbon matrix. This specific structure has demonstrated very high activity and selectivity towards CH
4 in CO
2 hydrogenation [
6]. Yet, understanding the nature of this structure and its impact on catalytic properties remains an open problem.
The active form of cobalt in conventional catalysts for CO
2 hydrogenation has been an ongoing subject of debate within the scientific community [
7]. Some researchers claim that it exists in a metallic form [
8], while others argue that it is present in an oxidized state [
9], which was also confirmed in the case of our plasma-prepared CoO-based catalyst [
6]. Moreover, there are assertions that both metallic and oxidized forms of Co coexist and are necessary for efficient catalytic performance [
10,
11]. Importantly, the interaction of Co metal or its oxide forms with other materials can significantly affect the catalytically active states. In addition, due to the interconversion of various forms under reaction conditions, identifying the exact nature of the catalytically active species is still a challenge [
12].
While cobalt-based conventional catalysts used for CO
2 methanation are mainly oxide-supported, recent studies have shown that carbon-based materials are gaining attention due to their high thermal and chemical stability, as well as the ability to enhance catalytic performance through metal–support interactions [
13]. It was reported that carbon, in the amorphous and nanostructured forms, participated in various ways to improve the activity and selectivity of the catalyst in CO
2 hydrogenation compared to the corresponding catalysts supported on traditional oxidic supports [
14]. Particularly, it could provide a surface that promotes the activation of H
2 molecules and the subsequent formation of hydrocarbons [
15,
16]. The presence of carbon also improved the dispersion and stability of Co metal nanoparticles or Co oxides, preventing the sintering and deactivation of the catalyst [
17]. It is therefore obvious that we should not forget about the possible role of the carbon matrix in our plasma-prepared catalysts.
In this work, we aimed to research the essential aspects of the structure of plasma-produced Co-based thin films, placing particular emphasis on the carbon matrix present in these films, and establishing connections with their catalytic activity in CO2 methanation. It is assumed that establishing such a relationship may have far-reaching consequences in the design of thin-film nanocatalysts for their desired properties.
2. Results
2.1. Catalytic Performance
The performance of different thin-film CoO-based catalysts, namely ad-CoO, TT- CoO, and PT-CoO, which are referred to as as-deposited (ad-); calcined at 400 °C in an argon atmosphere for 30 min (TT-); and argon plasma post-treated for 2 h (PT-), were evaluated in the CO2 methanation reaction. Additionally, solely carbon matrix thin films, as-deposited (ad-CM) and thermally treated (TT-CM) in the same way as the TT-CoO, were prepared and also tested. The comparative assessment was conducted in terms of CO2 conversion and CH4 selectivity over a temperature range of 250–400 °C.
As can be seen in
Figure 1a, the TT-CoO film is active at 250 °C with 84% selectivity toward methane, while both the ad-CoO and PT-CoO films were inactive at this temperature. As the temperature increases, the activity of all the tested catalysts appears, among which the ad-CoO and TT-CoO films achieve CH
4 selectivity of approximately 98% after reaching a temperature of 400 °C. On the other hand, the PT-CoO film apparently exhibits poorer performance. At 400 °C, both CO
2 conversion and CH
4 selectivity are lower by about 17% and 8%, respectively, compared to the best TT-CoO catalytic film.
The differences in the performance of the CoO-based films become evident when analyzing the time-on-stream catalytic test at 400 °C. According to the experimental findings depicted in
Figure 1b, the only catalyst that is stable over 24 h in the CO
2 methanation reaction is the thermally treated CoO-based thin film (TT-CoO). Both the CO
2 conversion and selectivity to CH
4 do not change over time. On the contrary, the as-deposited catalyst without any treatment (ad-CoO) demonstrates a gradual loss of activity over time. The PT-CoO film not only shows the lowest activity at 400 °C but also exhibits very poor stability. Over the course of 24 h, the CO
2 conversion achieved over this catalyst decreased from 66% to 46%, while the CH
4 selectivity decreased from 90% to 78%. The performance of the carbon matrix thin films (ad-CM, TT-CM) when subjected to the reaction mixture of CO
2 and H
2 is not shown in
Figure 1. This is because no products were detected by the GC analysis up to 350 °C, indicating no catalytic activity. However, at 400 °C, a small peak of CO appeared for ad-CM and TT-CM, suggesting some chemical processes occurring over both carbon matrix thin films at this temperature. The estimated conversion of CO
2 was approximately 1.5% for ad-CM and 2% for TT-CM, with a relative uncertainty of about 25% due to the low precision associated with the low concentration levels. During these catalytic tests, no water production was detected, which proves that this is not a CO
2 hydrogenation process.
2.2. Chemical Structure of CoO-Based Thin Films
The surface composition and molecular structure of the studied CoO-based catalysts, namely ad-CoO, TT-CoO, and PT-CoO, which exhibited different activities and stabilities in CO2 methanation, were investigated by X-ray photoelectron spectroscopy (XPS). Additionally, the spent samples of these catalysts obtained after subjecting them to a catalytic test at a temperature of 400 °C were examined to determine possible changes in the molecular structure of their surface during the CO2 methanation reaction.
As already mentioned in the Introduction, thin films prepared using the PECVD technique from organometallic precursors always contain, in addition to the metal-based phase, a carbon phase, which we call the carbon matrix. The XPS analysis of the elemental composition of the tested catalytic films revealed the presence of cobalt (Co), oxygen (O), and carbon (C) on their surface. The atomic ratios of carbon to cobalt (C/Co) and oxygen to cobalt (O/Co) are shown in
Table 1.
As can be seen, the fresh calcined films (TT-CoO) show very similar contents of both carbon and oxygen (relative to cobalt) compared to the fresh as-deposited films (ad-CoO). Meanwhile, in the case of the fresh PT-CoO films, a distinct decrease in the C/Co and O/Co values is revealed compared to the ad-CoO films. This can be considered to be the result of the argon plasma treatment inducing the preferential etching of carbon and oxygen from the film surface. After the catalytic process (spent catalysts), the carbon content (C/Co) in the ad-CoO film decreases clearly; in the TT-CoO film it remains at the same level; while in the PT-CoO film, this content increases significantly. These effects will be discussed further in
Section 3. On the other hand, no significant differences were observed in the oxygen content (O/Co) between the fresh and spent catalytic films.
To take a deeper look at the molecular structure of the studied catalytic films, the regions corresponding to Co 2p and C 1s in the XPS spectra were analyzed in detail.
Figure 2 shows the Co 2p spectra for the as-CoO, TT-CoO, and PT-CoO catalyst films before the catalytic process (fresh) and after this process (spent). These spectra consist of a 2p3/2 and 2p1/2 spin-orbital doublet, from which the more intense 2p3/2 spectrum is usually analyzed. Its numerical deconvolution allowed for the identification of three bands, which were assigned to the appropriate chemical states of cobalt [
6,
18,
19]: a narrow band (Co
0) in the range of 778.2–778.7 eV belonging to the Co–Co and Co–C bonds, a broad band in the range of 780.2–781.0 eV assigned to CoO (Co
2+), and a band in the range of 785.6–786.2 eV representing the Co
2+ shake-up satellite (sat.).
In turn,
Figure 3 shows the C 1s spectra for the above cases. As a result of the deconvolution of these spectra, we distinguished six bands which, based on the literature [
6,
19,
20,
21], were assigned in the order of increasing binding energy to cobalt carbide (Co–C) at 283.5 eV, sp
2 carbon (C=C) at 284.6 eV (used to calibrate the spectra), sp
3 carbon (C–C, C–H) at 285.2–285.4 eV, carbon single-bonded to oxygen (C–O) at 286.3–287.2 eV, carbon doubly bonded to oxygen (C=O) at 287.9–288.8 eV, and strongly oxidated carbon (C–OOR) at approx. 290 eV. The highest part of the carbon species in all the studied films is sp
2 and sp
3 carbon.
According to
Figure 2, in all the tested catalytic films, both before and after the CO
2 hydrogenation process, cobalt atoms occur in the form of CoO (Co
2+) and Co
0, which can be attributed to the metallic form (Co–Co bonds) or carbide form (Co–C bonds). The latter form occurs solely in the PT-CoO film, because only for this film the C 1s spectrum reveals the Co–C band (
Figure 3). Thus, in this case, a mixture of Co–Co and Co–C bonds may be present. However, a rough calculation of the content of the Co–C based on the C 1s and Co 2p spectra for PT-CoO (fresh) indicates that only cobalt carbide in the CoC form is present here. This unusual cobalt monocarbide form is already known, among others, as a product of the reaction of cobalt vapor and methane in a glow discharge [
22], which can be considered a situation similar to the plasma treatment we perform. A similar situation concerns the PT-CoO (spent) film, for which the Co
2+/Co
0 ratio is maintained within the error limits compared to PT-CoO (fresh) (
Table 1). However, the quantitative interpretation of the C 1s spectrum is limited here due to the sp
2 carbon deposit covering the surface of the film, which is produced during the catalytic process (
Figure 3). In all other cases, the Co
0 band should be associated with the metallic Co–Co form, the relation of which with the Co
2+ form (Co
2+/Co
0) is shown in
Table 1.
Analyzing the Co2+/Co0 ratio for the ad-CoO and TT-CoO films, it is easy to notice that the calcination of the ad-CoO films causes an approximately two-fold increase in this ratio, which should be associated with the rearrangement involving the oxidation of Co0 to CoO with the participation of oxygen initially bonded with carbon. The catalytic process causes the much more advanced oxidation of Co0 in the ad-CoO films, this time with the participation of the reaction mixture (CO2, H2O). Interestingly, however, for the TT-CoO films, no change in the Co2+/Co0 ratio is observed within the error limits, which proves the high stability of the chemical structure of these films.
The stability of the chemical structure of the TT-CoO films in the catalytic CO
2 hydrogenation process is also confirmed by the analysis of the ratio of sp
2 to sp
3 carbon content, which does not change between the fresh and spent films within the limits of error (
Table 1). On the contrary, for the ad-CoO film, there is a visible decrease in the C(sp
2)/C(sp
3) ratio and, as already mentioned, a simultaneous decrease in the carbon content on the film surface between the fresh and spent samples. This clearly confirms the instability of the chemical structure of such films during the catalytic process.
It should also be noted that the calcination process causes a significant increase in the sp
2 carbon content in relation to sp
3 carbon (ad-CoO fresh and TT-CoO fresh), which indicates the transformation of the carbon matrix into a more graphite-like structure. This was already noticed in our previous studies [
6]. In turn, plasma treatment of the ad-CoO film (PT-CoO fresh) causes a sharp increase in the sp
3 carbon content, which, apart from the previously mentioned preferential etching, is the result of the action of argon plasma on the carbon matrix [
23]. After the catalytic process, these films reveal a huge increase in the carbon content (C/Co), including sp
2 carbon (sp
2/sp
3 ratio increases), which, however, should not be associated with changes in the chemical structure of the film surface, but with the formation of the carbon deposit.
One more important result requires emphasizing, namely the comparison of the positions of the Co
2+ band maximum for different films, when all the XPS spectra were normalized to the same value (284.6 eV) for the C 1s sp
2 band. It is evident (
Figure 2) that for the TT-CoO, both the fresh and spent films, this maximum is shifted towards a lower binding energy (780.2 eV) compared to the ad-CoO and PT-CoO (fresh and spent) films, for which the Co
2+ band is approximately in the same position at 780.8–781.0 eV. Thus, the observed shift is about −0.7 eV.
2.3. Nanostructure of CoO-Based Thin Films
To better understand the reasons for the different catalytic performances exhibited by the plasma-prepared CoO-based thin films, their nanostructure was examined using X-ray diffraction (XRD). The raw XRD patterns for the ad-CoO and TT-CoO films, both fresh and spent, are shown in
Figure 4a. As one can see, the fresh ad-CoO film is completely amorphous, unlike the fresh TT-CoO film, where a clear nanocrystalline fraction formed during the calcination process is visible. This behavior of CoO-based films is known and has been analyzed in detail (in the analogous case of the calcination of these films in air and the formation of Co
3O
4 spinel nanocrystallites in them) in Ref. [
24]. After the catalytic process, the nanocrystalline structure in the TT-CoO film remains virtually unchanged, while in the spent ad-CoO film, a trace appearance of the nanocrystalline fraction in the amorphous matrix can be observed. A detailed analysis of the XRD pattern for the spent ad-CoO reveals its considerable similarity to the XRD patterns for the fresh and spent TT-CoO. As an example, the most intense band at 42.4 deg appropriately magnified for the spent ad-CoO is shown in
Figure 4b.
The numerical analysis of the XRD pattern for the TT-CoO films (
Figure 4c) clearly indicates (according to the ICDD-PDF card no. 48-1719) the most well-known cubic rock-salt structure of CoO, revealing reflections at 36.5, 42.4, 61.5, 73.8, and 77.4 deg. In this case, the size of the CoO nanocrystallites determined using the Scherrer equation is approximately 11 nm. The CoO nanocrystallite size estimated in the same way for the spent ad-CoO film based on
Figure 4b is approximately 3 nm.
It should be added that since Ar plasma treatment (PT-CoO) causes changes only in a very thin surface layer, much thinner (several nanometers) than the thickness of the film (several hundred nanometers), XRD tests (analyzing the entire mass of the film) are unjustified in this case.
2.4. Molecular and Electronic Structure of Carbon Matrix
As already presented above, the nature of plasma deposition from the cobalt organometallic precursor leads to the formation of films containing a carbon matrix with a CoO fraction. The molecular structure and nanostructure of such a system turned out to have a significant impact on the catalytic properties of these films (
Section 2.1,
Section 2.2 and
Section 2.3). To determine the role of the carbon matrix more precisely in the catalytic effect, films containing only this matrix (without Co) were investigated. These films were prepared under the same plasma process conditions as the ad-CoO films, using a mixture of cyclopentadiene and carbon monoxide in a 2:1 ratio as a precursor (the same ratio in which analogous ligands are present in the organometallic cobalt complex CpCo(CO)
2 used to deposit the ad-CoO films).
Figure 5 shows the XPS C 1s spectra for an as-deposited carbon film (ad-CM) and such a film thermally treated (TT-CM). The carbon species composition for the fresh films shows similarity with the corresponding chemical structure of the carbon matrix in CoO-based thin films (
Figure 3). A comparison of the C 1s spectra between ad-CM and TT-CM reveals that after the thermal treatment, the C(sp
2)/C(sp
3) ratio increases (
Table 2), analogously to what is observed in the case of changes between the TT-CoO and ad-CoO fresh films (
Table 1). Similarly to the ad-CoO spent film (
Table 1), after treatment with the reaction mixture of CO
2 and H
2 at 400 °C, an increase in the C(sp
3) content compared to C(sp
2) was observed in the ad-CM film, but also in the TT-CM film (
Figure 5 and
Table 2), which is not the case in the spent TT-CoO film (
Table 1).
Unfortunately, the ad-CM films treated in Ar plasma under the same conditions as the ad-CoO films, unlike them, were quickly etched and completely removed after the same treatment time of 2 h, making them impossible to study further. This result clearly indicates the role of the CoO fraction in stabilizing the carbon matrix in the plasma-treated catalytic films (PT-CoO).
To determine the electronic structure of the ad-CM and TT-CM carbon films, which constitute carbon matrices in the ad-CoO and TT-CoO catalytic films, respectively, ultraviolet photoelectron spectroscopy (UPS) and UV-VIS absorption spectroscopy studies were performed.
Figure 6a shows the measured UPS spectra for the ad-CM and TT-CM films. The interpretation of these spectra was carried out according to Ref. [
25]. On this basis, two important parameters from the point of view of the electronic structure of the films were determined. The first one is the valence band edge value (
EBE), which characterizes the energy distance of the upper edge of the valence band from the Fermi level. As can be seen from the inset in
Figure 6a, this value is virtually the same for both films and is 0.8 eV. The second parameter is the secondary electron cutoff (
SECO), the value of which was set at 16.7 eV and 16.3 eV for the TT-CM and ad-CM films, respectively. Using the determined parameters, it is possible to estimate the elements of the electronic structure, such as work function (
W) and ionization potential (
J), according to the following equations:
where
Ehν is the impinging photon energy (in our case, it is the He I emission line with an energy of 21.2 eV), and
To complete the band model of the electronic structure of the films, optical gap (
EOPT) values were determined from UV-VIS absorption measurements. For amorphous materials, which include the tested carbon films,
EOPT is most frequently calculated according to the Tauc relation [
26]:
where
α is the absorption coefficient,
hν is the energy of absorption light,
n is a parameter connected with the density-of-states (DOS) distribution in the band gap, and
B is a proportionality factor that also provides information about the DOS distribution.
The optical gap for the plasma-deposited semiconductor thin films is usually determined by assuming a parabolic function for the DOS distribution in the band tails (
n = 2 in Equation (3)), and in this case, it is assumed that no large error is made by taking the
EOPT approximately equal to the band gap (
EG) [
27].
Figure 6b, based on Equation (3), shows the measured UV-VIS optical absorption for the ad-CM and TT-CM films as a dependence of (
α hν)
1/2 on
hν. The
EOPT determined in this way is 1.6 eV and 1.2 eV, respectively.
Knowing the values of the band gap (
EG) and the ionization potential (
J), it is possible to calculate another important element of the electronic structure, which is the electron affinity (
χ):
In turn, by subtracting the measured EBE value from EOPT, we obtain the energy distance of the Fermi level from the lower edge of the conduction band.
Using the determined elements of the electronic structure, band models for the ad-CM and TT-CM films were proposed and presented in
Figure 7. While the electronic structure parameters of both films are similar and generally align with the values reported in the literature—such as the work function (from 4.55 to 4.85 eV, [
28]) and the optical gap (from 1.09 to 2.09 eV, [
29])—there exists a notable distinction between them. This distinction is characterized by the transition from an intrinsic semiconductor for the ad-CM film to the
n-type semiconductor for the TT-CM film, which is defined by the position of the Fermi level.
3. Discussion
Three types of plasma-deposited CoO-based films (ad-CoO, TT-CoO, and PT-CoO) were investigated for their catalytic performance in the CO
2 methanation process and the relationship between this performance and the composition and structure of the films. Among them, the TT-CoO films turned out to be the best in terms of CO
2 conversion, selectivity to CH
4, and stability during the process (
Figure 1). These films also demonstrated the complete stability in their molecular structure in the CO
2 methanation process, unlike the other two types of films (
Table 1).
In the ad-CoO films, following the catalytic process, the carbon content decreases, and at the same time the content of its sp
3 form increases compared to sp
2. Interestingly, maintaining these films at a temperature of 400 °C during the process causes only a slight change in the amorphous nanostructure composed of Co, C, and O atoms, consisting of the formation of a very small amount of CoO nanocrystallites with a size of about 3 nm (
Section 2.3). Meanwhile, by preparing TT-CoO films by the thermal treatment of ad-CoO films at the same temperature, however, not in the reaction mixture, but in an argon atmosphere, we create a stable structure containing a significant amount of CoO nanocrystallites with a size of approximately 11 nm in an amorphous carbon matrix. This means that in the case of ad-CoO films, the reaction mixture diffuses into their bulk and influences the structure transformation process. The phenomenon of the permeability of various gases, such as CO
2, through films of amorphous carbon produced by PECVD, is widely known [
30,
31], which confirms the correctness of the above assumption.
As a probable reaction, referring to the reverse Boudouard reaction [
32], an electrophilic attack of a carbon atom with a partial positive charge (δ+) in a CO
2 molecule on a double bond in the carbon matrix is proposed:
As a result of this reaction, which occurs simultaneously with the CO
2 methanation process, the carbon content in the matrix decreases by removing it as CO, and the concentration of carbon in the sp
3 form increases (
Table 1; compare fresh and spent ad-CoO films). The alternation of the carbon matrix in the ad-CoO film appears to be a plausible explanation for the substantial decrease in the formation of CoO nanocrystallites, which are readily produced at the same temperature (400 °C) in an argon atmosphere. It is noteworthy that the TT-CoO film, produced in this manner, maintains its structure unaffected by the reaction mixture (
Table 1; compare fresh and spent TT-CoO films). Contrastingly, the pure carbon matrix thermally treated at 400 °C (TT-CM) exhibits the evolution of CO under these conditions (
Section 2.1) and a rise in the Csp
3 content in comparison to Csp
2 (
Table 2), providing evidence of the reaction (5) progression. This clearly highlights the key role of CoO nanocrystallites in stabilizing the carbon matrix within TT-CoO.
Thus, a fundamental question arises: what role do CoO nanocrystallites play in stabilizing the structure of the TT-CoO film during the CO
2 methanation process, particularly as this structure demonstrates superior catalytic performance among all the tested films? The concept of nanoscale heterojunctions turns out to be crucial in addressing this query, having been previously invoked several times to elucidate catalytic phenomena in thermal catalysis processes [
33,
34,
35,
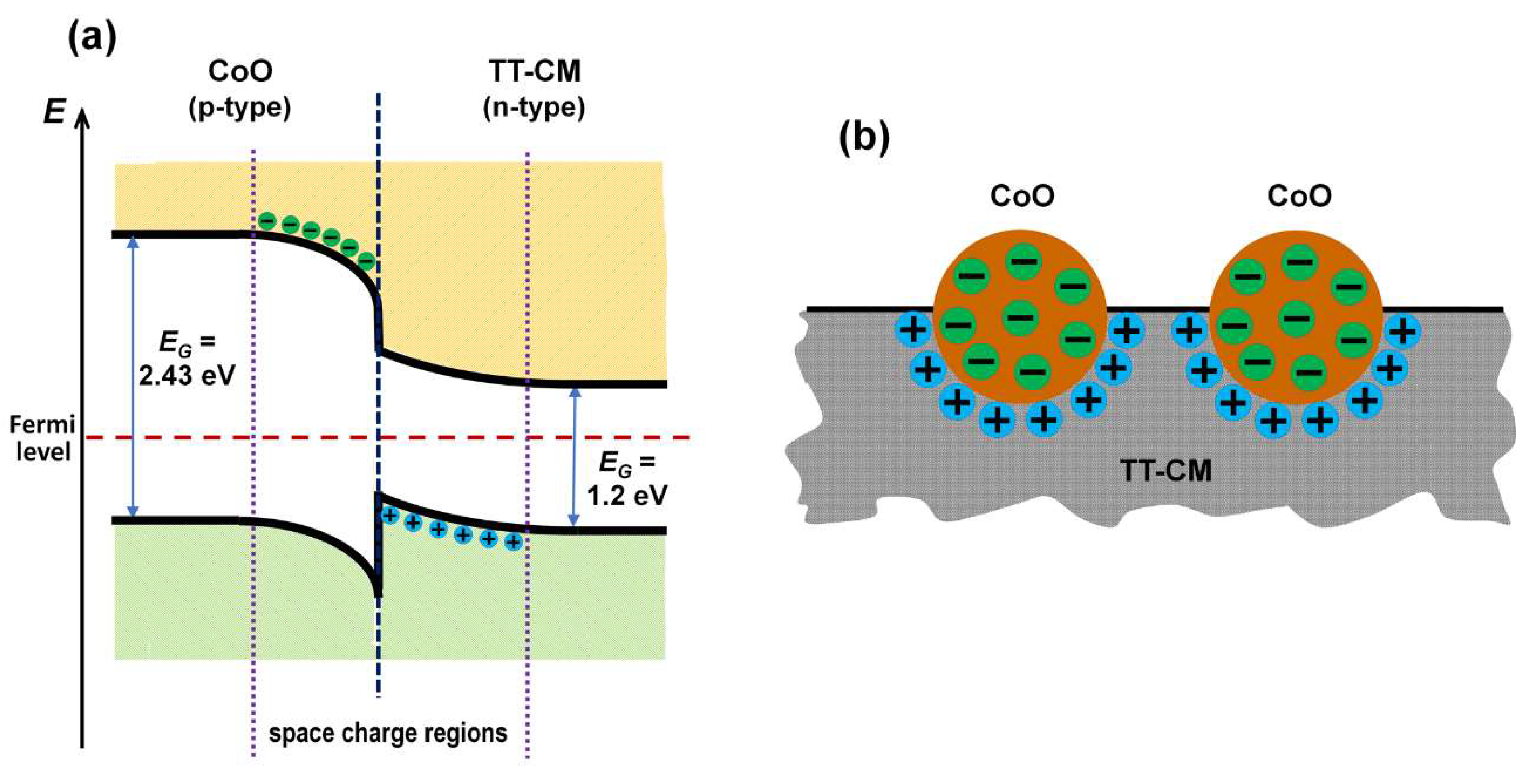
36]. In the case of the TT-CoO films under discussion, we hypothesize that such heterojunctions form between CoO nanocrystallites and the carbon matrix.
It has been extensively documented in the literature that undoped CoO nanocrystallites function as a p-type semiconductor [
37,
38], exhibiting a band gap within the range of 2.2–2.8 eV [
39,
40]. Based on the recent research by Zhai et al. [
41], who delved into the electronic structure of cubic CoO nanocrystallites using UPS and UV-VIS spectroscopy, very similar to our investigation of the electronic structure of the carbon matrix, we adopted their
EG = 2.43 eV and
EBE = 0.74 eV for further analysis. Taking these parameters of the electronic structure of CoO and the band model of the TT-CM carbon matrix (
Figure 7), a band model of the CoO–TM-CM heterojunction was proposed, which is shown in
Figure 8a. This heterojunction is of the straddling gap type (type I-2) [
42].
As a result of achieving thermodynamic equilibrium within the created heterojunction, marked by the alignment of the Fermi level, regions of negative and positive space charge are established on either side of the heterojunction, specifically in the p-type (CoO) and n-type (TT-CM) semiconductors, respectively. The dimensions of these space charge regions often span several dozen nanometers [
43]. Consequently, when nano-sized entities interact within the heterojunction, these space charge regions might entirely fill them (
Figure 8b), thereby altering their electronic characteristics. This transformation subsequently modifies the nature of the active catalytic centers on their surfaces, thereby influencing the catalytic properties.
The presence of space charge regions in the TT-CoO film has been confirmed by the X-ray photoelectron spectroscopy (XPS) studies, indicating a noticeable shift in the maximum position of the Co
2+ (2p3/2) band (
Section 2.2). The maximum position for both the fresh and spent ad-CoO and PT-CoO films remains virtually constant, at approximately 780.8–781.0 eV (
Figure 2). Moreover, according to the literature, the maximum of the Co
2+ band for the pure CoO nanocrystallites also has a very similar position, typically at 780.9 eV [
44] or 780.6 [
45]. However, within the TT-CoO film, where CoO nanocrystallites exist and presumed heterojunctions form between them and the carbon matrix, the Co
2+ band experiences a shift towards a lower binding energy (BE), settling at approximately 780.2 eV. The determined shift of about 0.7 eV is a combined effect of the Co
2+ band’s shift towards a lower binding energy and the expected shift of the sp
2 band towards a higher binding energy. However, the position of the sp
2 band remains fixed at 284.6 eV. These shifts result from the presence of a space charge, which, in the case of a negative charge, diminishes the binding energy, while in the case of a positive charge, elevates the binding energy of the photoelectron emitted from the atom during XPS measurements [
46].
The presence of a negative charge in the CoO nanocrystallites enhances their basic nature (catalytic basic sites), thus promoting CO
2 hydrogenation towards CH
4 [
47]. In turn, the positive charge on TT-CM inhibits the electrophilic attack of CO
2, effectively impeding the reaction (5), and thereby stabilizing the carbon matrix. This mechanism, reliant on the formation of nanoscale heterojunctions, adequately explains the differences in the catalytic performance and structural changes between the ad-CoO and TT-CoO films within the reaction mixture.
A distinct topic concerns plasma-treated film (PT-CoO). Contrary to the initial anticipation that subjecting ad-CoO films to argon plasma treatment would serve as an energy-efficient alternative to the more intensive calcination process, the PT-CoO films turned out to be unstable in the CO
2 hydrogenation process, revealing a clear decline in CO
2 conversion over time and a shift in selectivity towards CO production rather than CH
4 (
Figure 1). The XPS investigations revealed, similar to the ad-CoO films, the absence of heterojunctions in the PT-CoO film. However, the plasma treatment caused significant changes in the molecular structure of its surface, resulting in the formation of the monocarbide (CoC) form and a marked increase in the sp
3 carbon content compared to sp
2. This transformation likely contributes to the altered selectivity, favoring CO production. The resultant CO appears to be the source of carbon deposit formation [
36], as observed in this case (
Figure 3, spent PT-CoO), which impedes the conversion of CO
2. Nonetheless, due to the unattractive catalytic properties exhibited by the PT-CoO films, further comprehensive studies on them were not pursued.
In a summary of the discussion, the main findings from the conducted research are condensed in
Table 3.
4. Materials and Methods
4.1. Preparation of Thin Films
The plasma-enhanced chemical vapor deposition (PECVD) technique was employed to fabricate CoO-based and carbon matrix thin films. The deposition process took place in a parallel-plate radio-frequency (RF 13.56 MHz) plasma reactor, which is a home-made device. The reactor construction and film fabrication procedure are detailed in our previous papers [
6,
48]. For the preparation of CoO-based catalytic films, cyclopentadienyl (dicarbonyl) cobalt(I) (CpCo(CO)
2, Stream Chemicals, Newburyport, MA, USA) served as the deposition process precursor, with a vapor flow rate of 0.083 sccm. Argon (99.999%, Linde Gas, Cracow, Poland) was used as a carrier gas with a flow rate of 0.71 sccm. The reactor chamber maintained a total pressure of 4–5 Pa. The deposition was carried out at a discharge power of 60 W for 10–30 min, resulting in films with a thickness of approximately 280–850 nm.
Thin films with a pure carbon matrix were prepared using the same plasma process parameters as those for the CoO-based films: a discharge power of 60 W, an argon flow rate of 0.71 sccm, and a total pressure in the reactor chamber of 4–5 Pa. The precursor for the deposition process was a mixture of CO (99.99%, Linde Gas, Cracow, Poland) and cyclopentadiene (C5H6, Warchem, Poland) in a molar ratio of 2:1, corresponding to the ligand content in the CpCo(CO)2 complex. The flow rates of CO and C5H6 were set to 0.055 and 0.028 sccm, respectively. The films were deposited for 6–10 min, resulting in thicknesses of 190–320 nm.
Kanthal steel meshes, in the form of knitted wire gauze (TermTech, Warsaw, Poland), sized at 2.3 cm diameter with a 1 cm hole in the center, which corresponded to the size of the catalytic reactor, were utilized as supports for catalytic tests. Plates made of the same Kanthal steel, measuring 1.5 × 1.5 cm, were employed for studying the chemical structure of the films through XPS spectroscopy. Before plasma deposition, the Kanthal supports underwent calcination in air at 900 °C for 48 h to ensure high surface development [
6,
48]. For UV-VIS absorption measurements, the films were deposited on quartz wafers, while for XRD diffraction studies, zero background Si wafers (Si 510, Institute of Electronic Materials Technology—Łukasiewicz Research Network, Warsaw, Poland) were used as substrates.
Three types of CoO-based films were prepared for investigations: as-deposited films without any post-treatment (ad-CoO); as-deposited films thermally treated (calcined) in argon with a flow rate of 2 L/min and a heating rate of 18 °C/min, maintained at 400 °C for 30 min (TT-CoO); and as-deposited films treated in plasma at a constant argon flow rate of 0.71 sccm, with a discharge power of 80 W for 2 h. For the carbon matrix films, two forms were prepared for further studies: as-deposited films without any post-treatment (ad-CM) and calcined films (TT-CM), following the same procedure as the TT-CoO films.
4.2. Catalytic Tests
Catalytic tests were conducted at atmospheric pressure using a tube-in-tube fixed-bed quartz reactor, as described in detail in a previous paper [
35]. For each experiment, six circular pieces of catalyst (disc of mesh with OD = 2.3 cm and a hole ID = 1 cm), providing a geometric surface area of 38 cm
2, were loaded into the reactor. To maintain data comparability, the position of the meshes constituting a catalytic bed was kept the same in all experiments. After purging the lines and the reactor with helium (99.999%), a mixture of H
2 (99.999%) and CO
2 (99.99%) gases (all gases supplied by Linde Gas, Cracow, Poland) with a total flow rate of 25 sccm and the ratio of H
2/CO
2 = 4 was introduced into the reactor. The temperature was raised at a rate of 5 °C/min in stages of 50 °C from 250 °C to 400 °C. On the way of the outlet reaction mixture to a gas chromatography analyzer (GC Master, Dani Instruments, Cologno Monzese, Italy), a cold trap was inserted to collect the condensed water. The composition of outlet dry gas was analyzed using a thermal conductivity detector (TCD) and a ShinCarbon ST packed column (Restek, Bellefonte, PA, USA), with measurements taken at 10 and 20 min after reaching the set temperatures. At each temperature, the catalyst was kept for 30 min. Utilizing the calibration curves for CO
2, CO, and CH
4, the CO
2 conversion and selectivity to CO and CH
4 were determined. The stability of the catalyst was investigated over a period of 24 h at 400 °C.
4.3. Chemical Structure, Electronic Structure, and Nanostructure of the Films
The chemical structure of both the CoO-based and carbon matrix films was investigated by X-ray photoelectron spectroscopy (XPS) using a Kratos AXIS Ultra spectrometer (Kratos Analytical Ltd., Manchester, UK) with a monochromatic Al-Kα X-ray source (1486.6 eV). Spectra were obtained from an analysis area of 300 × 700 μm. The anode power was set at 180 W, and the hemispherical electron energy analyzer was operated at a pass energy of 20 eV for all high-resolution measurements. XPS spectra were analyzed using Kratos Vision 2.2.10 software, calibrated by setting the C 1s carbon peak assigned to the sp2 carbon at 284.6 eV. The Shirley algorithm was used for background subtraction.
The same Kratos AXIS Ultra spectrometer, equipped with a helium lamp with a He I line (21.21 eV), was used for ultraviolet photoelectron spectroscopy (UPS). Valence band spectra, providing information about the electronic structure of the films, were collected from 55 µm diameter spots on the sample surface at 50 meV resolution. Data analysis was performed using Kratos Vision 2 software (ver. 2.2.10, Kratos Analytical Ltd., Manchester, UK, 2012). To complement this information about the band gap value, optical absorption measurements were conducted using a Shimadzu UV-1800 spectrophotometer (Shimadzu Corporation, Kyoto, Japan). Measurements were made in the transmission mode in the wavelength range of 200–800 nm with a 1 nm sampling interval.
To determine the nanocrystalline structure of the films, the X-ray diffraction technique was employed. The measurement used an Empyrean diffractometer (Malvern Panalytical Ltd., Malvern, UK) operating with Cu Kα radiation (λ = 1.541874 Å) at 45 kV and 40 mA, utilizing a reflection Bragg–Brentano mode. The diffractometer was equipped with a PIXcel3D detector with 255 active channels. Diffractograms were obtained in the 2Theta range of 20–85 deg using a continuous scan mode with a step size of 0.0263 deg and a counting time of 176 s. Samples were rotated with a rotation time of 8 s.
5. Conclusions
Considering the vast potential of plasma-deposited thin-film CoO-based nanocatalysts for CO2 methanation, this work aimed to investigate the correlation between the molecular structure, nanostructure, and their catalytic activity. Three types of catalysts were analyzed: as-deposited (ad-CoO) films, films subjected to calcination in argon after plasma deposition (TT-CoO), and films post-treated with argon plasma (PT-CoO). The TT-CoO films exhibited the most favorable catalytic properties, showcasing the highest CO2 conversion rate, selectivity for CH4, and structural stability during the catalytic process.
The results indicate that the remarkable catalytic properties of the TT-CoO films arise from the formation of nanoscale heterojunctions between the CoO nanocrystallites and the carbon matrix. These heterojunctions create spatial charge regions—negative on the CoO nanocrystallites and positive on the carbon matrix—enhancing and stabilizing the catalytic activity towards CH4 production. These findings are in line with the traditional concept of acidic and basic catalytically active sites. Positively charged acidic sites are known to impede the conversion to CH4, while increased basicity promotes CH4 production. In our study, we have shown that a negative charge on CoO nanocrystallites, consistent with the concept of basic active centers, enhances the CO2 hydrogenation toward methane.
This work provides strong evidence for the key role of nanoscale heterojunctions in determining the properties of nanocatalysts. It emphasizes how this phenomenon can be used for a more rational design of novel catalytic systems for targeted products.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


