

Synthesis and Catalytic Activity of 1,2-Benzenediamine-Derived Organocatalysts Based on (1R,2R)-Cyclohexane-1,2-Diamine
 , , , and
, , , and 
Abstract
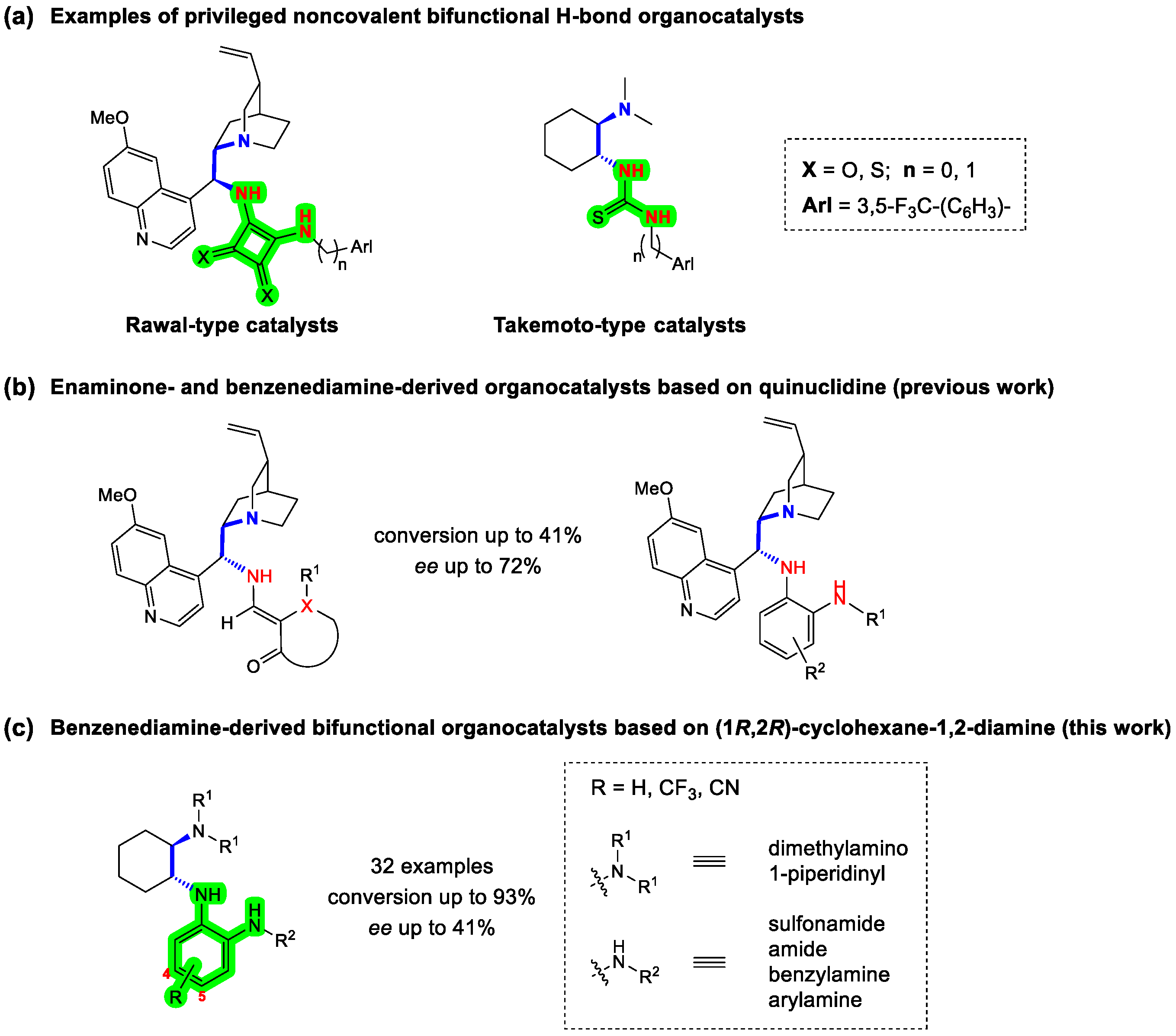
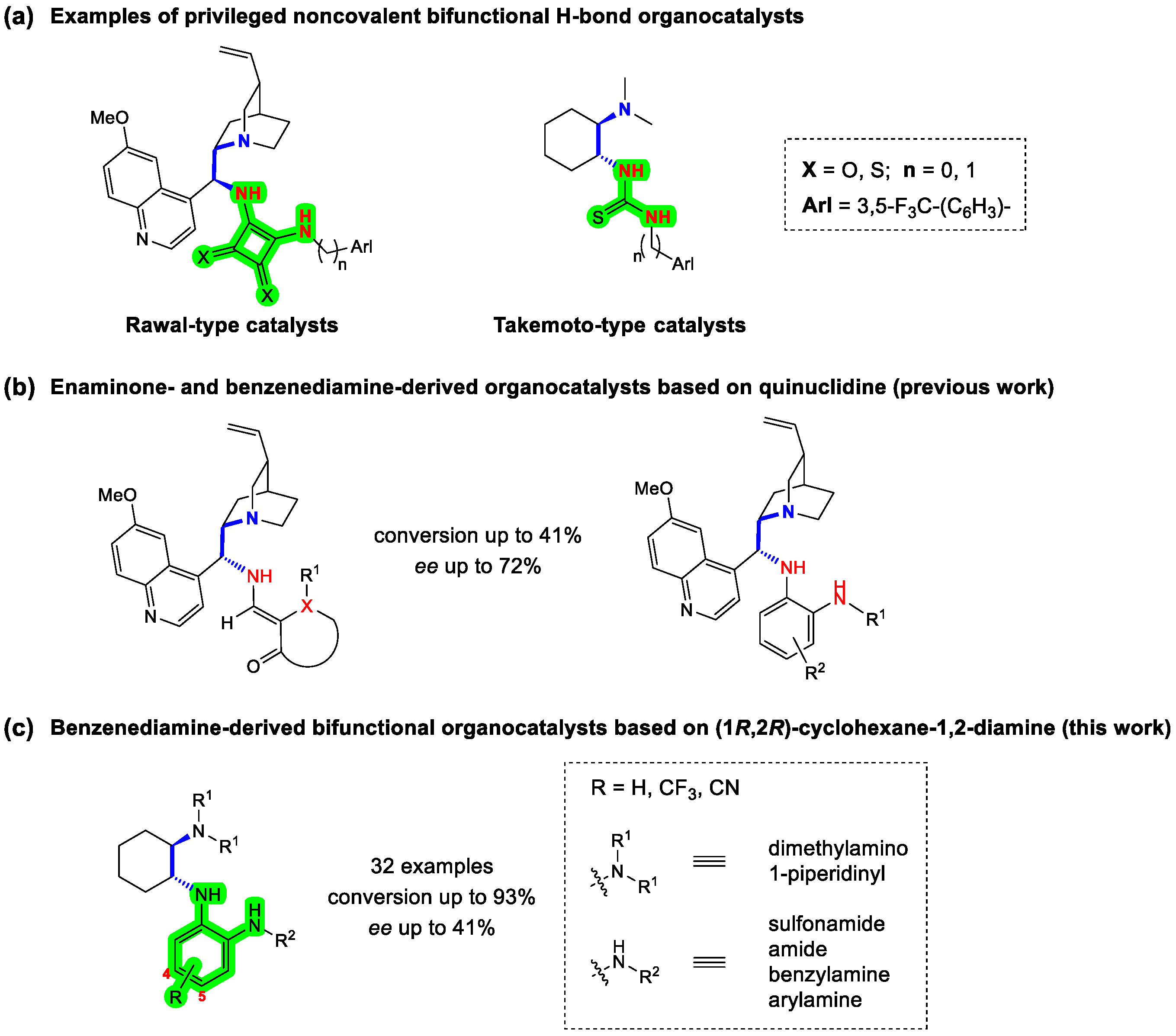
:1. Introduction
2. Results and Discussion
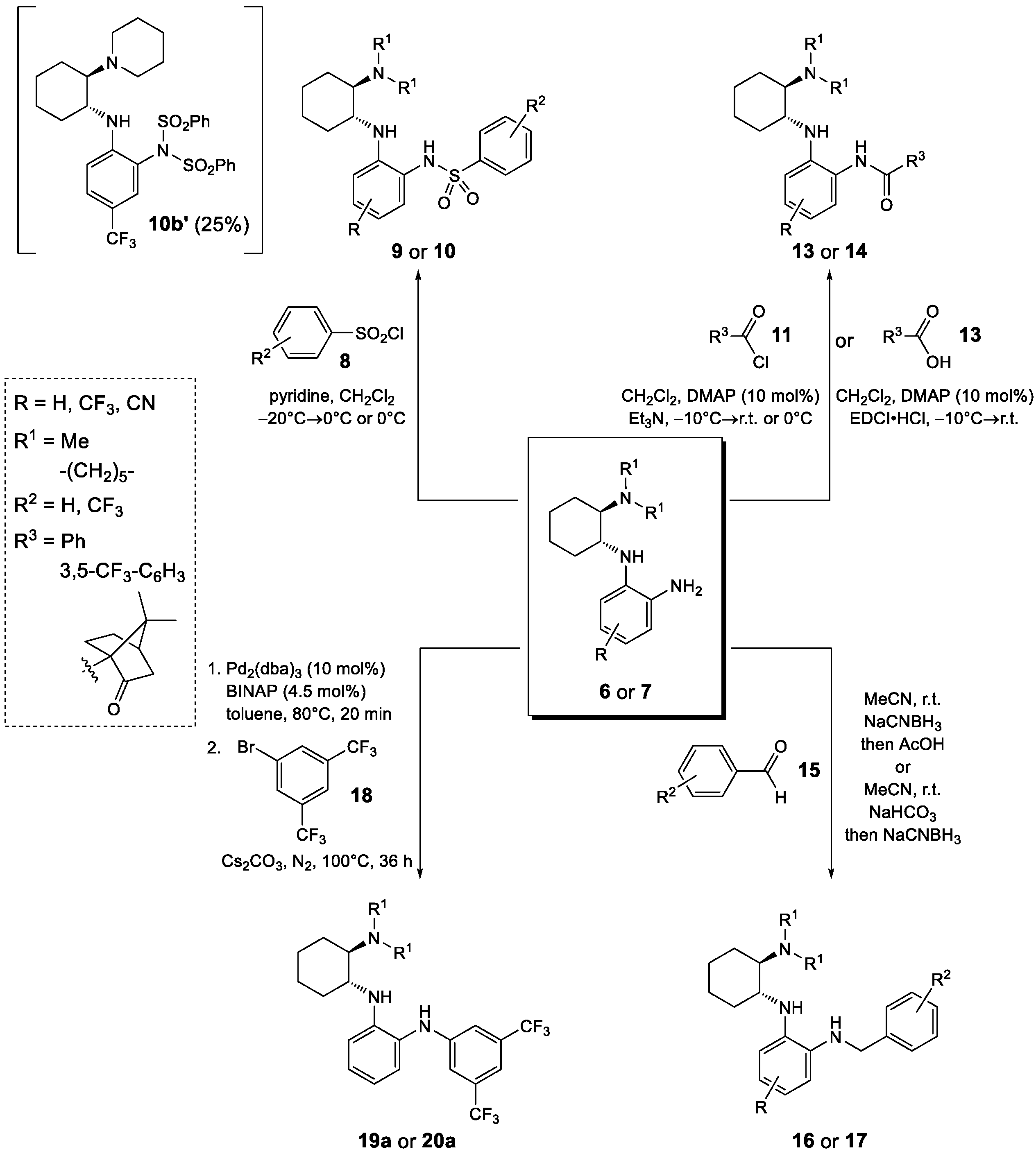
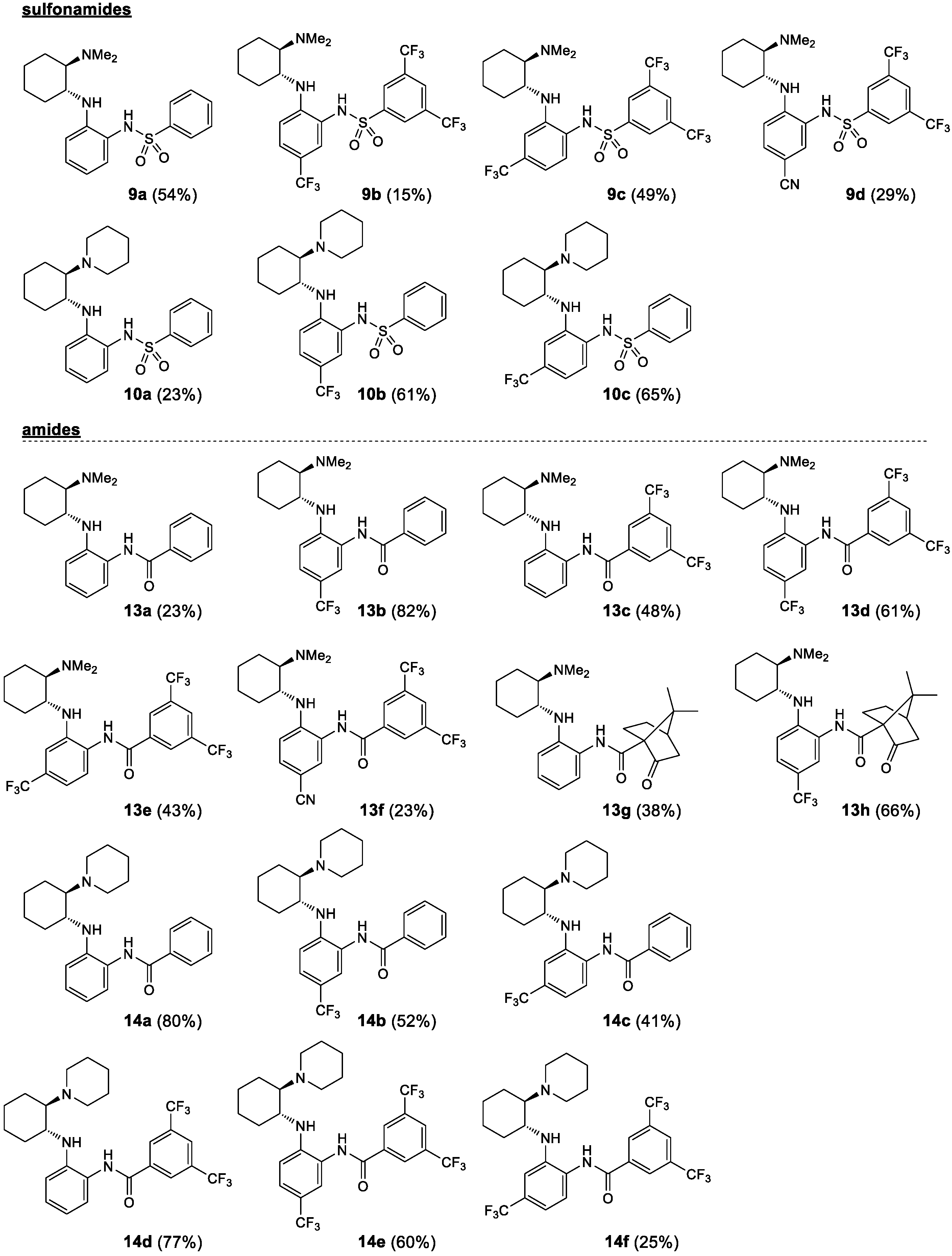
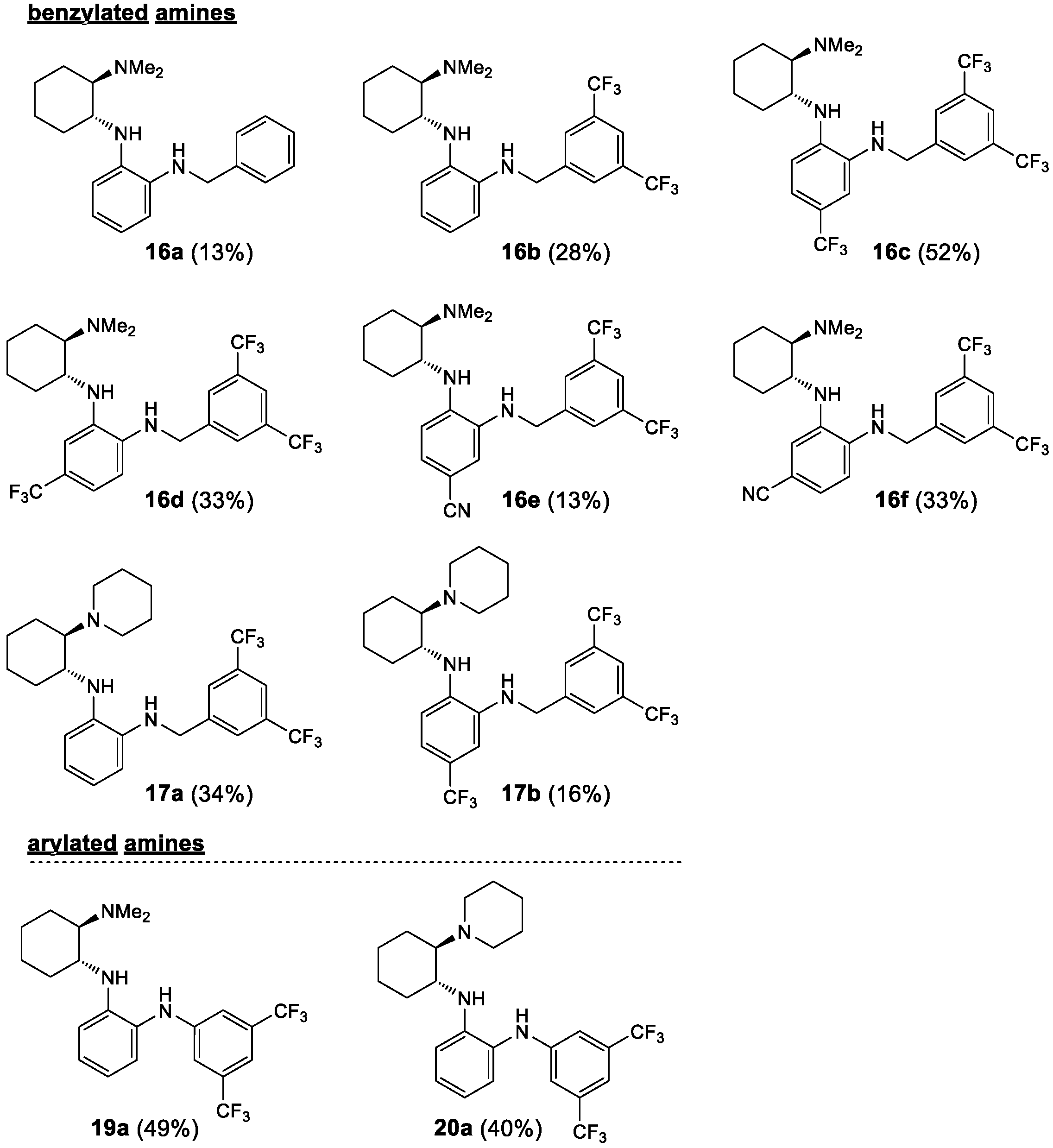
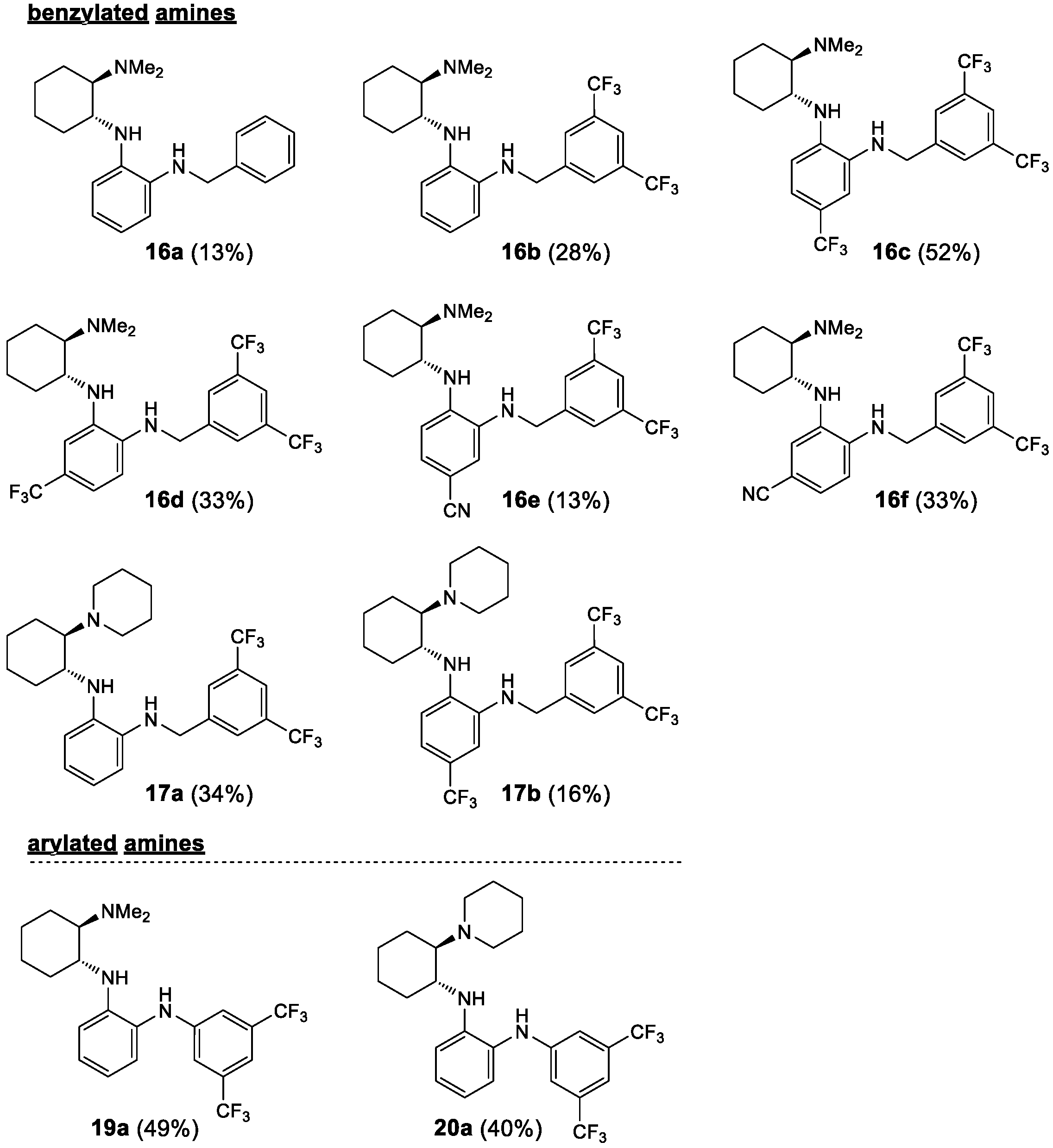
2.1. Synthesis
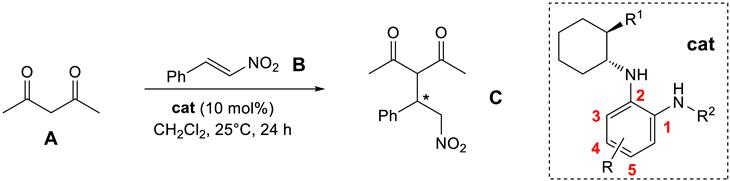
2.2. Organocatalytic Activity
3. Materials and Methods
3.1. Synthesis of (1R,2R)-N1-(2-nitrophenyl)cyclohexane-1,2-diamines 3—General Procedure 1 (GP1)
3.2. Synthesis of (1R,2R)-N1,N1-dimethyl-N2-(2-nitrophenyl)cyclohexane-1,2-diamines 4—General Procedure 2 (GP2)
3.3. Synthesis of 2-nitro-N-((1R,2R)-2-(piperidin-1-yl)cyclohexyl)aniline 5—General Procedure 3 (GP3)
3.4. Reduction of the Aromatic Nitro Group—General Procedure 4 (GP4)
3.5. Synthesis of Benzenesulfonamides 9 and 10—General Procedure 5 (GP5)
3.6. Acylation of the Primary Amine with Benzoyl Chloride—General Procedure 6 (GP6)
3.7. Acylation of the Primary Amine with EDCI-Activated Carboxylic Acid—General Procedure 7 (GP7)
3.8. Reductive Alkylation of the Primary Amine—General Procedure 8 (GP8)
3.9. Reductive Alkylation of the Primary Amine—General Procedure 9 (GP9)
3.10. Arylation of the Primary Amine—General Procedure 10 (GP10)
3.11. Organocatalyzed Addition of Acetylacetone to trans-β-nitrostyrene
3.12. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Jin, Y.; Zhao, R.; Wang, L.; Jin, Y. Organocatalytic Enantioselective Michael Reaction of Aminomaleimides with Nitroolefins Catalyzed by Takemoto’s Catalyst. Molecules 2022, 27, 7787. [Google Scholar] [CrossRef] [PubMed]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [PubMed]
- Rombola, M.; Sumaria, C.S.; Montgomery, T.D.; Rawal, V.H. Development of Chiral, Bifunctional Thiosquaramides: Enantioselective Michael Additions of Barbituric Acids to Nitroalkenes. J. Am. Chem. Soc. 2017, 139, 5297–5300. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.R. (Ed.) Stereoselective Organocatalysis: Bond Formation Methodologies and Activation Modes, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- List, B. Asymmetric Organocatalysis 1. Lewis Base and Acid Catalysis. In Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, Germany, 2012. [Google Scholar]
- Maruoka, K. Asymmetric Organocatalysis 2. Brønsted Base and Acid Catalysis. In Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, Germany, 2012. [Google Scholar]
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Etzenbach-Effers, K.; Berkessel, A. Noncovalent Organocatalysis Based on Hydrogen Bonding: Elucidation of Reaction Paths by Computational Methods. In Asymmetric Organocatalysis. Topics in Current Chemistry; List, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 291. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Prechtl, M.H.G.; Pombeiro, A.J.L. Non-Covalent Interactions in Enantioselective Organocatalysis: Theoretical and Mechanistic Studies of Reactions Mediated by Dual H-Bond Donors, Bifunctional Squaramides, Thioureas and Related Catalysts. Catalysts 2021, 11, 569. [Google Scholar] [CrossRef]
- Kótai, B.; Kardos, G.; Hamza, A.; Farkas, V.; Pápai, I.; Soós, T. On the Mechanism of Bifunctional Squaramide-Catalyzed Organocatalytic Michael Addition: A Protonated Catalyst as an Oxyanion Hole. Chem. Eur. J. 2014, 20, 5631–5639. [Google Scholar] [CrossRef] [PubMed]
- Cassani, C.; Martín-Rapún, R.; Arceo, E.; Bravo, F.; Melchiorre, P. Synthesis of 9-amino(9-deoxy)epi cinchona alkaloids, general chiral organocatalysts for the stereoselective functionalization of carbonyl compounds. Nat. Protoc. 2013, 8, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, Y.; Deng, L. Cinchona Alkaloids. In Privileged Chiral Ligands and Catalysts; Zhou, Q.-L., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Choong, E.S. (Ed.) Cinchona Alkaloids in Synthesis and Catalysis: Ligands, Immobilization and Organocatalysis; VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2009. [Google Scholar] [CrossRef]
- Kopyt, M.; Głowacki, M.P.; Kwiatkowski, P. trans-1,2-Diaminocyclohexane and Its Derivatives in Asymmetric Organocatalysis. In Chiral Building Blocks in Asymmetric Synthesis; Wojaczyńska, E., Wojaczyński, J., Eds.; Wiley-VCH GmbH: Weinheim, Germany, 2022. [Google Scholar] [CrossRef]
- Hirashima, S.-I.; Arai, R.; Nakashima, K.; Kawai, N.; Kondo, J.; Koseki, Y.; Miura, T. Asymmetric Hydrophosphonylation of Aldehydes using a Cinchona–Diaminomethylenemalononitrile Organocatalyst. Adv. Synth. Catal. 2015, 357, 3863–3867. [Google Scholar] [CrossRef]
- Arai, R.; Hirashima, S.-i.; Kondo, J.; Nakashima, K.; Koseki, V.; Miura, T. Cinchona–Diaminomethylenemalononitrile Organocatalyst for the Highly Enantioselective Hydrophosphonylation of Ketones and Enones. Org. Lett. 2018, 20, 5569–5572. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Taniguchi, Y.; Hayama, N.; Inokuma, T.; Takemoto, Y. A Powerful Hydrogen-Bond-Donating Organocatalyst for the Enantioselective Intramolecular Oxa-Michael Reaction of α,β-Unsaturated Amides and Esters. Angew. Chem. Int. Ed. 2013, 52, 11114–11118. [Google Scholar] [CrossRef]
- Maddox, S.M.; Dawson, G.A.; Rochester, N.C.; Ayonon, A.B.; Moore, C.E.; Rheingold, A.L.; Gustafson, J.L. Enantioselective Synthesis of Biaryl Atropisomers via the Addition of Thiophenols into Aryl-Naphthoquinones. ACS Catal. 2018, 8, 5443–5447. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, J.; Lu, Y. Highly Enantioselective Michael Addition of 2-Fluoro-1,3-diketones to Nitroalkenes. Eur. J. Org. Chem. 2015, 2015, 320–324. [Google Scholar] [CrossRef]
- Ding, M.; Zhou, F.; Liu, Y.-L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. Cinchona alkaloid-based phosphoramide catalyzed highly enantioselective Michael addition of unprotected 3-substituted oxindoles to nitroolefins. Chem. Sci. 2011, 2, 2035–2039. [Google Scholar] [CrossRef]
- Žabka, M.; Šebesta, R. Experimental and Theoretical Studies in Hydrogen-Bonding Organocatalysis. Molecules 2015, 20, 15500–15524. [Google Scholar] [CrossRef] [PubMed]
- Vera, S.; García-Urricelqui, A.; Mielgo, A.; Oiarbide, M. Progress in (Thio)urea- and Squaramide-Based Brønsted Base Catalysts with Multiple H-Bond Donors. Eur. J. Org. Chem. 2023, 26, e202201254. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Chai, Z.; Zhao, G. Novel bifunctional thiourea–ammonium salt catalysts derived from amino acids: Application to highly enantio-and diastereoselective aza-Henry reaction. Tetrahedron 2013, 69, 5104–5111. [Google Scholar] [CrossRef]
- Novacek, J.; Waser, M. Syntheses and Applications of (Thio) Urea-Containing Chiral Quaternary Ammonium Salt Catalysts. Eur. J. Org. Chem. 2014, 2014, 802–809. [Google Scholar] [CrossRef]
- Wang, H. Chiral Phase-Transfer Catalysts with Hydrogen Bond: A Powerful Tool in the Asymmetric Synthesis. Catalysts 2019, 9, 244. [Google Scholar] [CrossRef]
- Waser, M.; Winter, M.; Mairhofer, C. (Thio)urea containing chiral ammonium salt catalysts. Chem. Rec. 2023, 23, e202200198. [Google Scholar] [CrossRef]
- Ciber, L.; Požgan, F.; Brodnik, H.; Štefane, B.; Svete, J.; Grošelj, U. Synthesis and Catalytic Activity of Organocatalysts Based on Enaminone and Benzenediamine Hydrogen Bond Donors. Catalysts 2022, 12, 1132. [Google Scholar] [CrossRef]
- Boratyński, P.J.; Nowak, A.E.; Skarżewski, J. New Chiral Benzimidazoles Derived from 1,2-Diaminocyclohexane. Synthesis 2015, 47, 3797–3804. [Google Scholar] [CrossRef]
- Isobe, T.; Oriyama, T. Ring-opening reaction of aziridines with amines under the influence of dimethyl sulfoxide. Tetrahedron Lett. 2016, 57, 2849–2852. [Google Scholar] [CrossRef]
- Sutherlin, D.; Mckerrall, S.; Wilson, M.S.; Lai, K.W.; Bergeron, P.; Zhang, B. Therapeutic Compounds and Methods of Use Thereof. US 2018/0105504 A1, 19 April 2018. [Google Scholar]
- Gilli, P.; Pretto, L.; Bertolasi, V.; Gilli, G. Predicting Hydrogen-Bond Strengths from Acid−Base Molecular Properties. The pKa Slide Rule: Toward the Solution of a Long-Lasting Problem. Acc. Chem. Res. 2009, 42, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K.M.; Schreiner, P.R. (Thio)urea Organocatalyst Equilibrium Acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Li, X.; Cheng, J.-P. Equilibrium acidities of cinchona alkaloid organocatalysts bearing 6′-hydrogen bonding donors in DMSO. Org. Chem. Front. 2016, 3, 170–176. [Google Scholar] [CrossRef]
- Ni, X.; Li, X.; Wang, Z.; Cheng, J.-P. Squaramide Equilibrium Acidities in DMSO. Org. Lett. 2014, 16, 1786–1789. [Google Scholar] [CrossRef]
- Rostami, A.; Sadeh, E.; Ahmadi, S. Exploration of tertiary aminosquaramide bifunctional organocatalyst in controlled/living ring-opening polymerization of l-lactide. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2483–2493. [Google Scholar] [CrossRef]
- Dai, J.; Xiong, W.; Li, D.-Y.; Cai, Z.; Zhu, J.-B. Bifunctional thiourea-based organocatalyst promoted kinetic resolution polymerization of racemic lactide to isotactic polylactide. Chem. Commun. 2023, 59, 12731–12734. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, N.R.; Turner, P.; Todd, M.H. The First Catalytic, Enantioselective Aza-Henry Reaction of an Unactivated Cyclic Imine. Adv. Synth. Catal. 2012, 354, 2954–2958. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Finney, N.S. An efficient method for the preparation of N,N-disubstituted 1,2-diamines. Tetrahedron Lett. 2000, 41, 8431–8434. [Google Scholar] [CrossRef]
- Yin, H.; Lewis, A.J.; Williams, U.J.; Carroll, P.J.; Schelter, E.J. Fluorinated diarylamide complexes of uranium(iii, iv) incorporating ancillary fluorine-to-uranium dative interactions. Chem. Sci. 2013, 4, 798–805. [Google Scholar] [CrossRef]
- Das, T.; Mohapatra, S.; Mishra, N.P.; Nayak, S.; Raiguru, B.P. Recent Advances in Organocatalytic Asymmetric Michael Addition Reactions to α,β-Unsaturated Nitroolefins. ChemistrySelect 2021, 6, 3745–3781. [Google Scholar] [CrossRef]
- Kucharski, D.J.; Suchanek, R.; Kowalczyk, R.; Boratyński, P.J. Development of Mefloquine-Based Bifunctional Secondary Amine Organocatalysts for Enantioselective Michael and Friedel–Crafts Reactions. J. Org. Chem. 2024, 89, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Ričko, S.; Svete, J.; Štefane, B.; Perdih, A.; Golobič, A.; Meden, A.; Grošelj, U. 1,3-Diamine-Derived Bifunctional Organocatalyst Prepared from Camphor. Adv. Synth. Catal. 2016, 358, 3786–3796. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Furukawa, F.; Xu, X.; Takemoto, Y. Enantio- and Diastereoselective Michael Reaction of 1,3-Dicarbonyl Compounds to Nitroolefins Catalyzed by a Bifunctional Thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Du, D.-M. Chiral Squaramide-Catalyzed Highly Enantioselective Michael Addition of 2-Hydroxy-1,4-naphthoquinones to Nitroalkenes. Adv. Synth. Catal. 2011, 353, 1241–1246. [Google Scholar] [CrossRef]
- Narayanaperumal, S.; Rivera, D.G.; Silva, R.C.; Paixão, M.W. Terpene-Derived Bifunctional Thioureas in Asymmetric Organocatalysis. ChemCatChem 2013, 5, 2756–2773. [Google Scholar] [CrossRef]
- CrysAlis PRO; Agilent Technologies UK Ltd.: Yarnton, UK, 2011.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cristallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Catalyst | R | R1 | R2 | Conversion (%) ee (%) (a) | |
| 1 |  | 98 93 (R) | |||
| 2 | 9a | H | Me2N | Ph-SO2 | 82 26 (S) |
| 3 | 9b | 5-CF3 | Me2N | 3,5-(CF3)2-C6H3-SO2 | 11 41 (S) |
| 4 | 9c | 4-CF3 | Me2N | 3,5-(CF3)2-C6H3-SO2 | 14 26 (S) |
| 5 | 9d | 5-CN | Me2N | 3,5-(CF3)2-C6H3-SO2 | 13 19 (S) |
| 6 | 10a | H | 1-piperidyl | Ph-SO2 | 90 23 (S) |
| 7 | 10b | 5-CF3 | 1-piperidyl | Ph-SO2 | 16 23 (S) |
| 8 | 10b′ | 5-CF3 | 1-piperidyl | 2 × PhSO2 (b) | 0 - |
| 9 | 10c | 4-CF3 | 1-piperidyl | Ph-SO2 | 40 0 |
| 10 | 13a | H | Me2N | Ph-CO | 83 24 (S) |
| 11 | 13b | 5-CF3 | Me2N | Ph-CO | 84 17 (S) |
| 12 | 13c | H | Me2N | 3,5-(CF3)2-C6H3-CO | 85 26 (S) |
| 13 | 13d | 5-CF3 | Me2N | 3,5-(CF3)2-C6H3-CO | 86 32 (S) |
| 14 | 13e | 4-CF3 | Me2N | 3,5-(CF3)2-C6H3-CO | 83 32 (S) |
| 15 | 13f | 5-CN | Me2N | 3,5-(CF3)2-C6H3-CO | 50 33 (S) |
| 15 | 13g | H | Me2N |  | 13 27 (S) |
| 17 | 13h | 5-CF3 | Me2N |  | 9 23 (S) |
| 18 | 14a | H | 1-piperidyl | Ph-CO | 76 14 (S) |
| 19 | 14b | 5-CF3 | 1-piperidyl | Ph-CO | 22 20 (S) |
| 20 | 14c | 4-CF3 | 1-piperidyl | Ph-CO | 15 28 (S) |
| 21 | 14d | H | 1-piperidyl | 3,5-(CF3)2-C6H3-CO | 93 13 (S) |
| 22 | 14e | 5-CF3 | 1-piperidyl | 3,5-(CF3)2-C6H3-CO | 13 11 (S) |
| 23 | 14f | 4-CF3 | 1-piperidyl | 3,5-(CF3)2-C6H3-CO | 36 22 (S) |
| 24 | 16a | H | Me2N | Ph-CH2 | 93 3 (S) |
| 25 | 16b | H | Me2N | 3,5-(CF3)2-C6H3-CH2 | 56 3 (S) |
| 26 | 16c | 5-CF3 | Me2N | 3,5-(CF3)2-C6H3-CH2 | 36 11 (S) |
| 27 | 16d | 4-CF3 | Me2N | 3,5-(CF3)2-C6H3-CH2 | 20 1 (S) |
| 28 | 16e | 5-CN | Me2N | 3,5-(CF3)2-C6H3-CH2 | 11 17 (S) |
| 29 | 16f | 4-CN | Me2N | 3,5-(CF3)2-C6H3-CH2 | 27 9 (S) |
| 30 | 17a | H | 1-piperidyl | 3,5-(CF3)2-C6H3-CH2 | 12 - (c) |
| 31 | 17b | 4-CF3 | 1-piperidyl | 3,5-(CF3)2-C6H3-CH2 | 11 6 (S) |
| 32 | 19a | H | Me2N | 3,5-(CF3)2-C6H3 | 38 27 (S) |
| 33 | 20a | H | 1-piperidyl | 3,5-(CF3)2-C6H3 | 39 27 (S) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciber, L.; Klemenčič, K.; Golob, A.; Brodnik, H.; Požgan, F.; Svete, J.; Štefane, B.; Grošelj, U. Synthesis and Catalytic Activity of 1,2-Benzenediamine-Derived Organocatalysts Based on (1R,2R)-Cyclohexane-1,2-Diamine. Catalysts 2024, 14, 274. https://doi.org/10.3390/catal14040274
Ciber L, Klemenčič K, Golob A, Brodnik H, Požgan F, Svete J, Štefane B, Grošelj U. Synthesis and Catalytic Activity of 1,2-Benzenediamine-Derived Organocatalysts Based on (1R,2R)-Cyclohexane-1,2-Diamine. Catalysts. 2024; 14(4):274. https://doi.org/10.3390/catal14040274
Chicago/Turabian StyleCiber, Luka, Klara Klemenčič, Ana Golob, Helena Brodnik, Franc Požgan, Jurij Svete, Bogdan Štefane, and Uroš Grošelj. 2024. "Synthesis and Catalytic Activity of 1,2-Benzenediamine-Derived Organocatalysts Based on (1R,2R)-Cyclohexane-1,2-Diamine" Catalysts 14, no. 4: 274. https://doi.org/10.3390/catal14040274