
Kinetic and Mechanistic Study of Aldose Conversion to Functionalized Furans in Aqueous Solutions
by
 ,
,
Stefan S. Warthegau
1,
Magnus Karlsson
2,
Robert Madsen
1,
Pernille Rose Jensen
2 and
Sebastian Meier
1,* 1
Department of Chemistry, Technical University of Denmark, Kemitorvet, Building 206, 2800 Kgs. Lyngby, Denmark
2
Department of Health Technology, Technical University of Denmark, Elektrovej 349, 2800 Kgs. Lyngby, Denmark
*
Author to whom correspondence should be addressed.
Catalysts 2024, 14(3), 199; https://doi.org/10.3390/catal14030199
Submission received: 8 February 2024
/
Revised: 9 March 2024
/
Accepted: 12 March 2024
/
Published: 18 March 2024
(This article belongs to the Special Issue Novel Catalytic Strategies for the Synthesis of Furans and Their Derivatives)
Abstract
:Reaction mixtures of naturally abundant aldoses and CH nucleophiles allow for the formation of functionalized furan precursors using low temperatures and metal-free catalysis in aqueous solutions of dilute base catalysts. We employ in situ NMR assays to clarify the mechanism and kinetics of the conversion. Catalysis serves a double role in ring-opening of stable aldoses such as glucose and xylose and facilitating the subsequent reactions with CH acids such as malononitrile or cyanoacetamide. Resultant acyclic products are shown to convert quickly to a monocyclic product prior to the slower formation of a more stable bicyclic intermediate and dehydration to tri-functionalized furan. Especially the reversible 5-exo-dig ring closure entailing oxygen attack onto a nitrile carbon is surprisingly fast with an equilibrium vastly towards the cyclic state, sequestering reactive groups and allowing the selective conversion to tri-functionalized furan. The reaction hinges on the fast formation of intermediates without CH acidity and competes with the oligomerization of CH nucleophiles. Insight derived from in situ NMR analysis shows the prowess of high-resolution in situ spectroscopy in clarifying the interplay between catalysts and reactants. Such insight will be vital for the optimization of reactions that upgrade biorenewables under benign conditions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Sustainable future production of fuels and materials requires strides towards including renewable carbon from biomass into organic chemicals. The room for improvement in this endeavor is enormous, as approximately 95% of organic chemicals in the chemical industry remain sourced from fossil resources [1]. The target chemicals that are reasonably easily accessible by biological or chemical processes often include acids [2,3,4,5,6,7,8,9], alcohols [10,11,12,13,14,15,16,17,18,19,20], and furanic scaffolds [21,22,23,24,25,26,27,28]. The conversion of carbohydrates to furanic compounds by acid-catalyzed dehydration has been known for over a century [29,30]. Furfural and 2,5-disubstituted analogs (including 5-(hydroxymethyl)furan-2-carbaldehyde, 5-(methyl)furan-2-carbaldehyde, and 2,5-furandicarboxylic acid) [31,32] accordingly have been investigated by both academic and industrial partners as prospective precursors for materials and energy applications.
As an alternative to the direct conversion of carbohydrates as sole reactants, strategies based on the use of reactant mixtures for the upgrading of aldoses have been devised [33]. Reactions of the carbohydrate backbone with CH acidic nucleophilic compounds have shown promise in the formation of various substance classes, including C-glycosidic ketones, oligomeric cyclic compounds, and polyfunctional furfural derivatives [34,35,36,37,38,39,40,41]. These reactions are mainly catalyzed by either Lewis acids or Brønsted bases in either aqueous solutions or alcohols and can lead to important heterocyclic precursors, including enzyme inhibitors, pharmaceuticals, and fine chemicals [38]. Considering the promise of sustainable aldose upgrading with CH acidic nucleophiles in benign solvents [42,43,44] under mild conditions, detailed experimental insight into the reaction mechanisms would be desirable, but such insight is currently sparse.
It has previously been shown that state-of-the-art in situ NMR spectroscopy can clarify the kinetics and mechanisms of carbohydrate conversion in reactions relevant to sustainable chemistry [45,46,47,48,49,50]. Here, we employ various in situ 1H and 13C NMR assays to advance the insight into the upgrading of carbohydrates to chain-elongated furanic compounds [51]. Owing to its large chemical shift range and detection of sharp singlet signals, 13C NMR especially provides sufficient resolution and structural information to offer detailed insight into the formation and conversion of functional groups. Special emphasis is placed on the mild conversion of glucose and xylose with malononitrile to a trisubstituted furanic derivative bearing amino, nitrile, and polyol functionality. This reaction has recently attracted significant attention [36,38]. Proposed mechanisms initially included reactions to compound 3 of Scheme 1 in competition to aromatization to 4 [38]. Alternatively, previous DFT calculations suggested a pathway that proceeded through ketenimine intermediates [36]. The formation of the intermediate 2 was not considered. Using conventional and hyperpolarized 13C NMR spectroscopy, we seek to clarify the mechanism and kinetics of this transformation.
2. Results and Discussion
2.1. Stable Aldoses Can Be Converted in a Surprisingly Fast Reaction via Two Intermediates, One of Them Previously Not Anticipated
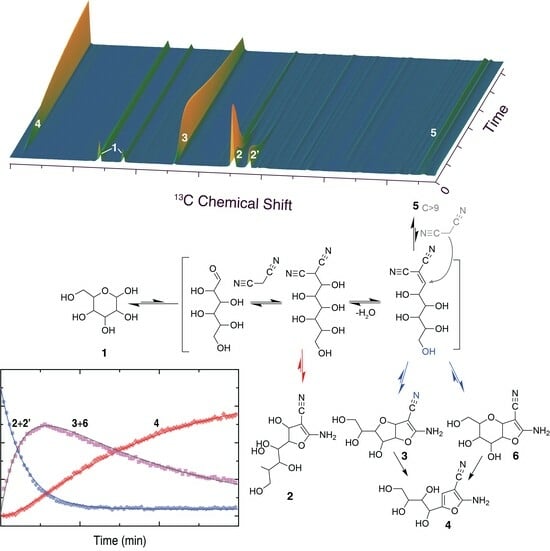
As a starting point for this study, we chose to focus on the conversion of abundant aldohexoses and aldopentoses, in particular glucose and xylose, with malononitrile under homogeneous organic base catalysis in water [38]. To this end, we employed triethylamine, which had previously been suggested as a particularly efficient base catalyst for the chain elongation of aldoses with malononitrile at room temperature over 1.5–4 h [37]. Surprisingly, in situ NMR observations using 13C NMR spectroscopy indicated full conversion of the reactant within 5 min when using 1.0 base equivalents at 293 K. A trisubstituted furan product was formed in the reaction and was fully assigned by NMR spectroscopy. This product carried an amino–, cyano-, and polyol-functionality on the furan ring, as previously described for Lewis acid and Brønsted base-catalyzed reaction of aldose and malononitrile [38,52]. It has been suggested that the precursor of the trisubstituted furan product comprises a mixture of bicyclic intermediates containing a tetrahydrofuran- (compound 3) or a tetrahydropyran-derived ring deriving from the aldose reactant [38]. We subsequently repeated the reaction at 288 K and 0.15 equivalents triethylamine to slow down the reaction relative to the initial conditions (Figure 1). These conditions still yielded near-complete conversion of the aldose reactant within the experiment dead time prior to NMR acquisition in our setup (five minutes), albeit residual carbohydrate signal could be observed at the beginning of the time series, prior to the sequential accumulation of two major intermediates and the expected product, all of which showed nine signals of identical kinetic profile for the conversion of glucose and eight signals of identical kinetic profile for the conversion of xylose. Only the second intermediate resembled a bicyclic structure, and the chemical shifts deriving from C3–C6 of glucose specifically indicated the presence of a tetrahydrofuran ring formed from the glucose moiety in the bicyclic intermediate 3.
2.2. Assignment of the Detected Species in a Complex Reaction Mixture
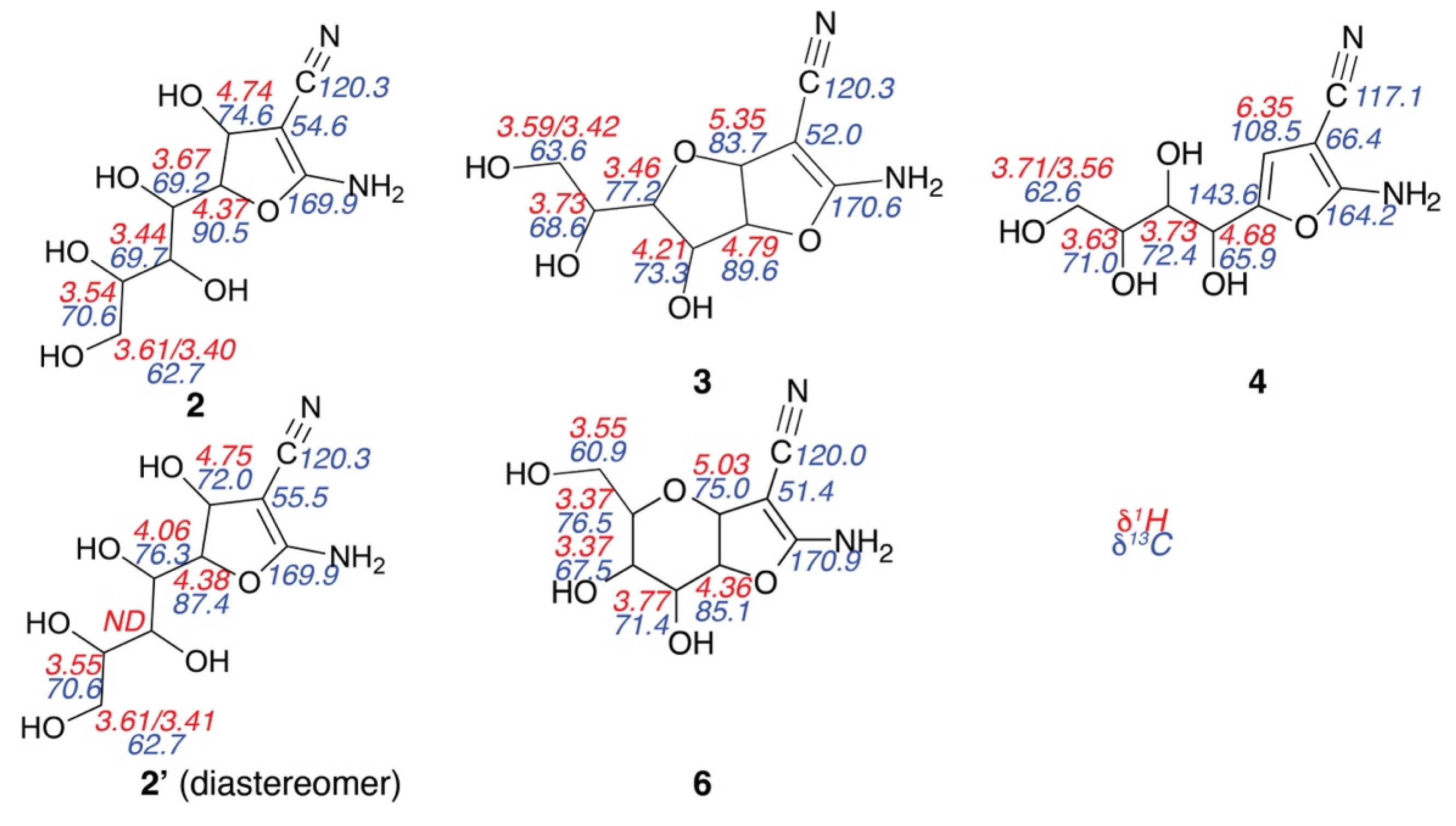
To identify the nature of the main intermediates en route to densely functionalized furans, the reaction was further slowed down by mixing all reactants and the diluted catalyst (0.05 equivalents triethylamine) on ice prior to acquiring two-dimensional assignment spectra for the characterization of all detected species. To this end, heteronuclear (1H-13C HSQC, H2BC, HMBC, and HSQC-TOCSY) and homonuclear (1H-1H COSY, TOCSY, and ROESY) spectra were optimized to afford data acquisition within few minutes. The resultant spectra allowed unambiguous assignments of the two main intermediates, structures 2 and 3, in Scheme 1 and Figure 2. We hence suggest revising previously anticipated mechanisms [36,38] to include the rapid formation of monocyclic intermediate 2 upon Knoevenagel addition and 5-exo-dig ring closure, prior to slower dehydration and concerted 5-exo-dig ring closure and oxa-Michael addition to complete the Knoevenagel condensation into the bicyclic compound 3. The acyclic structure rather than the formation of a six-membered ring in compound 2 is corroborated by the low chemical shift (70.6 ppm) deriving from the C5 position in glucose (Figure 2) and by the absence of 1H-13C HMBC and 1H-1H ROESY correlations between the positions deriving from C1H1 and C5H5 in glucose (Figures S1 and S2). By contrast, the cyclic structure in compound 3 could be validated by the characteristic deshielding of C4 (77.2 ppm) and by the presence of 1H-13C HMBC correlations across the bridging oxygen (Figure S2). In addition, compound 3 exhibited diffusion at 288 K with a translational diffusion constant of 2.17 × 10−10 m2/s, which compared to 4 with a translational diffusion constant of 2.61 × 10−10 m2/s reflected a more compact structure in the bicyclic intermediate than in the monocyclic product.
2.3. Isotope Tracking to Validate Assignments and Clarify Mechanisms
Subsequently, we validated the assignments using [1-13C]glucose as the site-specifically labeled glucose reactant for tracking the anomeric carbon using a time series of 13C NMR spectra. The resultant time series is shown in Figure 3 and validates the rapid conversion of glucose (1, signals at 96.8 and 92.3 ppm) to an intermediate 2 with signals at 74.6 ppm deriving from the glucose anomeric carbon, prior to conversion to a second major intermediate with signals at 83.7 ppm deriving from the glucose anomeric carbon (intermediate 3) and conversion to a sp2 hybridized position that is deshielded to 108.5 ppm in a stable product 4 (Figure 3). Additional minor species were detectable in this experiment due to the 100-fold sensitivity gain from isotope enrichment with 13C. Surprising was the formation of a set of signals from the glucose anomeric carbon near 45 ppm. These signals indicate that the glucose anomeric carbon, which results in the only non-substituted position in the furan ring, can transiently add to additional carbon nucleophiles, which may pave the way to fully substituted heterocycles upon suitable optimizations. This observation resembles the formation of oligomeric structures in base-catalyzed Knoevenagel reactions with benzaldehyde [53].
We subsequently extended the reaction tracking studies to the aldotetrose [2-13C]erythrose as the reactant to validate that the formation of a six-membered ring is not an essential on-pathway reaction in the conversion of aldoses to tri-functionalized furans. The reaction of [2-13C]erythrose with malononitrile showed a rapid influx of erythrose into intermediates where the aldose-derived C2 position is part of structures with chemical shifts near 90 ppm, as expected for C7 analogs of intermediates 2 and 3 formed from glucose. Hence, these intermediates were formed, albeit aldoses are too short to form pyranose rings, and such pyranose ring formation was no prerequisite en route to a furan derivative product [36]. The signals at 89–90 ppm showed line broadening and thus indicated dynamic exchange on the milliseconds time scale. Subsequently, the product was formed with a 13C chemical shift near 145 ppm, as expected (Figure 4). During in situ NMR experiments using C4-C6 aldoses as reactants, the C2 position remained protonated in the conversions to intermediates 2 and 3 (or shorter analogs for C4 or C5 aldoses) when conducting reactions in D2O, thus excluding a mechanism entailing a C1-C2 enediol intermediate and deuteration of the C2 position.
2.4. DNP NMR Investigates Rapid Steps and Shows the Transient Nature of Acyclic Intermediates
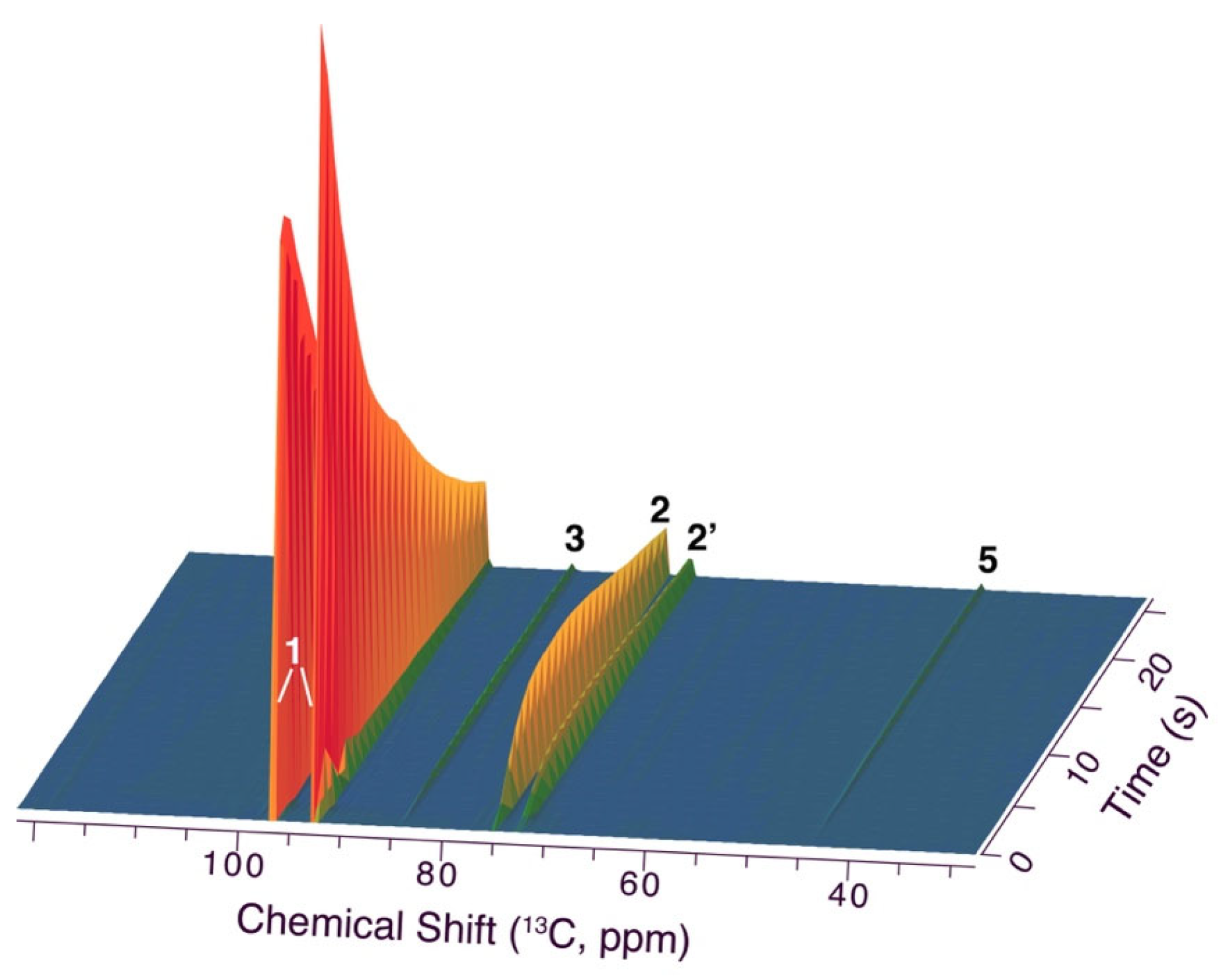
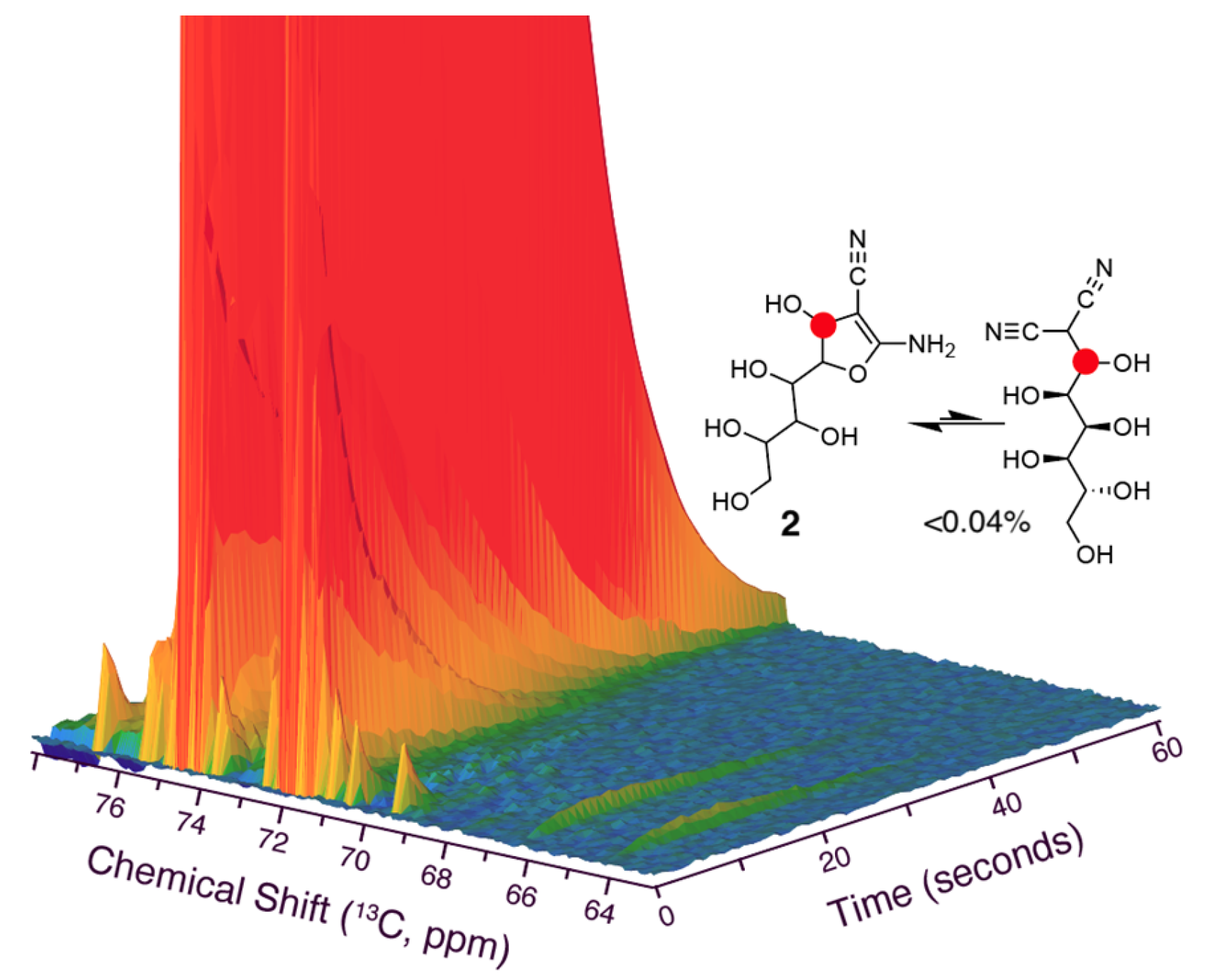
Considering the rapid initial conversion of glucose, we further pursued rapid injection NMR methodology to evaluate whether additional information on the initial steps and first intermediates in the reaction cascade could be derived, as the first detected intermediate 2 plausibly is formed after initial steps involving a Knoevenagel addition to yield an acyclic species prior to its cyclization to 2. Rapid injection NMR was further enhanced by dissolution Dynamic Nuclear Polarization (dDNP), a method that temporarily redistributes spin states and can lead to an enhancement of nuclear magnetization for reactants of interest by more than four orders of magnitude [54,55]. This enhanced magnetization approaches equilibrium with a time constant of T1, and the T1 time thus defines the time scale during which enhanced signal can be detected. The T1 time of carbon sites can be extended by deuteration. Hence, [1-d,1-13C]glucose was chosen as the glucose isotopologue of choice. The rapid injection of isotope and spin-polarization-enhanced glucose yielded the time series of Figure 5, where 13C NMR spectra were acquired each second. The assay showed that glucose can be predominantly converted to the initial intermediate 2 (signal near 75 ppm) within 35 s. Conversion to intermediate 3 (signal near 85 ppm) and a species forming a non-oxygenated, sp3 hybridized carbon due to the addition of two nucleophiles [53] to the anomeric glucose carbon (signal near 45 ppm) was also detected using dDNP. The initial Knoevenagel addition converts the anomeric glucose carbon to a secondary acyclic alcohol site or, upon dehydration, to a conjugated olefinic site, with expected chemical shifts in the 60–70 ppm and the 160–170 ppm 13C chemical shift ranges, respectively. Upon close inspection of the dDNP NMR experiment, no signal near 160–170 ppm was found to accumulate from the anomeric position, while weak signals in the 60–70 ppm chemical shift region could be detected. As these signals were formed transiently and at low levels, structural assignments of these species were not possible. These species account for less than 0.04% of the intermediate pool of 2, and the formation of higher populations of non-cyclized Knoevenagel adduct or its dehydrated form with lifetimes on the milliseconds to seconds time scale can be excluded (Figure 6). The prevalent and rapid cyclization to five-membered rings somewhat resembles the fast cyclization of carbohydrates, resulting in low non-cyclized equilibrium fractions below 0.1% [56,57].
2.5. Role of Aldose Ring Opening and Correlation of Reactivity with Open Populations
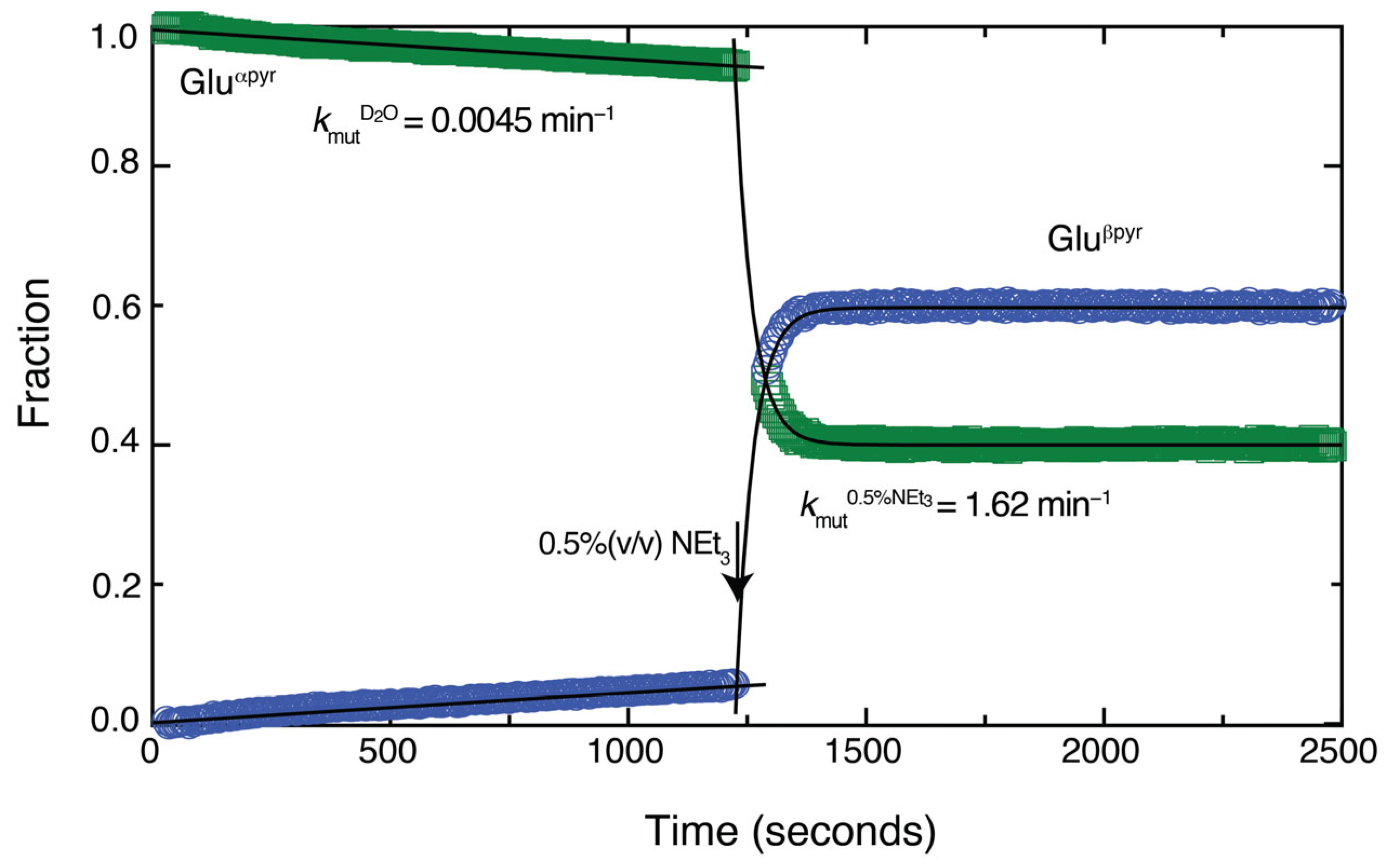
Considering the experimental observations, we propose that the conversion of aldoses to triple functionalized furan rings commences with a rapid Knoevenagel addition prior to cyclization to more long-lived species. The rapidity of the Knoevenagel addition is surprising considering that mutarotation (and hence ring opening) of stable aldopyranose forms like those of glucose and xylose proceeds on a time scale considerably slower than the C–C formation observed herein. We hence concluded that a seminal role of triethylamine is the catalysis of ring opening in the aldose, consistent with known effects of base on ring opening [58]. To substantiate this suspicion, we dissolved pure α-D-glucopyranose (1 M) in water and tracked mutarotation by using 1H NMR spectra to follow the equilibration to a mixture of approximately 60:40 α-pyranose/β-pyranose form. Compared to mutarotation in pure D2O, the addition of only 0.5% (v/v; corresponding to 0.015 equivalents) triethylamine accelerated mutarotation by a factor of 360 (Figure 7), thus underlining the effect of the catalyst on the ring opening kinetics for the Knoevenagel additions to unprotected aldoses.
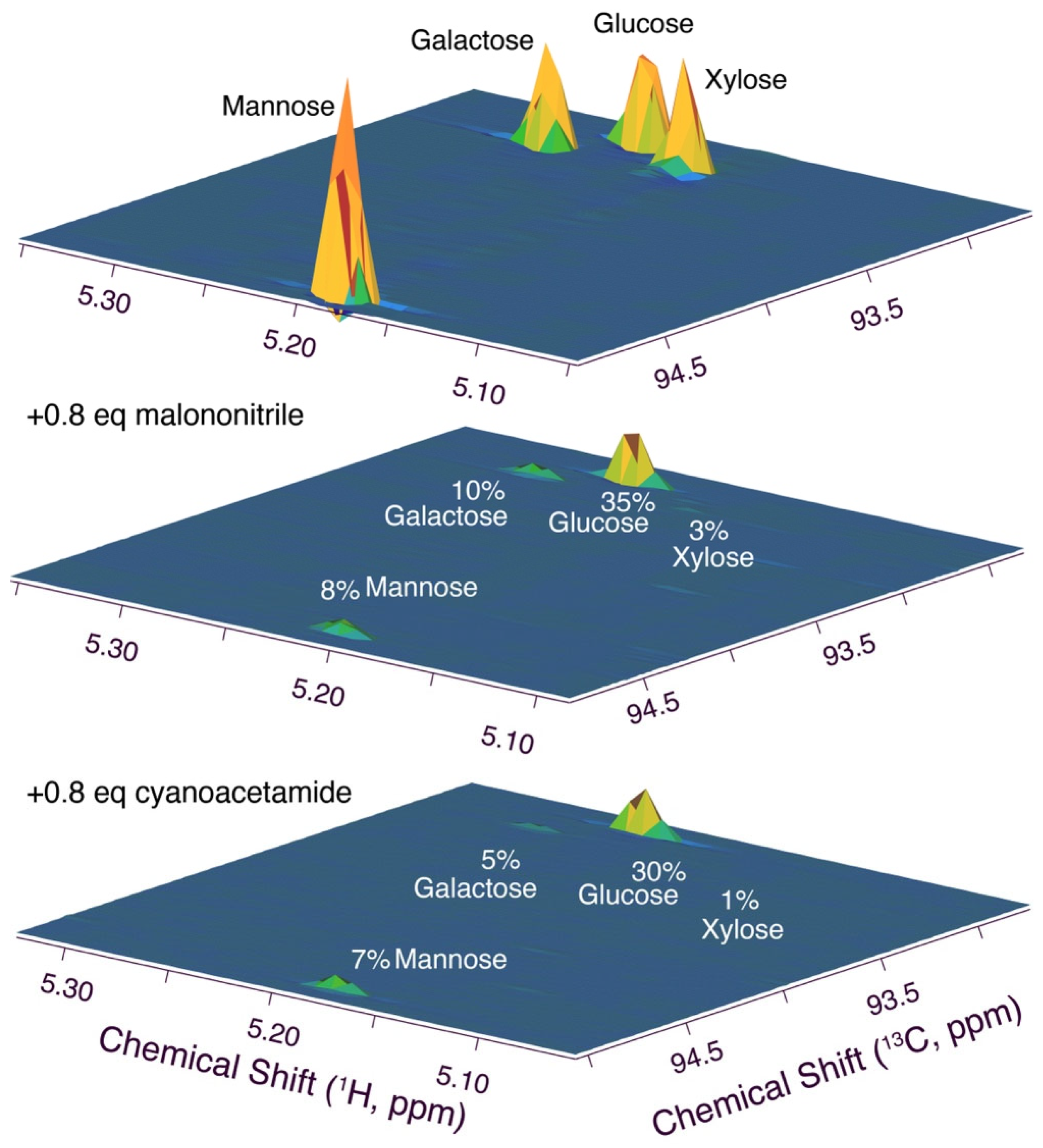
Due to the importance of aldose ring opening in the Knoevenagel addition of malononitrile to aldoses, we then devised a competitive assay to evaluate the relative reactivity of various aldoses. An equimolar mixture of some of the naturally most abundant monosaccharide aldoses (glucose, xylose, mannose, and galactose) with sub-stochiometric amounts of malononitrile (0.8 equivalents) was divided into a reference sample and a sample containing triethylamine base (0.15 equivalents). Subsequently, high-resolution 1H-13C HSQC spectra [59] were acquired on both samples to evaluate the reactivity scale of the carbohydrates. Conversion decreased in the order xylose > mannose, galactose > glucose in this competitive assay. This reactivity was corroborated by the corresponding reaction using cyanoacetamide as the nucleophile (Figure 8; a time course of the reaction with glucose is shown in Figure S3). The overall trends are consistent with the higher stability of glucose cyclic forms relative to mannose and galactose as well as pentoses, and the reactivity correlated with the expected fraction of acyclic forms in aqueous solution [56,57]. Hence, base-catalyzed ring opening in naturally abundant carbohydrates affords an even more efficient and facile conversion of other naturally abundant monosaccharides as compared to glucose.
2.6. Plausible Mechanism and Kinetic Analysis Are Mutually Consistent
Experimental observations with various NMR assays lead us to propose the mechanism shown in Scheme 2. Aldoses rapidly react to fast-exchanging and/or low-populated initial intermediates in a Knoevenagel addition that hinges on initial ring opening via base catalysis. From the high-energy intermediates formed upon Knoevenagel addition, intermediate 2 appears to form reversibly under kinetic control, while the thermodynamically more stable intermediate 3 is formed after completion of the Knoevenagel condensation by dehydration. This bicyclic intermediate 3 appears to be formed in concerted ring closures by oxa-Michael addition and 5-exo-dig ring closure. The bicyclic intermediate population is finally converted to the stable, trisubstituted furan product 4 (Scheme 2). Diastereomeric forms of 2 were formed due to the installment of a stereogenic center at the anomeric carbon upon Knoevenagel addition and the subsequent cyclization to yield 2. Slower dehydration and cyclization, as well as oxa-Michael addition by nucleophilic attack of the C4 or C5 hydroxide, yielded 3 and 6, respectively (Scheme 2, inset). Opposite to intermediate 2, intermediate 3 is only formed in one detectable isomeric form, and the conversion of 2 to 3 thus plausibly proceeds upon opening of the heterocycle and abolishing stereochemistry at the position deriving from the anomeric carbon by dehydration. The bicyclic forms 3 and 6 are formed with identical kinetics (Figures S4 and S5), indicating their formation in competing reactions upon Knoevenagel condensation (Scheme 2). Their further conversion with identical kinetic reaction progress to product 4 indicates that equilibration between 3 and 6 could be faster than conversion to product 4.
Hypothetically, the direct conversion of intermediate 2 by base-catalyzed dehydration and subsequent tautomerization to 4 or oxa-Michael addition to 3 and 6 could explain the conversion to 3, 4, and 6 (indicated in Scheme 2 as a cyclic path with rate constant k2*). Kinetic data indicates, however, that 2 is not converted to 3, 4, and 6 in competing reactions but in sequential reactions leading from 2 to 3/6 and to 4. Hence, we propose the overall mechanism shown in Scheme 2. This mechanism especially emphasizes rapid cyclization upon Knoevenagel addition to 2 and indicates that the concerted formation of the bicyclic rings 3/6 occurs prior to eliminative aromatization to 4 rather than in parallel to this eliminative aromatization.
Taking inspiration from the proposed mechanism of Scheme 2, the sequential formation of compounds 2, 3, and 4 upon the initial addition of malononitrile to the aldose anomeric center was fitted to a model of sequential elementary steps. The resultant fits for the conversion of glucose and xylose, respectively, are shown in Figure 9. Kinetic data of progress in the glucose conversion reaction indicates that the conversion of 2 to 3 with rate constant k2 and of 3 to 4 with rate constant k3 is very well described by a sequential mechanism upon influx of reactant into 2. Kinetic fitting indicates that the apparent rate constants for cyclization (e.g., from 2 to 3 and 6) are approximately one order of magnitude larger than the rate constants for dehydration (e.g., from 3 to 4). Corresponding rates for xylose conversion were slightly higher than the rates for glucose conversion. dDNP data were used to gain indications that 2 is an off-pathway intermediate that can be formed as the kinetic product from a minor and transient pool of open-chain forms formed from Knoevenagel addition and condensation. This model was indeed consistent with the experimental dDNP data showing the kinetically favored influx of a transient intermediate pool into 2 in competition with almost an order of magnitude slower influx into species such as 3 and 5 (Figure S6).
2.7. Kinetics of Desired Product Formation and of Some Competing Reactions
Finally, we wanted to clarify the kinetics of product formation from glucose as the naturally most abundant carbohydrate, under conditions that were previously described as optimal reaction conditions (room temperature, 1 mmol glucose, 1.1 mmol malononitrile, 1 equivalent base) [38]. These conditions yielded a rapid influx of the reactant into intermediates 3 and 6 upon mixing, prior to the rate-limiting conversion of the bicyclic intermediates to the triple-substituted furan 4. The formation of 4 under these conditions reached 90% of its maximum within a reaction time of 1.5 h in the absence of stirring. Tracking the formation of 4 indicated that product formation proceeded with biexponential kinetics (1/kapp of 35.3 min and 94.8 min) rather than with simple first- or second-order kinetics (Figure 10). This observation indicates that various pathways contributed to the conversion of intermediates to the trisubstituted furan product, and product formation continued to proceed noticeably after more than 6 h. The presence of minor species was also evident from in situ NMR experiments and included the formation of intermediates where two malononitrile molecules add to the anomeric carbon.
Lastly, we note that the reaction conditions induce the dimerization of malononitrile (100% conversion over 15 h, Figure S7). By contrast, the mild reaction conditions in dilute aqueous base elicited only limited aldose to ketose isomerization (9% conversion over 15 h, Figure S7) of glucose in control incubations in the absence of CH acidic nucleophile. Hence, the success of carbohydrate upgrading via Knoevenagel condensation at the anomeric center of aldoses critically depends on their rapid ring opening, C–C bond formation, and cyclization to thermodynamically stable intermediates and products in competition to reactions that lead to higher aggregates of the CH acidic nucleophile alone or with the carbohydrate.
3. Materials and Methods
3.1. Chemicals
Triethylamine (≥99.5%) and malononitrile (99%) were purchased from Sigma Aldrich (St. Louis, MO, USA), while cyanoacetamide (99%) was purchased from Thermo Scientific (Waltham, MA, USA). Glucose (96%), mannose (≥99%), xylose (≥99%), and galactose (≥99%) were purchased from Sigma Aldrich. Isotope enriched [1-13C]glucose was purchased from Sigma Aldrich, while [2-13C]erythrose (0.2 M aqueous solution) was purchased from Omicron Biochemicals Inc. (South Bend, IN, USA), and [1-d,1-13C]glucose was purchased from Cambridge Isotope Laboratories (Tewksbury, MA, USA). Deuterated water (D2O) was purchased from Sigma Aldrich.
3.2. Reaction Procedure
In a typical reaction procedure, aldose was weighed into a 1.5 mL Safe-lock Eppendorf (Hamburg, Germany) microcentrifuge tube to yield a final concentration of 2 M (in a 0.5 mL reaction) alongside 1.1 equivalents of malononitrile and 350 μL D2O or 300 μL deionized H2O and 50 μL D2O. After whirl-mixing, the reactant mixture was kept on ice. The reaction was started with the desired amount of triethylamine. For in situ NMR observations, the mixture was rapidly transferred to a 5 mm NMR sample tube prior to tuning and matching, pulse calibration, and manual shimming (approximately 5 min dead time) prior to NMR spectroscopic observations. Control reactions employing only either reactant were tracked using either only glucose or malononitrile in the presence of 0.15 equivalents of triethylamine. Reactions employing isotope-labeled [1-13C]glucose were conducted on a scale of 175 μL in 3 mm NMR tubes. Reactions employing isotope-labeled [2-13C]erythrose were conducted on a scale of 175 μL in 3 mm NMR tubes by dissolving malononitrile in the 0.2 M aqueous substrate solution and conducting the reaction at elevated temperature (313 K) due to lower concentrations.
Mutarotation was followed in a sample of 1 M α-D glucose (500 μL in D2O; 288 K), initially in the absence of base (20 min) and subsequently upon addition of 0.5% (v/v) triethylamine. The mutarotation was followed using a 1D 1H NMR experiment employing excitation (zgesgp pulse sequence) sculpting that was implemented as a pseudo-2D data set with two transients per time point, an acquisition time of 1.3 s and an inter-scan relaxation delay of 1 s, resulting in a time resolution of 4.7 s. The anomeric proton of α-glucopyranose and the H2 of β-glucopyranose were used to follow the approach of equilibrium with 60% β-glucopyranose and 40% α-glucopyranose.
To establish relative reactivities of different naturally abundant aldoses, 1 mmol of glucose, xylose, galactose, and mannose was dissolved in 1.5 mL D2O and incubated at room temperature for 24 h. This mixture was split into stock solutions, to which 0.8 equivalents of either malononitrile or cyanoacetamide were added. A 1H-13C HSQC spectrum was acquired on the stable samples. The reaction was started by the addition of 0.3 equivalents of triethylamine. 1H-13C HSQC spectra were acquired until no further conversion of the carbohydrate mixture was observed to yield the data in Figure 8.
3.3. NMR Spectroscopy
All spectra were acquired on an 800 MHz Bruker (Fällanden, Switzerland) Avance III instrument equipped with an 18.7 T magnet and a 5 mm TCI cryoprobe. A time series of 1D 13C NMR spectra (zgig30) was implemented as a pseudo-2D experiment. An Inter-scan relaxation delay d1 of 1.5 s was used and 32–160 transients (depending on the sensitivity required) were accumulated of the FID sampled during an acquisition time of 0.68 s. This approach results in a time resolution of 1.18–5.9 min. Homo-and heteronuclear 2D NMR assignment spectra employed included 1H-1H COSY (cosygpmfphpp), 1H-1H TOCSY (mlevphpr.2), multiplicity edited 1H-13C HSQC (hsqcedetgpsisp.2), 1H-13C HMBC (hmbcgplpndprqf) and 1H-13C H2BC (h2bcetgpl3). Non-uniform sampling was used to accelerate data acquisitions for instable samples to warrant acquisitions of most 2D experiments within 10 min of experiment time each. A 1H DOSY spectrum was acquired using the LED bipolar gradients (ledbpgp2s) pulse sequence. All spectra were acquired using Bruker Topspin 3.5 pl6. The resultant NMR data were processed in the same software, where integrations were also performed.
3.4. dDNP-NMR Experiments
A substrate sample stock solution was prepared from [1-d,1-13C]glucose (30 mg, 164.7 μmol) in 23.7 mg Milli-Q water and was doped with trityl radical OX063 (GE Health-care, Mw = 1426.77 g/mol, 2.2 mg) and Gd-complex Gadoteridol (Mw = 558.69 g/mol, 1.1 mg of a solution of the Gd-complex at 100 μmol Gd-complex per 1 g Milli-Q water). An amount of 29 mg of the [1-d,1-13C]glucose/radical solution was hyperpolarized to equilibrium polarization in a Spin-Aligner 6.7 T polarizer (Polarize ApS, Frederiksberg, Denmark). After complete hyperpolarization build-up of 1 h, the sample was dissolved in 5 mL Milli-Q water. The concentration of [1-d,1-13C]glucose after dissolution was 15 mM. The dissolved hyperpolarized [1-d,1-13C]glucose was collected in a 50 mL receiver container at the polarizer, and 1 mL was drawn into a 1 mL syringe. This hyperpolarized substrate solution was rapidly transferred to the NMR magnet where the hyperpolarized solution was manually injected into the NMR tube via an inlet line. For reaction tracking on the seconds time scale, 300 μL hyperpolarized solution was injected to 0.129 mmol malononitrile and 25 μL triethylamine in 250 μL Milli-Q water (Merck Millipore, Burlington, MA, USA) incubated in the NMR instrument at 310 K. The NMR data were recorded on a Bruker 500 MHz AVANCE NEO spectrometer equipped with a 5 mm DCH cryoprobe. After injecting the hyperpolarized substrate solution into a solution of malononitrile (8.52 mg, 0.129 mmol) and triethylamine (25 µL, 0.179 mmol) in 250 µL Milli-Q water, a time series of 13C NMR spectra was recorded using a nominal pulse angle of 15° and 1 s time resolution. Each 13C NMR spectrum sampled 21,084 complex data points during an acquisition time of 0.7 s in a single transient.
3.5. Fitting
Kinetic data of reaction tracking using conventional NMR were fitted using pro Fit 7 (Quantum Soft, Uetikon am See, Switzerland). The simplified model used for fitting dDNP data was based on differential equations for unidirectional conversion of glucose and a transient pool of open chain intermediates (compiled as 1) to 2, 3, and 5 as depicted in Scheme 2, while a sequential reaction of 3 to 4 was included. Multiple fits of the dDNP data for compounds 1–5 to a single model were consistent with this model, encompassing the formation of 2, 3, and 5 competitively for the substrate and high-energy intermediate pool. The model was programmed and fitted in Python 3.9.18 via the Anaconda Navigator using the SciPy 1.11.4 pack.
4. Conclusions
In conclusion, state-of-the-art in situ NMR provided insight into the mechanism of upgrading naturally abundant aldoses with CH-acidic nitrile-containing nucleophiles. The unanticipated mechanism entails a rapid Knoevenagel addition that is facilitated by the rapid ring opening in the presence of the Brønsted base. Prior to dehydration, the reactive intermediate formed by Knoevenagel addition cyclizes reversibly via 5-exo-dig ring closure entailing attack of the oxygen on C2 of the aldose onto nitrile carbon. Slower dehydration to complete the Knoevenagel condensation allows the formation of more stable intermediates formed upon concerted 5-exo-dig ring closure and oxa-Michael addition. The resultant bicyclic species mainly consist of two fused furanic rings formed with high stereoselectivity (just one stereoisomer was detected), while the ring closure of the carbohydrate-derived moiety to a six-membered ring and its conversion occurred in parallel to the formation and conversion of the main intermediate. Isotope tracking studies employing both 13C and deuterated solvent supported the mechanism, while previous theoretical considerations had favored a different pathway [36].
Relative rates of the central steps could be derived and could be fitted closely to a sequential model, where subsequently slower reaction steps were observed in the successive conversions of aldose to (i) cyclized Knoevenagel adduct, (ii) bicyclic intermediates formed via concerted Knoevenagel condensation and oxa-Michael addition, and (iii) amine-, nitrile- and polyol-bearing furan product. Advanced NMR methods, including hyperpolarized NMR, indicated that the non-cyclized intermediates deriving from Knoevenagel addition and Knoevenagel condensation do not form species that are stable on the milliseconds time scale at levels of more than 0.04% of the main intermediates containing at least one ring, somewhat resembling the low populations observed of acyclic carbohydrates. Cyclization contributes to the resilience of carbohydrates in sustainable chemistry conversions but appears to play a beneficial role in the chain extension by malononitrile by both protecting reactive nitrile groups and by converting the CH acidic position deriving from malononitrile to a quaternary site. The successful formation of trisubstituted furan competes with minor glucose-to-fructose isomerization in the presence of triethylamine base, with the more critical dimerization of malononitrile, and with the addition of two nucleophiles to the C1 position of the aldose. The reactivity for other naturally abundant aldoses (xylose, mannose, and galactose) was even larger than the reactivity of glucose.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/catal14030199/s1. Figure S1: 1H-1H ROESY acquired rapidly on a reaction conducted with 0.05 equivalents triethylamine; Figure S2: Overlay of 1H-13C HSQC (blue) and 1H-13C HMBC (green) acquired rapidly on a reaction conducted with 0.05 equivalents triethylamine; Figure S3: Time series of 1D 13C NMR spectra in the conversion of glucose and cyanoacetamide; Figure S4: Minor forms formed in parallel to intermediates 2 and 3; Figure S5: Kinetic profiles of signal areas for the bicyclic intermediates show similar kinetic profiles consistent with competing formation and conversion; Figure S6: Fit of dDNP data to a kinetic model; Figure S7: Stability of the individual reactants under reaction conditions.
Author Contributions
Conceptualization, S.M.; methodology, S.S.W., M.K., P.R.J. and S.M.; validation, S.M.; formal analysis, S.S.W., M.K., R.M., P.R.J. and S.M.; investigation, S.S.W., M.K., R.M., P.R.J. and S.M.; resources, P.R.J. and S.M.; data curation, P.R.J. and S.M.; writing—original draft preparation, S.M.; writing—review and editing, S.S.W., R.M., P.R.J. and S.M.; visualization, S.M.; supervision, R.M., P.R.J. and S.M.; project administration, S.M.; funding acquisition, R.M., P.R.J. and S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Independent Research Fund Denmark (grants 0217-00277A and 2035-00119B) and the Villum Foundation (Villum experiment, Project no. 57925). The 800 MHz NMR spectra were recorded at the NMR Center DTU, supported by the Villum Foundation. dDNP NMR data were acquired with equipment partially funded by the Novo Nordisk Foundation (NNF 19OC0055825).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Christensen, C.H.; Rass-Hansen, J.; Marsden, C.C.; Taarning, E.; Egeblad, K. The Renewable Chemicals Industry. ChemSusChem 2008, 1, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Tolborg, S.; Meier, S.; Sádaba, I.; Elliot, S.G.; Kristensen, S.K.; Saravanamurugan, S.; Riisager, A.; Fristrup, P.; Skrydstrup, T.; Taarning, E. Tin-Containing Silicates: Identification of a Glycolytic Pathway via 3-Deoxyglucosone. Green Chem. 2016, 18, 3360–3369. [Google Scholar] [CrossRef]
- Holm, M.S.; Saravanamurugan, S.; Taarning, E. Conversion of Sugars to Lactic Acid Derivatives Using Heterogeneous Zeotype Catalysts. Science 2010, 328, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Taarning, E.; Sádaba, I.; Jensen, P.R.; Meier, S. Discovery and Exploration of the Efficient Acyclic Dehydration of Hexoses in Dimethyl Sulfoxide/Water. ChemSusChem 2019, 12, 5086–5091. [Google Scholar] [CrossRef] [PubMed]
- Elliot, S.G.; Tolborg, S.; Sádaba, I.; Taarning, E.; Meier, S. Quantitative NMR Approach to Optimize the Formation of Chemical Building Blocks from Abundant Carbohydrates. ChemSusChem 2017, 10, 2990–2996. [Google Scholar] [CrossRef]
- Zhang, Z.; Huber, G.W. Catalytic Oxidation of Carbohydrates into Organic Acids and Furan Chemicals. Chem. Soc. Rev. 2018, 47, 1351–1390. [Google Scholar] [CrossRef] [PubMed]
- Dörsam, S.; Fesseler, J.; Gorte, O.; Hahn, T.; Zibek, S.; Syldatk, C.; Ochsenreither, K. Sustainable Carbon Sources for Microbial Organic Acid Production with Filamentous Fungi. Biotechnol. Biofuels 2017, 10, 242. [Google Scholar] [CrossRef]
- Wang, M.; Ma, J.; Liu, H.; Luo, N.; Zhao, Z.; Wang, F. Sustainable Productions of Organic Acids and Their Derivatives from Biomass via Selective Oxidative Cleavage of C–C Bond. ACS Catal. 2018, 8, 2129–2165. [Google Scholar] [CrossRef]
- De Clippel, F.; Dusselier, M.; Van Rompaey, R.; Vanelderen, P.; Dijkmans, J.; Makshina, E.; Giebeler, L.; Oswald, S.; Baron, G.V.; Denayer, J.F.M.; et al. Fast and Selective Sugar Conversion to Alkyl Lactate and Lactic Acid with Bifunctional Carbon–Silica Catalysts. J. Am. Chem. Soc. 2012, 134, 10089–10101. [Google Scholar] [CrossRef]
- Gupta, A.; Verma, J.P. Sustainable Bio-Ethanol Production from Agro-Residues: A Review. Renew. Sustain. Energy Rev. 2015, 41, 550–567. [Google Scholar] [CrossRef]
- Ghosh, D.; Dasgupta, D.; Agrawal, D.; Kaul, S.; Adhikari, D.K.; Kurmi, A.K.; Arya, P.K.; Bangwal, D.; Negi, M.S. Fuels and Chemicals from Lignocellulosic Biomass: An Integrated Biorefinery Approach. Energy Fuels 2015, 29, 3149–3157. [Google Scholar] [CrossRef]
- Sheldon, R.A. Green and Sustainable Manufacture of Chemicals from Biomass: State of the Art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- Dagle, R.A.; Winkelman, A.D.; Ramasamy, K.K.; Lebarbier Dagle, V.; Weber, R.S. Ethanol as a Renewable Building Block for Fuels and Chemicals. Ind. Eng. Chem. Res. 2020, 59, 4843–4853. [Google Scholar] [CrossRef]
- Iroegbu, A.O.; Hlangothi, S.P. Furfuryl Alcohol a Versatile, Eco-Sustainable Compound in Perspective. Chem. Afr. 2019, 2, 223–239. [Google Scholar] [CrossRef]
- Sannelli, F.; Jensen, P.R.; Meier, S. In-Cell NMR Approach for Real-Time Exploration of Pathway Versatility: Substrate Mixtures in Nonengineered Yeast. Anal. Chem. 2023, 95, 7262–7270. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Han, J.; Lee, J. Renewable Butanol Production via Catalytic Routes. Int. J. Environ. Res. Public Health 2021, 18, 11749. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.G.; Meylemans, H.A. The Role of Butanol in the Development of Sustainable Fuel Technologies. J. Chem. Technol. Biotechnol. 2011, 86, 2–9. [Google Scholar] [CrossRef]
- Park, S.-H.; Hahn, J.-S. Development of an Efficient Cytosolic Isobutanol Production Pathway in Saccharomyces Cerevisiae by Optimizing Copy Numbers and Expression of the Pathway Genes Based on the Toxic Effect of α-Acetolactate. Sci. Rep. 2019, 9, 3996. [Google Scholar] [CrossRef]
- Ayoub, M.; Abdullah, A.Z. Critical Review on the Current Scenario and Significance of Crude Glycerol Resulting from Biodiesel Industry towards More Sustainable Renewable Energy Industry. Renew. Sustain. Energy Rev. 2012, 16, 2671–2686. [Google Scholar] [CrossRef]
- Monteiro, M.R.; Kugelmeier, C.L.; Pinheiro, R.S.; Batalha, M.O.; Da Silva César, A. Glycerol from Biodiesel Production: Technological Paths for Sustainability. Renew. Sustain. Energy Rev. 2018, 88, 109–122. [Google Scholar] [CrossRef]
- Binder, J.B.; Blank, J.J.; Cefali, A.V.; Raines, R.T. Synthesis of Furfural from Xylose and Xylan. ChemSusChem 2010, 3, 1268–1272. [Google Scholar] [CrossRef]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; López Granados, M. Furfural: A Renewable and Versatile Platform Molecule for the Synthesis of Chemicals and Fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Meier, S.; Hansen, A.R.; Jensen, P.R. Probing the Role of Individual OH Sites in Carbohydrate Conversion Suggests Strategies for Increasing Product Selectivity and Avoiding Humins. ACS Sustain. Chem. Eng. 2023, 11, 1027–1036. [Google Scholar] [CrossRef]
- Jaswal, A.; Singh, P.P.; Mondal, T. Furfural—A Versatile, Biomass-Derived Platform Chemical for the Production of Renewable Chemicals. Green Chem. 2022, 24, 510–551. [Google Scholar] [CrossRef]
- Xu, C.; Paone, E.; Rodríguez-Padrón, D.; Luque, R.; Mauriello, F. Recent Catalytic Routes for the Preparation and the Upgrading of Biomass Derived Furfural and 5-Hydroxymethylfurfural. Chem. Soc. Rev. 2020, 49, 4273–4306. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, C.; Pong, F.; Sen, A. Chemical Conversion Pathways for Carbohydrates. Green Chem. 2015, 17, 40–71. [Google Scholar] [CrossRef]
- Abou-Yousef, H.; Hassan, E.B. Efficient Utilization of Aqueous Phase Bio-Oil to Furan Derivatives through Extraction and Sugars Conversion in Acid-Catalyzed Biphasic System. Fuel 2014, 137, 115–121. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, K.; Xu, H.; Zhu, L.; Wang, S. A Critical Review of Recent Advances in the Production of Furfural and 5-Hydroxymethylfurfural from Lignocellulosic Biomass through Homogeneous Catalytic Hydrothermal Conversion. Renew. Sustain. Energy Rev. 2021, 139, 110706. [Google Scholar] [CrossRef]
- Brownlee, H.J.; Miner, C.S. Industrial Development of Furfural. Ind. Eng. Chem. 1948, 40, 201–204. [Google Scholar] [CrossRef]
- Garrett, E.R.; Dvorchik, B.H. Kinetics and Mechanisms of the Acid Degradation of the Aldopentoses to Furfural. J. Pharm. Sci. 1969, 58, 813–820. [Google Scholar] [CrossRef]
- Chen, C.; Wang, L.; Zhu, B.; Zhou, Z.; El-Hout, S.I.; Yang, J.; Zhang, J. 2,5-Furandicarboxylic Acid Production via Catalytic Oxidation of 5-Hydroxymethylfurfural: Catalysts, Processes and Reaction Mechanism. J. Energy Chem. 2021, 54, 528–554. [Google Scholar] [CrossRef]
- Lange, J.-P.; van der Heide, E.; van Buijtenen, J.; Price, R. Furfural-A Promising Platform for Lignocellulosic Biofuels. ChemSusChem 2012, 5, 150–166. [Google Scholar] [CrossRef]
- Monrad, R.N.; Madsen, R. Modern Methods for Shortening and Extending the Carbon Chain in Carbohydrates at the Anomeric Center. Tetrahedron 2011, 67, 8825–8850. [Google Scholar] [CrossRef]
- Rodrigues, F.; Canac, Y.; Lubineau, A. A Convenient, One-Step, Synthesis of β-C-Glycosidic Ketones in Aqueous Media. Chem. Commun. 2000, 20, 2049–2050. [Google Scholar] [CrossRef]
- Sato, S.; Naito, Y.; Aoki, K. Scandium Cation-Exchanged Montmorillonite Catalyzed Direct C-Glycosylation of a 1,3-Diketone, Dimedone, with Unprotected Sugars in Aqueous Solution. Carbohydr. Res. 2007, 342, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Ronaghi, N.; Jones, E.V.; Moon, H.J.; Park, S.J.; Jones, C.W.; France, S. Selective Conversion of Malononitrile and Unprotected Carbohydrates to Bicyclic Polyhydroxyalkyl Dihydrofurans Using Magnesium Oxide as a Recyclable Catalyst. ACS Sustain. Chem. Eng. 2022, 10, 5966–5975. [Google Scholar] [CrossRef]
- Zhang, R.; Eronen, A.; Du, X.; Ma, E.; Guo, M.; Moslova, K.; Repo, T. A Catalytic Approach via Retro-Aldol Condensation of Glucose to Furanic Compounds. Green Chem. 2021, 23, 5481–5486. [Google Scholar] [CrossRef]
- Lambu, M.R.; Judeh, Z.M.A. Efficient, One-Step, Cascade Synthesis of Densely Functionalized Furans from Unprotected Carbohydrates in Basic Aqueous Media. Green Chem. 2019, 21, 821–829. [Google Scholar] [CrossRef]
- Ronaghi, N.; Fialho, D.M.; Jones, C.W.; France, S. Conversion of Unprotected Aldose Sugars to Polyhydroxyalkyl and C -Glycosyl Furans via Zirconium Catalysis. J. Org. Chem. 2020, 85, 15337–15346. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, B. Recent Advances in the Chemical Synthesis of C -Glycosides. Chem. Rev. 2017, 117, 12281–12356. [Google Scholar] [CrossRef]
- Scherrmann, M.-C. Knoevenagel Reaction of Unprotected Sugars. In Carbohydrates in Sustainable Development II; Rauter, A.P., Vogel, P., Queneau, Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1–18. ISBN 978-3-64215-161-3. [Google Scholar]
- Gröger, H.; Gallou, F.; Lipshutz, B.H. Where Chemocatalysis Meets Biocatalysis: In Water. Chem. Rev. 2022, 123, 5262–5296. [Google Scholar] [CrossRef] [PubMed]
- Kitanosono, T.; Masuda, K.; Xu, P.; Kobayashi, S. Catalytic Organic Reactions in Water toward Sustainable Society. Chem. Rev. 2018, 118, 679–746. [Google Scholar] [CrossRef]
- Simon, M.-O.; Li, C.-J. Green Chemistry Oriented Organic Synthesis in Water. Chem. Soc. Rev. 2012, 41, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Carraher, J.M.; Fleitman, C.N.; Tessonnier, J.-P. Kinetic and Mechanistic Study of Glucose Isomerization Using Homogeneous Organic Brønsted Base Catalysts in Water. ACS Catal. 2015, 5, 3162–3173. [Google Scholar] [CrossRef]
- Meier, S.; Karlsson, M.; Jensen, P.R. Detecting Elusive Intermediates in Carbohydrate Conversion: A Dynamic Ensemble of Acyclic Glucose–Catalyst Complexes. ACS Sustain. Chem. Eng. 2017, 5, 5571–5577. [Google Scholar] [CrossRef]
- Meier, S. Mechanism and Malleability of Glucose Dehydration to HMF: Entry Points and Water-Induced Diversions. Catal. Sci. Technol. 2020, 10, 1724–1730. [Google Scholar] [CrossRef]
- Zhao, E.W.; Liu, T.; Jónsson, E.; Lee, J.; Temprano, I.; Jethwa, R.B.; Wang, A.; Smith, H.; Carretero-González, J.; Song, Q.; et al. In Situ NMR Metrology Reveals Reaction Mechanisms in Redox Flow Batteries. Nature 2020, 579, 224–228. [Google Scholar] [CrossRef]
- Blasco, T. Insights into Reaction Mechanisms in Heterogeneous Catalysis Revealed by in Situ NMR Spectroscopy. Chem. Soc. Rev. 2010, 39, 4685. [Google Scholar] [CrossRef]
- Bertz, S.H.; Cope, S.; Murphy, M.; Ogle, C.A.; Taylor, B.J. Rapid Injection NMR in Mechanistic Organocopper Chemistry. Preparation of the Elusive Copper(III) Intermediate. J. Am. Chem. Soc. 2007, 129, 7208–7209. [Google Scholar] [CrossRef]
- Voigt, B.; Matviitsuk, A.; Mahrwald, R. Organocatalyzed Knoevenagel-Addition—Simple Access to Carbon Chain-Elongated Branched Carbohydrates. Tetrahedron 2013, 69, 4302–4310. [Google Scholar] [CrossRef]
- Eger, K.; Storz, T.; Spätling, S. The First Condensation Product of Malononitrile with Ribose. Liebigs Ann. Chem. 1989, 1989, 1049. [Google Scholar] [CrossRef]
- Van Beurden, K.; De Koning, S.; Molendijk, D.; Van Schijndel, J. The Knoevenagel Reaction: A Review of the Unfinished Treasure Map to Forming Carbon–Carbon Bonds. Green Chem. Lett. Rev. 2020, 13, 349–364. [Google Scholar] [CrossRef]
- Ardenkjær-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in Signal-to-Noise Ratio of >10,000 Times in Liquid-State NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef]
- Karlsson, M.; Jensen, P.R.; Duus, J.Ø.; Meier, S.; Lerche, M.H. Development of Dissolution DNP-MR Substrates for Metabolic Research. Appl. Magn. Reson. 2012, 43, 223–236. [Google Scholar] [CrossRef]
- Drew, K.N.; Zajicek, J.; Bondo, G.; Bose, B.; Serianni, A.S. 13C-Labeled Aldopentoses: Detection and Quantitation of Cyclic and Acyclic Forms by Heteronuclear 1D and 2D NMR Spectroscopy. Carbohydr. Res. 1998, 307, 199–209. [Google Scholar] [CrossRef]
- Zhu, Y.; Zajicek, J.; Serianni, A.S. Acyclic Forms of [1-13C]Aldohexoses in Aqueous Solution: Quantitation by 13C NMR and Deuterium Isotope Effects on Tautomeric Equilibria. J. Org. Chem. 2001, 66, 6244–6251. [Google Scholar] [CrossRef] [PubMed]
- Isbell, H.S.; Pigman, W. Mutarotation of Sugars in Solution: Part II. In Advances in Carbohydrate Chemistry and Biochemistry; Wolfrom, M.L., Tipson, R.S., Horton, D., Eds.; Academic Press: Cambridge, MA, USA, 1969; Volume 24, pp. 13–65. ISBN 0065-2318. [Google Scholar]
- Petersen, B.O.; Hindsgaul, O.; Meier, S. Profiling of Carbohydrate Mixtures at Unprecedented Resolution Using High-Precision 1H-13C Chemical Shift Measurements and a Reference Library. Analyst 2013, 139, 401–406. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Incorporation of malononitrile and aldose (1) carbons into heterocyclic compounds upon Knoevenagel condensation. A red circle tracks the aldose C1 position, while a grey square tracks the aldose C2 position. The nucleophilic CH acidic methylene group in malononitrile is sequestered as a quaternary carbon in 2, 3, and 4.
Scheme 1.
Incorporation of malononitrile and aldose (1) carbons into heterocyclic compounds upon Knoevenagel condensation. A red circle tracks the aldose C1 position, while a grey square tracks the aldose C2 position. The nucleophilic CH acidic methylene group in malononitrile is sequestered as a quaternary carbon in 2, 3, and 4.

Figure 1.
Time series of 13C NMR spectra showing the conversion of glucose (top) or xylose (bottom) along similar pathways, judging from similar signal positions. Reaction conditions: 1 mmol aldose, 1.1 mmol malononitrile, 0.15 equivalents triethylamine, 288 K, 0.35 mL H2O/D2O (90:10).
Figure 1.
Time series of 13C NMR spectra showing the conversion of glucose (top) or xylose (bottom) along similar pathways, judging from similar signal positions. Reaction conditions: 1 mmol aldose, 1.1 mmol malononitrile, 0.15 equivalents triethylamine, 288 K, 0.35 mL H2O/D2O (90:10).
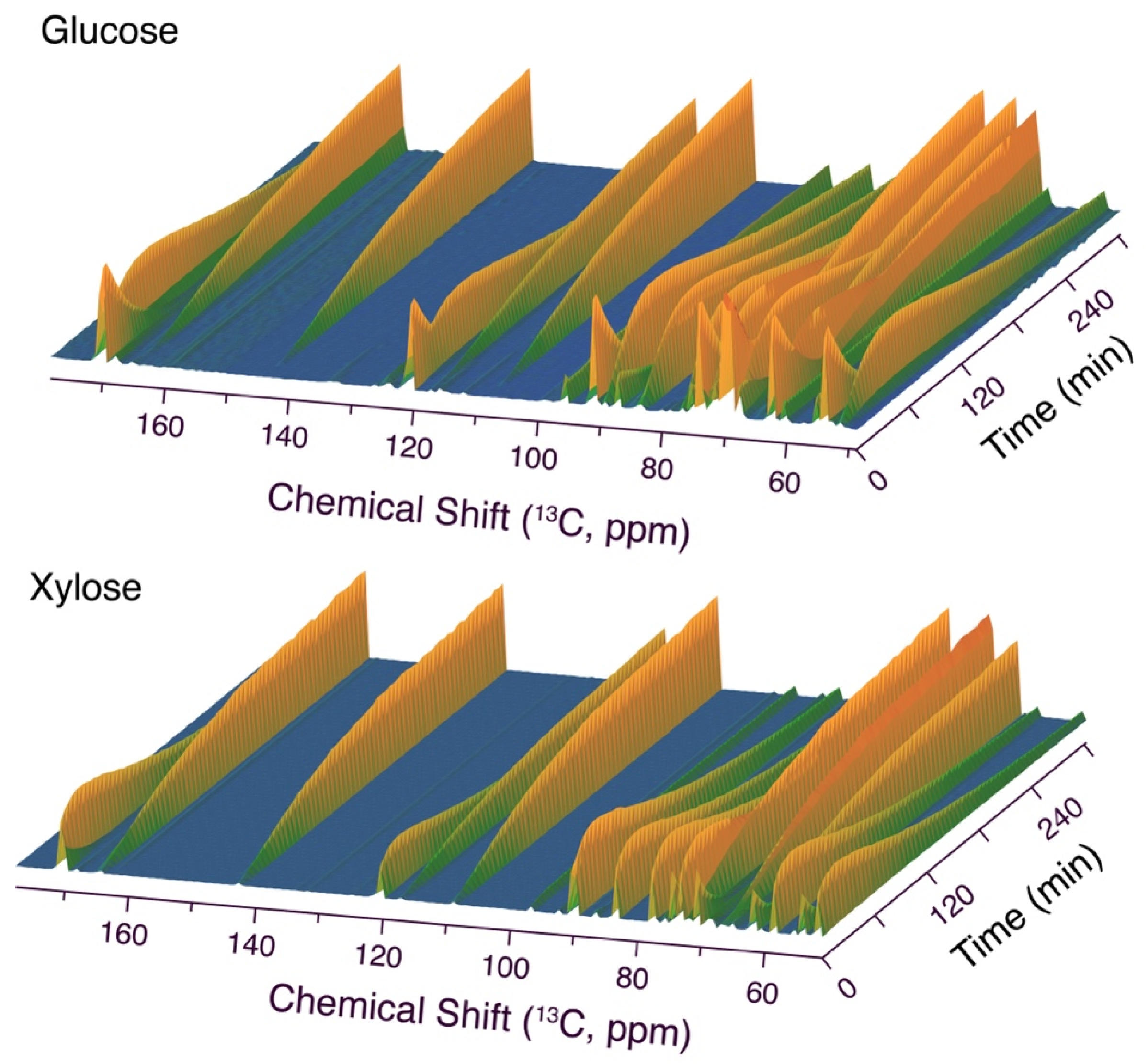
Figure 2.
NMR chemical shift assignments of intermediates and products formed in the reaction of Figure 1. 1H chemical shifts (red) are given above 13C chemical shifts for protonated carbons (blue), ND = not determined.
Figure 2.
NMR chemical shift assignments of intermediates and products formed in the reaction of Figure 1. 1H chemical shifts (red) are given above 13C chemical shifts for protonated carbons (blue), ND = not determined.
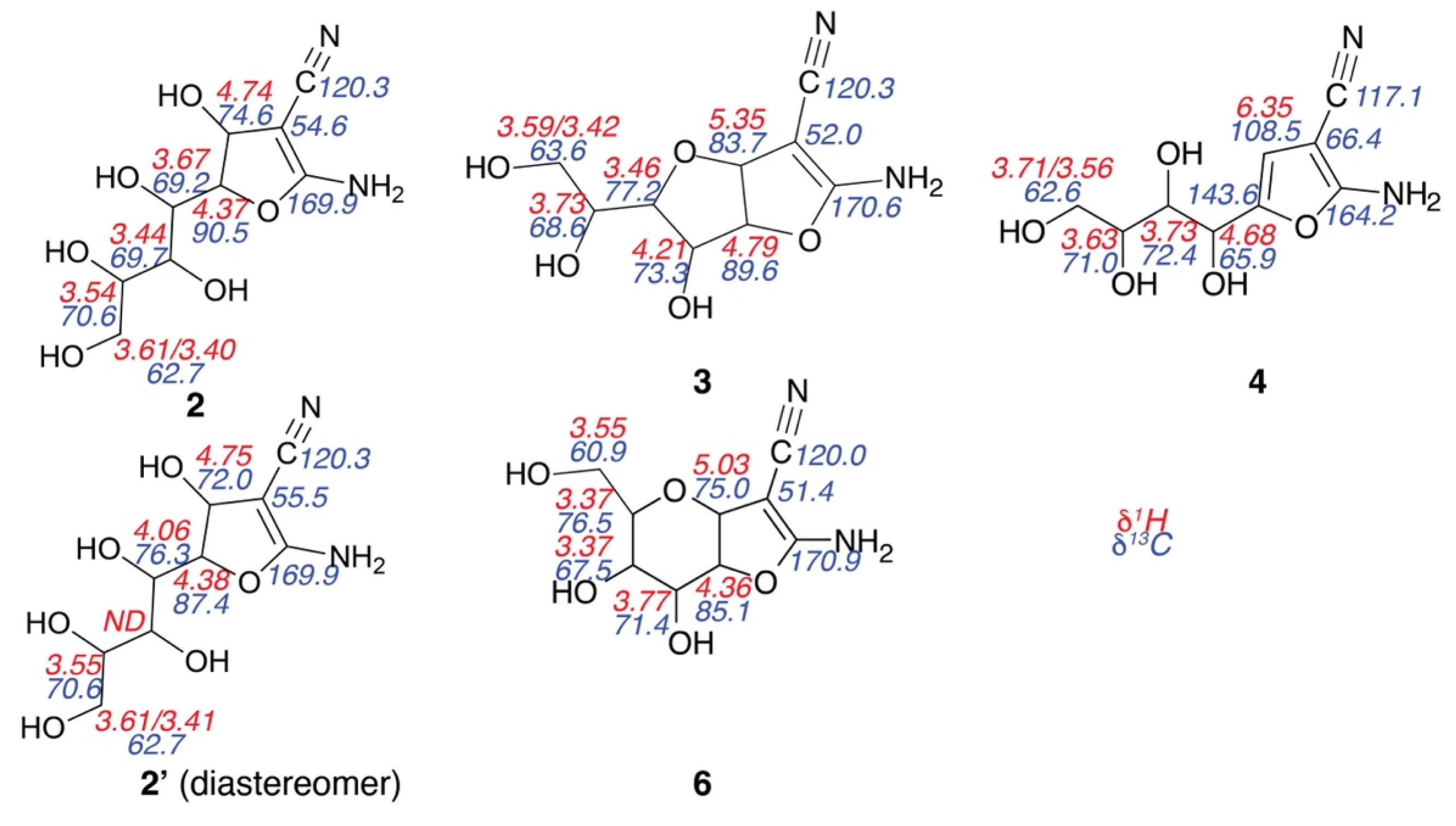
Figure 3.
Time series of 1D 13C NMR spectra in the conversion of [1-13C]glucose (1), validating the assigned positions in 2 (two diastereomeric forms), 3 (formed with high stereoselectivity), and 4. Reaction conditions: 0.35 mmol [1-13C]glucose, 0.33 mmol malononitrile, 0.15 equivalents triethylamine, 288 K, 0.15 mL H2O/0.025 mL D2O.
Figure 3.
Time series of 1D 13C NMR spectra in the conversion of [1-13C]glucose (1), validating the assigned positions in 2 (two diastereomeric forms), 3 (formed with high stereoselectivity), and 4. Reaction conditions: 0.35 mmol [1-13C]glucose, 0.33 mmol malononitrile, 0.15 equivalents triethylamine, 288 K, 0.15 mL H2O/0.025 mL D2O.
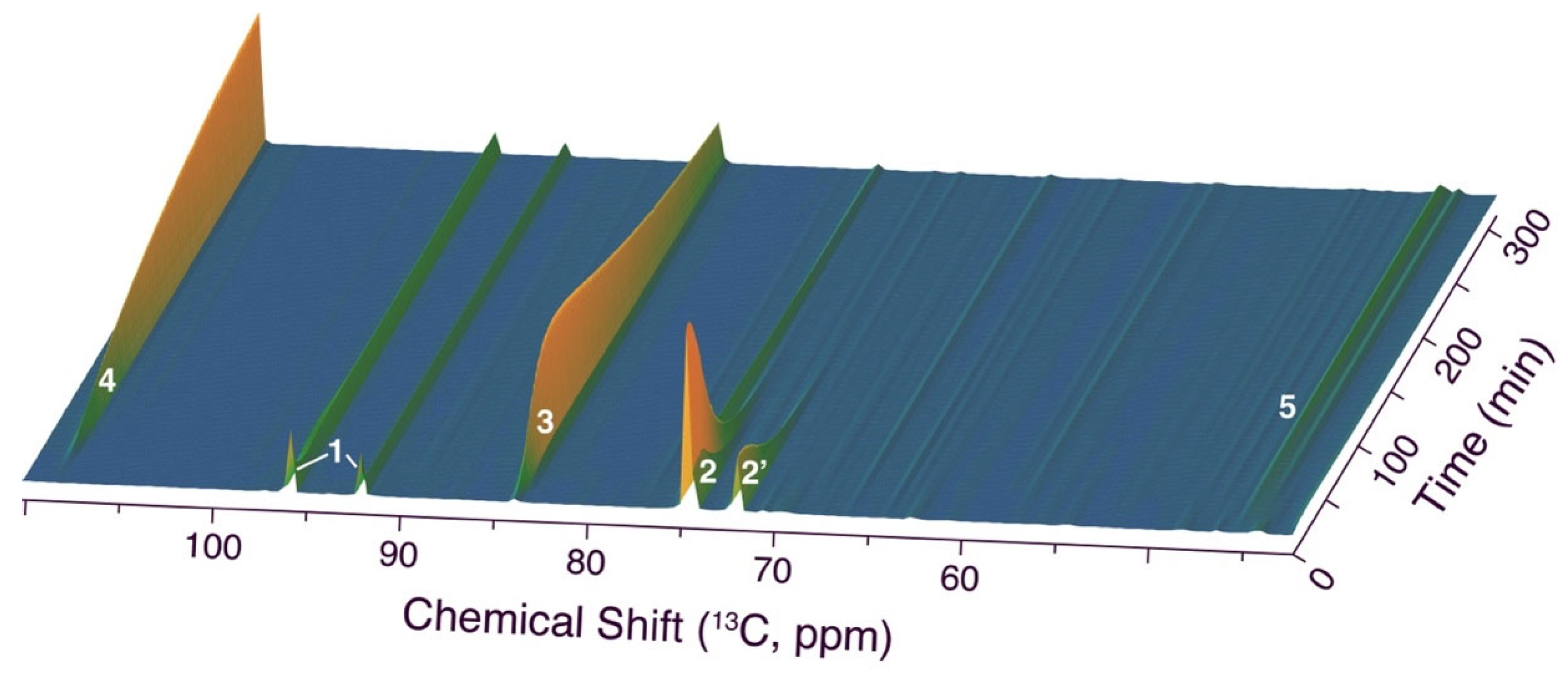
Figure 4.
Time series of 1D 13C NMR spectra in the conversion of [2-13C]erythrose, validating the assigned positions in the main species of Scheme 1. Reaction conditions: 0.03 mmol [2-13C]erythrose, 1.1 equivalents malononitrile, 0.15 equivalents triethylamine, 313 K, 0.15 mL H2O/0.025 mL D2O, 313 K.
Figure 4.
Time series of 1D 13C NMR spectra in the conversion of [2-13C]erythrose, validating the assigned positions in the main species of Scheme 1. Reaction conditions: 0.03 mmol [2-13C]erythrose, 1.1 equivalents malononitrile, 0.15 equivalents triethylamine, 313 K, 0.15 mL H2O/0.025 mL D2O, 313 K.
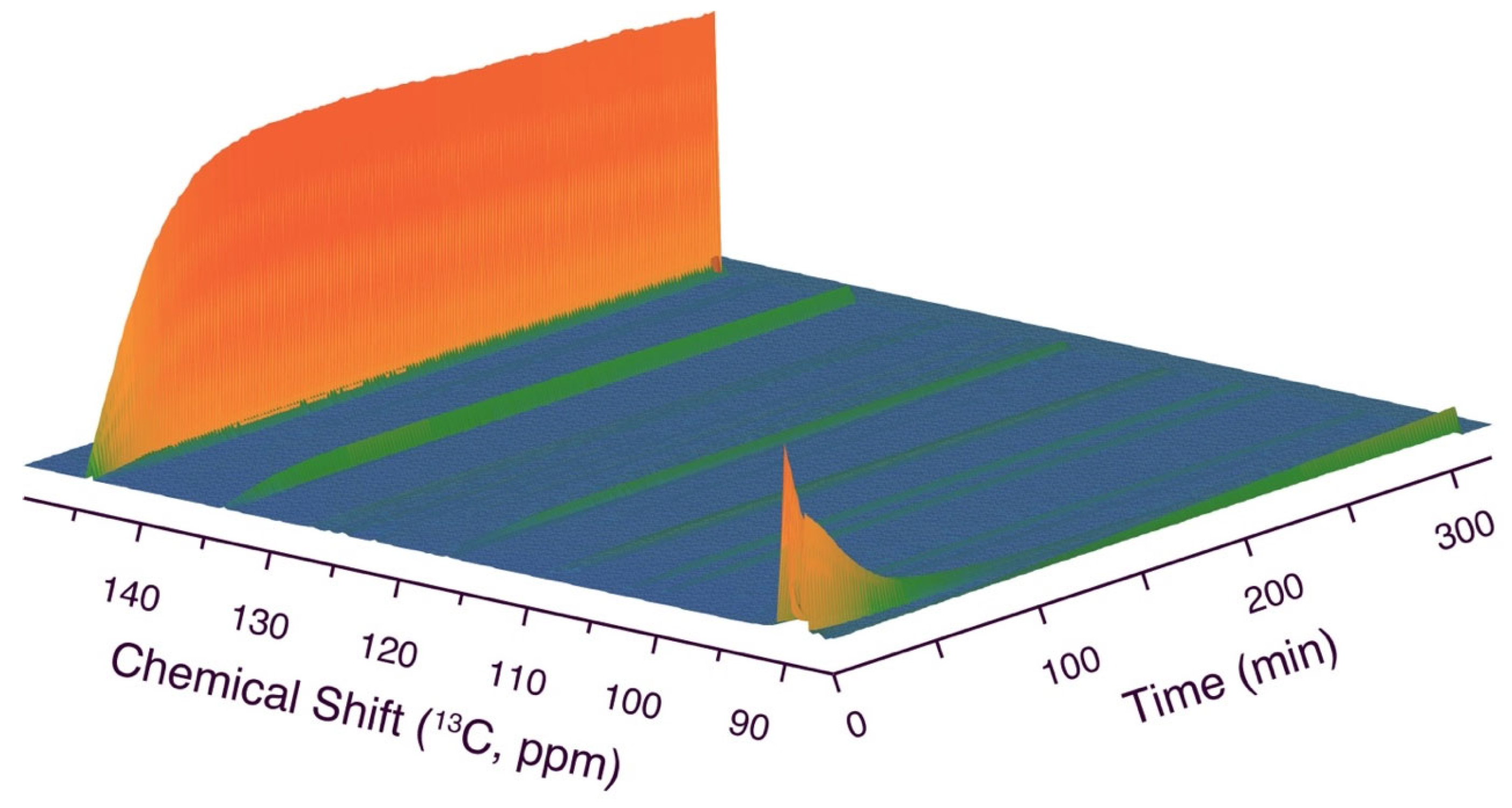
Figure 5.
dDNP NMR spectra in the conversion of hyperpolarized [1-d,1-13C]glucose, validating the rapid formation of 2, its diastereomer 2’ and non-oxygenated, sp3 hybridized carbon from the glucose C1 position(5) in addition to a later intermediate 3. Reaction conditions: Injection of 300 μL 15 mM hyperpolarized [1-d,1-13C]glucose into a solution of 0.13 mmol malononitrile and 25 μL triethylamine in 0.25 mL MilliQ water, 310 K.
Figure 5.
dDNP NMR spectra in the conversion of hyperpolarized [1-d,1-13C]glucose, validating the rapid formation of 2, its diastereomer 2’ and non-oxygenated, sp3 hybridized carbon from the glucose C1 position(5) in addition to a later intermediate 3. Reaction conditions: Injection of 300 μL 15 mM hyperpolarized [1-d,1-13C]glucose into a solution of 0.13 mmol malononitrile and 25 μL triethylamine in 0.25 mL MilliQ water, 310 K.
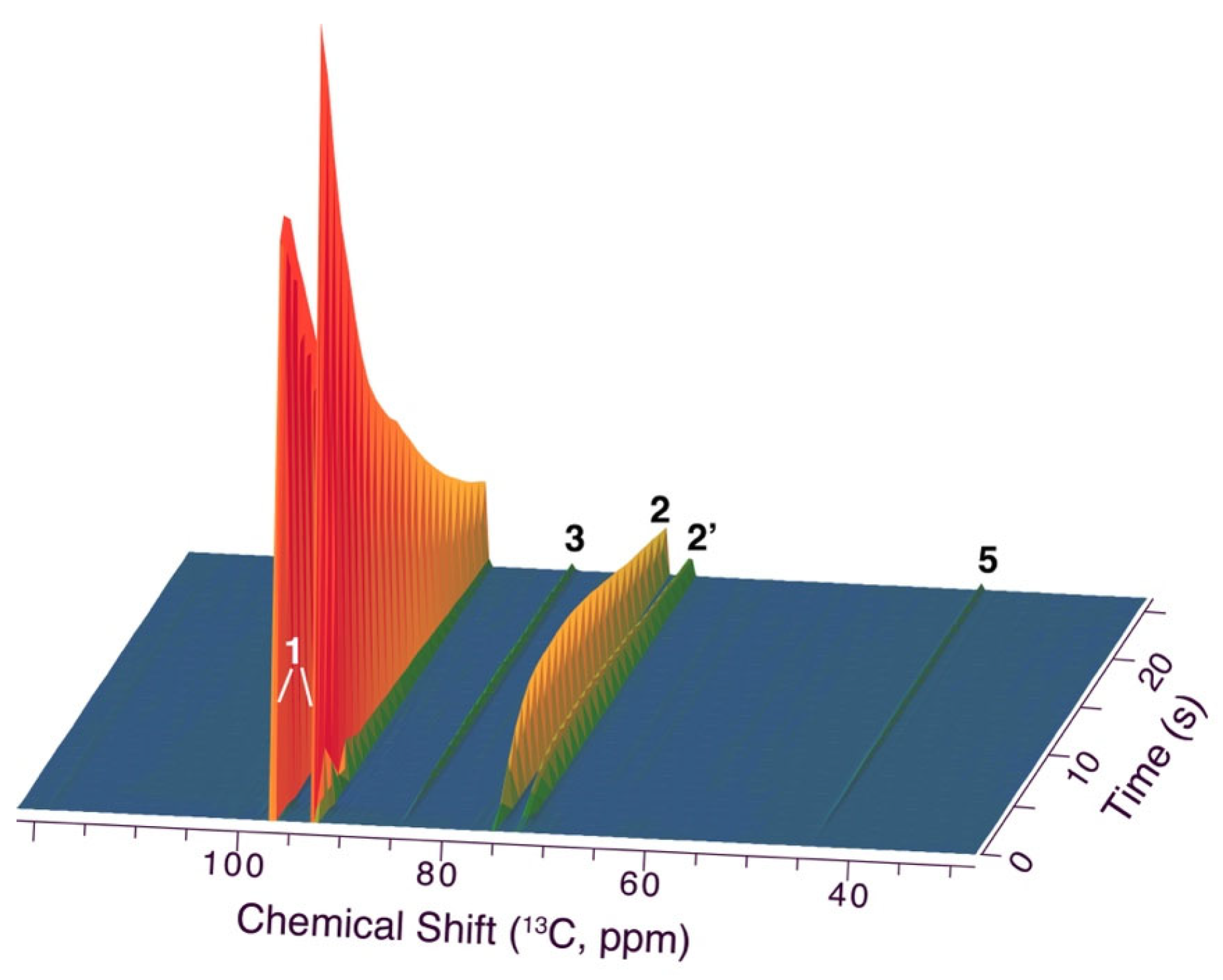
Figure 6.
Zoom onto minor signals in dDNP NMR spectra of Figure 5, showing only very minor alcoholic signals and excluding the formation of acyclic products to more than 0.04% of intermediate 2. Reaction conditions are the same as in Figure 5.

Figure 7.
Addition of 0.5% (v/v) triethylamine accelerates mutarotation and hence ring opening by 360-fold for glucose.
Figure 7.
Addition of 0.5% (v/v) triethylamine accelerates mutarotation and hence ring opening by 360-fold for glucose.
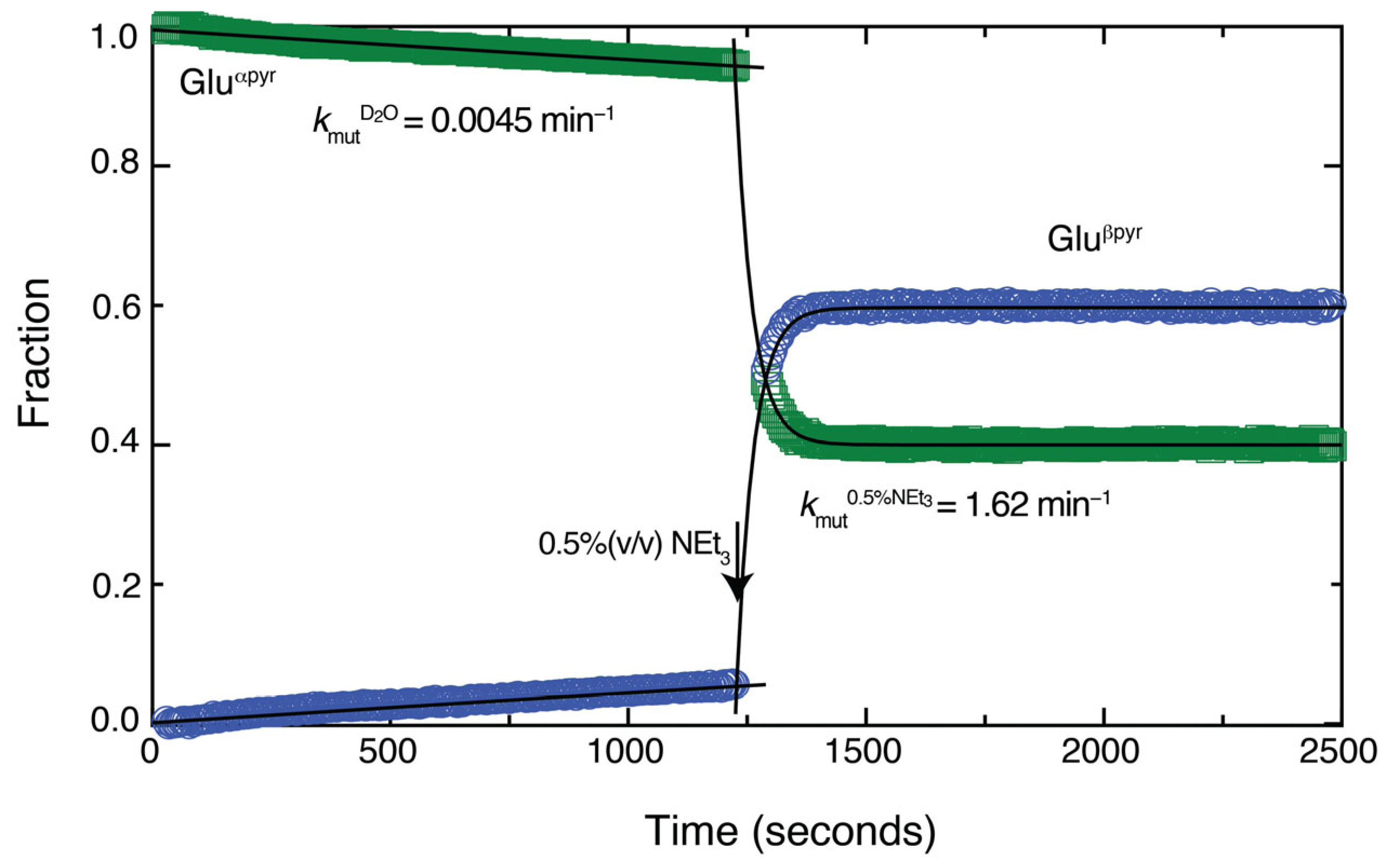
Figure 8.
Competitive assay for ranking the reactivity of naturally abundant aldoses (xylose > galactose, mannose > glucose). Reaction conditions: 0.25 mmol glucose, 0.25 mmol galactose, 0.25 mmol xylose, 0.25 mmol mannose, 0.8 mmol malononitrile, 0.3 equivalents triethylamine, 0.45 mL H2O/0.05 mL D2O, 298 K for malononitrile reaction, 313 K for cyanoacetamide reaction.
Figure 8.
Competitive assay for ranking the reactivity of naturally abundant aldoses (xylose > galactose, mannose > glucose). Reaction conditions: 0.25 mmol glucose, 0.25 mmol galactose, 0.25 mmol xylose, 0.25 mmol mannose, 0.8 mmol malononitrile, 0.3 equivalents triethylamine, 0.45 mL H2O/0.05 mL D2O, 298 K for malononitrile reaction, 313 K for cyanoacetamide reaction.
Scheme 2.
Plausible mechanism for the conversion of glucose and malononitrile to trisubstituted furan based on NMR experimental data. The reaction includes a rapid, reversible 5-exo-dig ring closure to 2 upon Knoevenagel addition, prior to a slower dehydration and intramolecular oxa-Michael addition. CH acidic sites are rapidly sequestered via cyclization and tautomerization.
Scheme 2.
Plausible mechanism for the conversion of glucose and malononitrile to trisubstituted furan based on NMR experimental data. The reaction includes a rapid, reversible 5-exo-dig ring closure to 2 upon Knoevenagel addition, prior to a slower dehydration and intramolecular oxa-Michael addition. CH acidic sites are rapidly sequestered via cyclization and tautomerization.

Figure 9.
(a) Kinetic analysis for the sequential conversion of 2 (blue) to 3 + 6 (purple) and 4 (red) in consecutive steps from glucose. (b) Kinetic analysis for the sequential conversion of xylose to C8 analogs of 2 (blue) to 3 + 6 (purple) and 4 (red).
Figure 9.
(a) Kinetic analysis for the sequential conversion of 2 (blue) to 3 + 6 (purple) and 4 (red) in consecutive steps from glucose. (b) Kinetic analysis for the sequential conversion of xylose to C8 analogs of 2 (blue) to 3 + 6 (purple) and 4 (red).
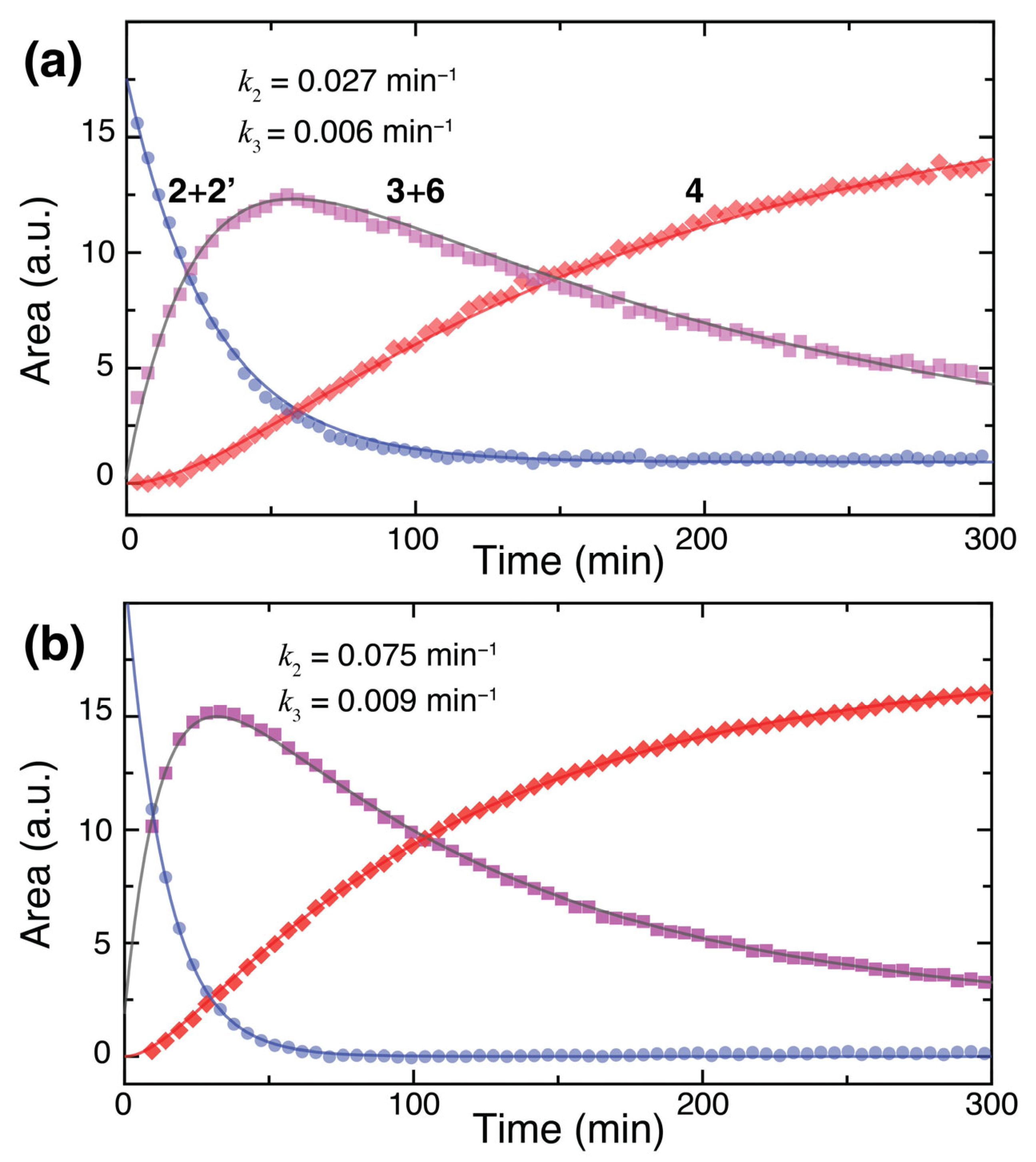
Figure 10.
Formation of product 4 at room temperature using 1.0 equivalents of base as the catalyst indicates biexponential kinetics. Fits to exponential (1. order kinetics) and hyperbolic (2. order kinetics) are shown for comparison. Reaction conditions: 1 mmol aldose, 1.1 mmol malononitrile, 1.0 equivalents triethylamine, 293 K.
Figure 10.
Formation of product 4 at room temperature using 1.0 equivalents of base as the catalyst indicates biexponential kinetics. Fits to exponential (1. order kinetics) and hyperbolic (2. order kinetics) are shown for comparison. Reaction conditions: 1 mmol aldose, 1.1 mmol malononitrile, 1.0 equivalents triethylamine, 293 K.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Warthegau, S.S.; Karlsson, M.; Madsen, R.; Jensen, P.R.; Meier, S. Kinetic and Mechanistic Study of Aldose Conversion to Functionalized Furans in Aqueous Solutions. Catalysts 2024, 14, 199. https://doi.org/10.3390/catal14030199
AMA Style
Warthegau SS, Karlsson M, Madsen R, Jensen PR, Meier S. Kinetic and Mechanistic Study of Aldose Conversion to Functionalized Furans in Aqueous Solutions. Catalysts. 2024; 14(3):199. https://doi.org/10.3390/catal14030199
Chicago/Turabian StyleWarthegau, Stefan S., Magnus Karlsson, Robert Madsen, Pernille Rose Jensen, and Sebastian Meier. 2024. "Kinetic and Mechanistic Study of Aldose Conversion to Functionalized Furans in Aqueous Solutions" Catalysts 14, no. 3: 199. https://doi.org/10.3390/catal14030199
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.