1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a class of volatile organic compounds (VOCs), that when released into the atmosphere have been identified as detrimental to the environment and harmful to health [
1,
2]. Since 1998, PAHs have also been specifically listed as persistent organic pollutants, and identified as compounds that must be eliminated from the atmosphere [
3]. As a result, legislation has been put in place to reduce their emissions through mechanisms such as European Union Directives, and similar control measures also exist in many other regions of the world. PAHs are released from many combustion processes, and the broader classification of VOCs are emitted even more widely from a very large number of sources. A number of techniques are available to control the release of organic atmospheric pollutants, amongst these catalytic oxidation offers a number of significant advantages over competing options, as it provides a selective and low energy pathway to produce benign products [
4]. Both metal oxide and noble metal-based catalysts have been widely investigated for total oxidation of VOCs. However, more specifically the total oxidation of PAHs have not been studied so extensively, but there are a number of studies in the literature, which have investigated the total oxidation of naphthalene as a model PAH [
5]. Naphthalene is recognised as a suitable model PAH as it is the major PAH produced in many combustion processes and it is easy to handle due to its relatively low toxicity.
Previously, we have investigated naphthalene total oxidation over a range of catalysts and a number of simple metal oxides have demonstrated high activity. For example, recently manganese oxide has been identified as a potentially effective catalyst [
6]. However, the most active metal oxide catalysts identified were those based around ceria. These can be prepared relatively simply using precipitation with urea, and activity structure relationships have been established that nanocrystalline ceria with a small crystallite size, high surface area and a high concentration of oxygen defects leads to high activity for naphthalene total oxidation [
7,
8]. Ceria can also form the basis of high activity PAH oxidation catalysts, by preparation using nanocasting to prepare high surface area mesoporous structures with improved accessibility [
9]. Whilst modification of ceria by incorporation of another element, such as zirconium [
10] or copper [
11] into the ceria lattice also improve catalyst activity for naphthalene oxidation.
Early studies on catalytic total oxidation of PAHs showed that metal-supported catalysts were more effective than metal oxide catalysts available at the time. For the metal-based catalysts Pt was identified as being more active than Pd and Ru, with Pt-supported on γ-Al
2O
3 being the most active catalyst [
12,
13]. Considering the identification of Pt as the most active component for naphthalene total oxidation and the high activity exhibited by nanocrystalline ceria, it may be thought that Pt supported on nanocrystalline ceria would be particularly effective. However, the addition of Pt on to nanocrystalline ceria actually suppresses the total oxidation of naphthalene [
14]. These findings demonstrate that the interaction between the support and the Pt is an important factor in controlling the total oxidation of naphthalene. In a previous study of naphthalene oxidation we investigated the role of the support for a 0.5 wt.% Pt-based catalyst [
15]. The supports investigated were SIO
2, γ-Al
2O
3, CeO
2, TiO
2 and SnO
2, and the SiO
2 supported catalyst was the most active, and remains one of the most active catalysts reported in the literature for naphthalene total oxidation. The metal-support interaction (MSI) of Pt and silica was relatively weak, and surface Pt existed in the metallic and oxidised states. CO chemisorption studies revealed that the Pt crystallite size was relatively large, with a low dispersion. Considering the other supports investigated, it was found that silica was the only support which formed a significant concentration of surface metallic Pt in combination with oxidised Pt. For example, Pt/CeO
2 displayed predominantly surface oxidised Pt species and was amongst the least active of the Pt catalysts investigated in the study.
Hence, the present work aims to build on our previous study of the preparation and activity of 0.5 wt.% Pt/SiO2. In the current work both the influence of Pt loading and the variation of calcination temperature have been systematically studied and their influence on catalyst activity established.
2. Results and Discussion
The steady-state oxidation of naphthalene to CO
2 as a function of temperature over the SiO
2 supported catalysts containing varying wt.% Pt are shown in
Figure 1. The blank experiment for oxidation of 100 vppm naphthalene using an empty reactor tube with a total gas flow of 50 mL min
−1 has been measured previously [
16]. Naphthalene conversion was not detectable below 350 °C, whilst it was <2% at 350 °C and increased to around 20% at 400 °C. Experiments with the silica-based catalysts in this study have been carried out up to 300 °C, and therefore conversion of naphthalene is attributed to surface initiated reactions, with the contribution from purely gas phase homogeneous reactions negligible.
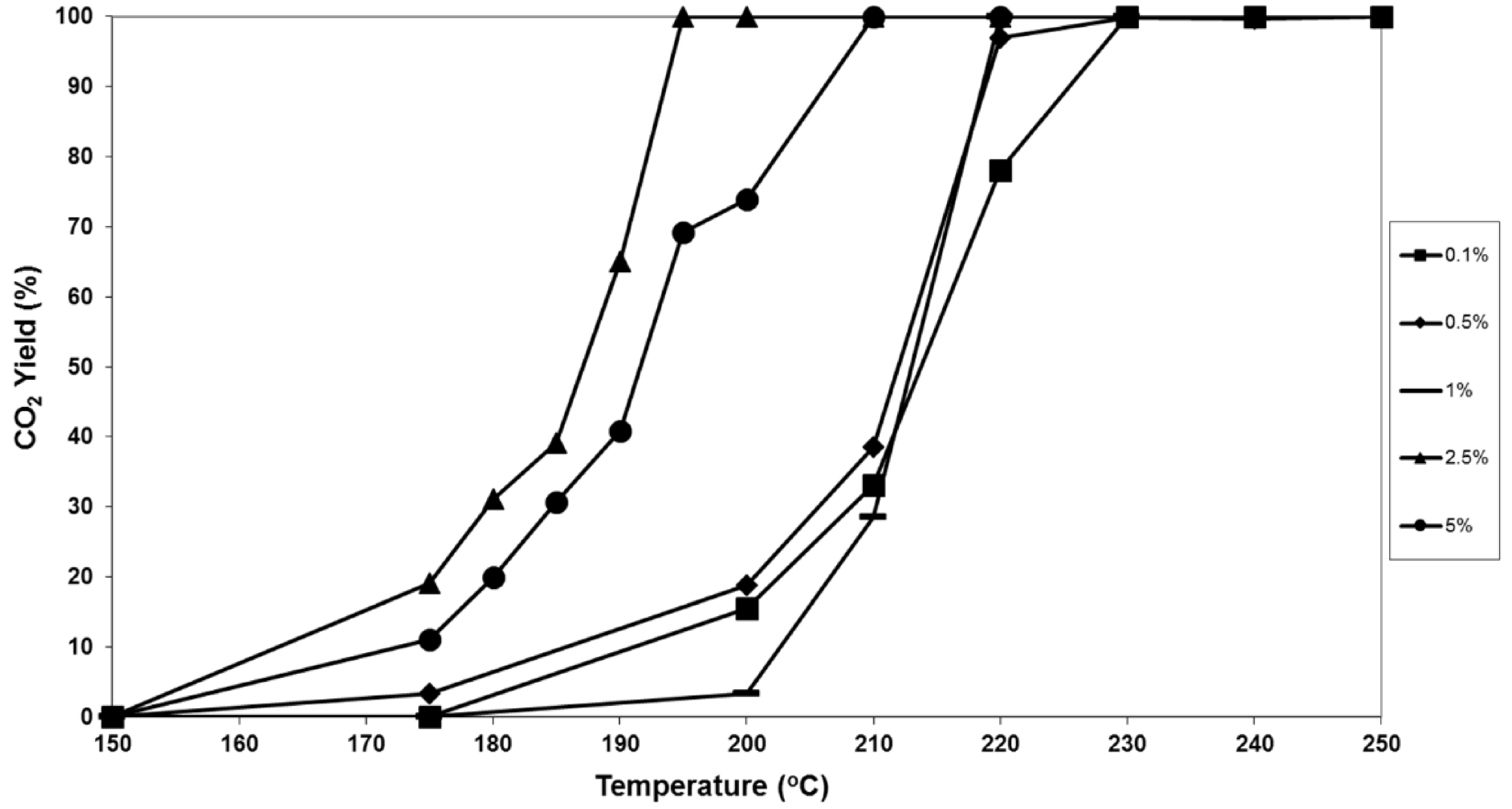
Figure 1.
Steady-state conversion of naphthalene to CO2 as a function of temperature for catalysts with varying platinum loadings on a silica support: ■ 0.1 wt.% Pt; ♦ 0.5 wt.% Pt; ▬ 1.0 wt.% Pt; ▲ 2.5 wt.% Pt; ● 5.0 wt.% Pt.
Figure 1.
Steady-state conversion of naphthalene to CO2 as a function of temperature for catalysts with varying platinum loadings on a silica support: ■ 0.1 wt.% Pt; ♦ 0.5 wt.% Pt; ▬ 1.0 wt.% Pt; ▲ 2.5 wt.% Pt; ● 5.0 wt.% Pt.
The data in
Figure 1 shows the conversion of naphthalene to CO
2 rather than the naphthalene conversion. The reason for using the CO
2 yield is that naphthalene is a polycyclic aromatic molecule, which can be readily adsorbed onto the surface of the catalyst, potentially leading to over estimation of oxidation activity, especially at lower temperatures [
17]. Furthermore, the use of total Naphthalene conversion may also lead to erroneous conclusions due to the possible formation of by-products [
13]. Hence, the yield to CO
2 and not the total conversion of naphthalene is a better parameter to establish the total oxidation activity of the catalysts. However, with the Pt/SiO
2 catalysts used in this study the carbon balances were always close to 100%, and the conversion to CO
2 and naphthalene conversion were virtually the same. These observations indicate that selectivity to CO
2 from naphthalene oxidation was around 100%, and there was little or no formation of partially oxidized products under our reaction conditions.
The Pt loading had a significant influence on the catalyst activity for naphthalene total oxidation (
Figure 1). The catalysts were not active below 150 °C, but started to show trace conversion to CO
2 at 150 °C. The catalysts containing 0.1, 0.5 and 1.0 wt.% Pt all showed broadly similar naphthalene oxidation activity, and these catalysts were significantly less active than the 2.5 and 5.0 wt.% catalysts. The 0.5 and 1.0 Pt wt.% catalysts demonstrated total (or near total) conversion of naphthalene to CO
2 around 220 °C. The 0.1 Pt wt.% catalyst, with the lowest Pt loading, exhibited some mass transport limited characteristics at higher conversion (as conversion did not rise so steeply to 100%), and total naphthalene conversion was only observed at 230 °C. This observation could be related to the higher surface area of the catalyst, as it most likely has the highest micropore volume. The most active catalyst was 2.5 wt.% Pt, followed closely by 5.0 wt.%. Total oxidation of naphthalene to CO
2 was achieved at 190 °C for 2.5 wt.% Pt, whilst it was around 210 °C for the 5.0 wt.% catalyst, as it exhibited some mass transport limitations at higher naphthalene conversion, the reasons for this are unclear at this stage. The order of catalyst activity can be summarised as:
Table 1 shows a summary of the characterisation and activity data (T
50 and T
100, representing temperatures for 50% and 100% naphthalene conversion to CO
2 respectively, for the range of silica supported catalysts with varying Pt loading. The BET surface area increased slightly as a low loading of Pt was added to the SiO
2 support, and this is associated with the process of aqueous impregnation and drying. As the loading of Pt was increased there was a decrease of the catalyst surface area, and this would be expected as a consequence of some pore filling by the deposition of the Pt. The Pt dispersion of the catalysts was relatively low, and this is consistent with previous findings for Pt/SiO
2 catalysts prepared by the same method [
15]. The greatest Pt dispersion of 28% was observed for the 0.1 Pt/SiO
2 catalyst, and as Pt loading was increased there was a decrease of Pt dispersion. The catalysts containing 2.5 and 5.0 wt.% Pt had very low Pt dispersion of 4 and 3% respectively. CO chemisorption data were also used to calculate average Pt particles sizes, and in accordance with the decrease of dispersion with increasing Pt loading, average particle sizes also increased as Pt loading increased. The average Pt size increased from around 40 Å to 90 Å as Pt loading increased from 0.1 to 1.0 wt.%. When the Pt loading was increased to 2.5 and 5.0 wt.% the average Pt particle size increased significantly to around 280 Å and 740 Å respectively. The number of surface Pt sites also changed as a function of Pt loading, increasing from 0.1 wt.% to a maximum for the 1.0 wt.% Pt/SiO
2 catalyst. When the Pt loading was increased above 1 wt.% there was a decrease of the number of Pt surface sites.
It is interesting to consider the activity of the catalysts in relation to the data for Pt dispersion, average particle size and the number of Pt surface sites. There is no simple relationship between the number of Pt surface sites and total oxidation activity of naphthalene. As Pt loading increased from 0.1 to 1.0 wt.% activity increased, as did the number of surface Pt sites. However the number of surface sites for the 1.0 wt.% catalyst was significantly greater than for the 0.5 wt.% catalyst, but activity was very similar. Furthermore, the number of surface Pt sites decreased as the Pt loading increased from 1.0 to 2.5 and then 5.0 wt.%, but the naphthalene total oxidation activity increased markedly. Significantly the more active 2.5 and 5.0 wt.% catalysts have far larger average Pt particle sizes than the lower loading catalysts, indicating that relatively large Pt particle sizes are required for greater activity.
Table 1.
Summary of characterisation data and naphthalene oxidation performance for Pt/SiO2 catalysts with varying Pt loading prepared by calcination in static air at 550 °C for 6 h (ramp rate 10 °C min−1).
Table 1.
Summary of characterisation data and naphthalene oxidation performance for Pt/SiO2 catalysts with varying Pt loading prepared by calcination in static air at 550 °C for 6 h (ramp rate 10 °C min−1).
| Pt content/wt.% | BET surface area/m2g−1 | Pt dispersion a/% | Average Pt particle size a/Å | No. Pt sites (×1018) a/g | Surface Pt0:Pt2+:Pt4+ ratio b | Catalytic performance |
|---|
| T50 c/°C | T100 c/°C |
|---|
| 0 | 431 | - | - | - | - | - | - |
| 0.1 | 450 | 28 | 41 | 0.88 | Not Detected | 213 | 230 |
| 0.5 | 443 | 19 | 61 | 2.63 | 1.8:1.1:1.0 | 211 | 230 |
| 1 | 430 | 12 | 91 | 3.88 | 1.4:2.3:1.0 | 212 | 220 |
| 2.5 | 436 | 4 | 276 | 3.44 | 9.1:1.6:1.0 | 187 | 195 |
| 5 | 417 | 3 | 736 | 2.38 | 6.2:1.2:1.0 | 191 | 210 |
The nature of the surface Pt species, and how the relative proportions of different species may vary as a function of Pt loading, can also influence the activity of the catalyst. Accordingly the range of catalysts were characterised using X-ray photoelectron spectroscopy (XPS) and data are summarised in
Table 1. For catalysts containing low loadings (<0.5 wt.%) of Pt it was not possible to analyse the Pt species present due to their very low surface concentration. For the catalysts for which it was possible to measure the Pt surface species, it was evident that Pt was present in a range of oxidation states, comprising Pt
0, Pt
2+ and Pt
4+. These general findings are consistent with our previous work [
15], as we identified the presence of significant concentrations of metallic Pt along with oxidised Pt on the surface of a Pt/SiO
2 catalyst. Conversely, Pt catalysts prepared by the same method, but supported on TiO
2, CeO
2 and γ-Al
2O
3 only exhibited oxidised surface Pt. The Pt/SiO
2 catalyst was considerably more active than the catalysts prepared using the other supports, suggesting that the presence of metallic Pt, possibly in combination with oxidised Pt is important for naphthalene total oxidation. For the 0.5 and 1.0 wt.% Pt/SiO
2 catalysts the majority of the Pt measured was in the +2 and +4 oxidised states, whilst for the higher loadings of 2.5 and 5.0 wt.% the majority of Pt was in the metallic state, although significant concentrations of Pt
2+ and Pt
4+ were also detected. The relative concentration of Pt
0 was greater for the 2.5 wt.% catalyst when compared to the 5.0 wt.% catalyst. These data indicate that the presence of metallic Pt is again important, as the most active catalyst with 2.5 wt.% had the highest concentration of surface Pt
0.
It is difficult to draw too many definitive conclusions between the catalyst structure and activity for the catalysts with different Pt loadings, as Pt dispersion, total number of surface Pt sites, Pt particle size, and the relative and absolute number of surface metallic and oxidised Pt sites all change as a function of loading. However, it is clear that the more active catalysts contain higher loadings of Pt, and these catalysts have very low Pt dispersion, relatively large Pt particles and greater surface concentrations of metallic Pt. Hence, these characteristics are required for more effective naphthalene total oxidation catalysts. In order to try and develop a stronger link between the characteristics of the Pt nanoparticles and naphthalene oxidation, future studies focusing on a range of catalysts containing between 1.0 and 2.5 wt.% Pt would be informative.
In order to try to investigate further the relationship between catalyst structure and activity for naphthalene total oxidation, a range of catalysts were prepared by varying the calcination temperature. As the 2.5 wt.% Pt catalyst was the most active from the study varying the Pt loading, this loading was selected and catalysts were prepared by calcining from 450 to 750 °C.
The steady-state oxidation of naphthalene to CO
2 as a function of temperature over the SiO
2 supported catalysts containing 2.5 wt.% Pt prepared by calcination at different temperatures are shown in
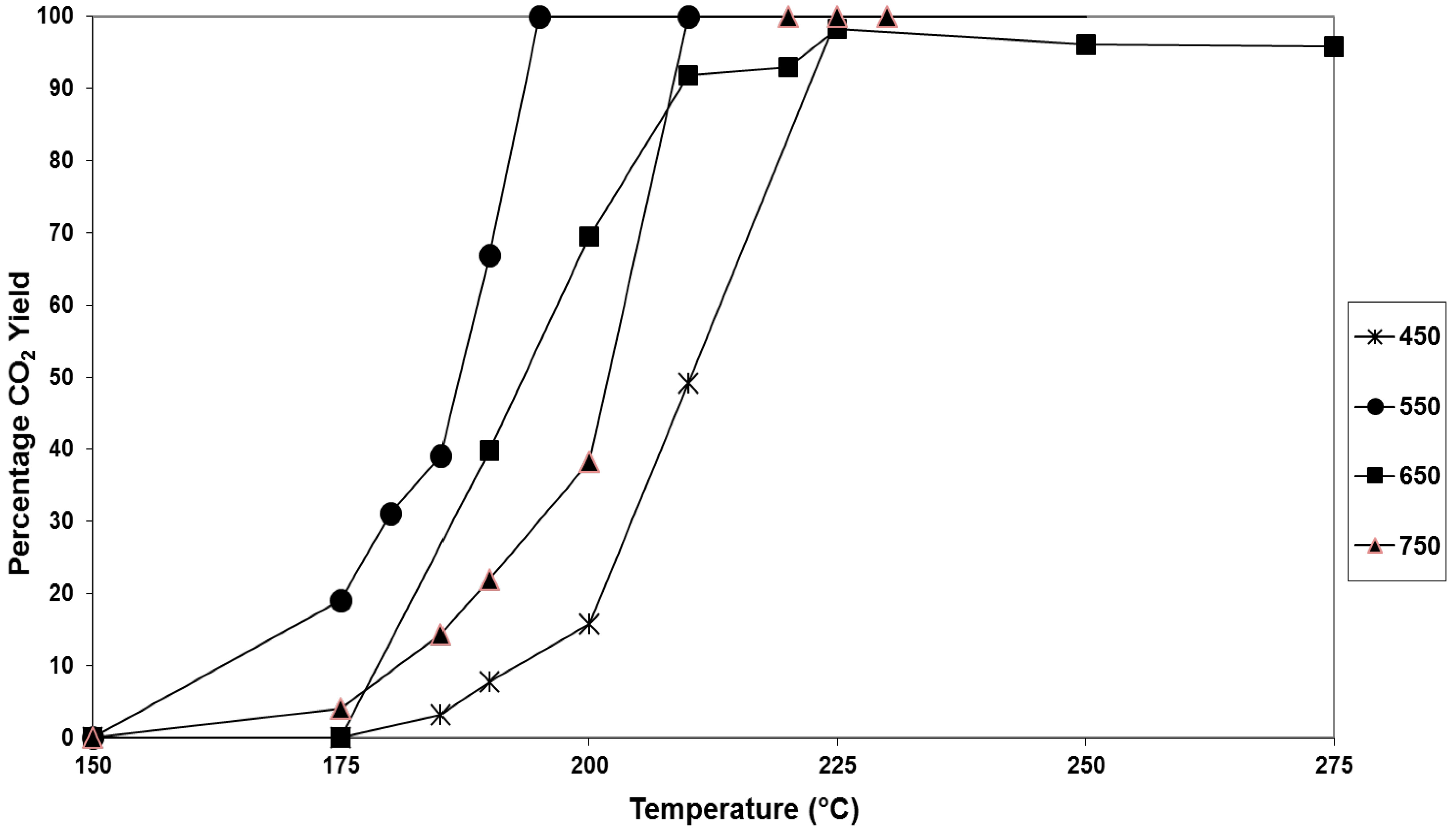
Figure 2. Increasing the calcination temperature from 450 to 550 °C resulted in an increase of catalyst activity. Calcination at 550 °C produced a catalyst with optimum activity, as the calcination temperature was increased to 650 and then 750 °C there was successive decreases of catalyst activity. These trends of activity can also be seen by consideration of the T
50 values (
Table 2).
Figure 2.
Steady-state conversion of naphthalene to CO2 as a function of temperature for 2.5 wt.% Pt/SiO2 catalyst prepared at varying calcination temperatures: ✱ 450 °C; ● 550 °C; ■ 650 °C; ▲ 750 °C.
Figure 2.
Steady-state conversion of naphthalene to CO2 as a function of temperature for 2.5 wt.% Pt/SiO2 catalyst prepared at varying calcination temperatures: ✱ 450 °C; ● 550 °C; ■ 650 °C; ▲ 750 °C.
Table 2.
Summary of characterisation data and naphthalene oxidation performance for 2.5 wt.% Pt/SiO2 catalysts calcined in static air for 6 h at varying temperatures.
Table 2.
Summary of characterisation data and naphthalene oxidation performance for 2.5 wt.% Pt/SiO2 catalysts calcined in static air for 6 h at varying temperatures.
| Calcination temperature/°C | BET surface area/m2g−1 | Pt dispersion a/% | Average Pt particle size a/Å | No. Pt sites (×1018) a/g | Surface Pt0:Pt2+:Pt4+ ratio b | Catalytic performance |
|---|
| T50 c/°C | T100 c/°C |
|---|
| 450 | 479 | 5 | 203 | 5.00 | 6.5:1.3:1.0 | 210 | 225 |
| 550 | 436 | 4 | 276 | 3.44 | 9.1:1.6:1.0 | 187 | 195 |
| 650 | 432 | 4 | 316 | 3.23 | 6.8:2.1:1.0 | 193 | 225 |
| 750 | 400 | 2 | 417 | 0.81 | 8.4:1.5:1.0 | 202 | 210 |
A summary of the catalysts characteristics are presented in
Table 2. The BET catalyst total surface area decreased as the calcination temperature was increased. This was an expected trend as sintering of the silica support will be more extensive at higher temperature. The Pt dispersion remained relatively low, ranging from 5% at 450 °C to 2% at 750 °C. The dispersions for the 2.5 wt.% catalysts, regardless of calcination temperature, were much lower than catalysts calcined at 550 °C with lower Pt content (
Table 1). These data indicate that the dispersion of Pt using our preparation method is strongly influenced by the Pt loading and not the calcination temperature. The decrease of Pt dispersion with increasing calcination temperature also results in an increase of the calculated average Pt particle size. This observation is also consistent with the decrease of total surface area, as increasing the size of Pt particles would be expected to increase pore blocking of the silica support. The number of surface Pt sites was greatest for the catalyst calcined at 450 °C, and decreased as the calcination temperature was raised. Hence, as observed for the series of catalysts with varying Pt loading, there was no simple relationship between the number of Pt surface sites and naphthalene total oxidation activity. Rather, it seems there is a more complex relationship with the number of surface sites and Pt particle size, but it is again evident that low dispersion and large Pt particles are required for more active naphthalene total oxidation catalysts.
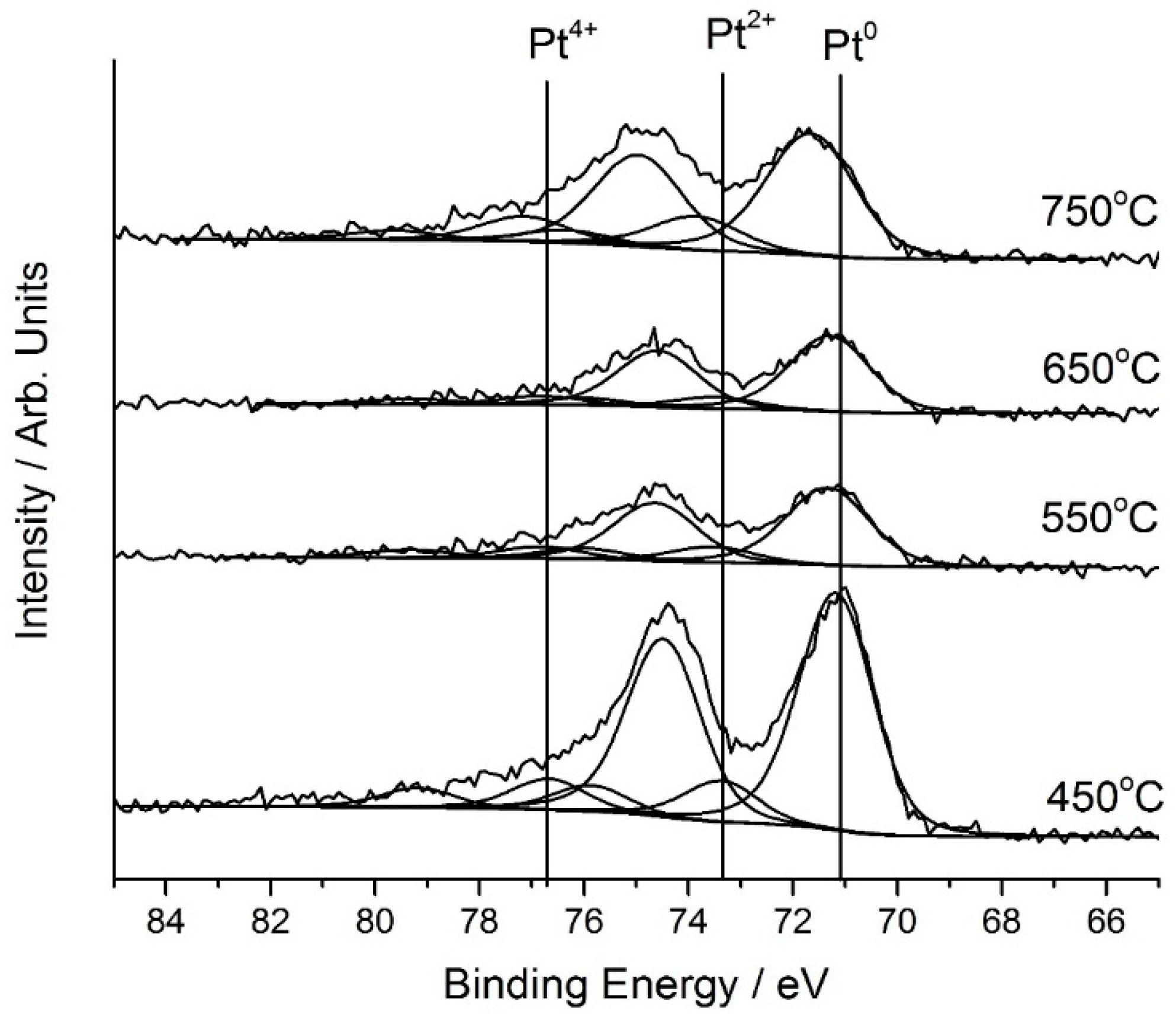
Analysis results from XPS for catalysts prepared with varying calcination temperature are shown in
Table 2 and
Figure 3. There was no general simple trend between metallic Pt surface concentration and calcination temperature. In discussion above and in our previous work [
15], we consider that the presence of Pt in a combination of oxidised and metallic states is important for enhanced naphthalene total oxidation. The catalyst calcined at 450 °C has the lowest concentration of Pt
0, the concentration increased to a maximum for the catalyst calcined at 550 °C, which was also more active. Therefore it is tempting to conclude that a higher Pt
0 concentration is beneficial for total oxidation, but there is also an effect of Pt dispersion influencing the number of Pt surface sites and Pt particle size. Accordingly, it is interesting to consider the Pt oxidation states for the catalysts calcined at 550 and 650 °C, as these catalysts have similar surface areas, Pt dispersion and Pt average particle sizes. The more active 550 °C calcined catalyst has around 40% more surface Pt in the metallic state, indicating that a higher concentration of Pt
0 in combination with oxidised Pt is an important factor for greater activity.
Figure 3.
Pt(4f) core-level spectra for a 2.5 wt.% Pt/SiO2 catalyst showing the effect of increasing calcination temperature.
Figure 3.
Pt(4f) core-level spectra for a 2.5 wt.% Pt/SiO2 catalyst showing the effect of increasing calcination temperature.
The mechanism of naphthalene total oxidation over Pt-based catalysts remains a subject of debate. A kinetic study of naphthalene oxidation over 1% Pt/γ-Al
2O
3 by Zhang
et al. showed that the overall rate of reaction could be expressed using a Langmuir-Hinshelwood kinetic model [
13]. The reaction was first order with respect to naphthalene and oxygen. Shie
et al. also investigated a similar Pt/γ-Al
2O
3 catalyst to study the total oxidation of naphthalene [
18]. However, from their kinetic analysis they concluded that oxidation could best be described by an Eley-Rideal mechanism. In contrast, Radic
et al. performed kinetic experiments for the total oxidation of the related molecule toluene, over a Pt/Al
2O
3 catalyst [
19], and the kinetics were best described by the Mars van Krevelen mechanism. It was reported that the rate of oxygen chemisorption increased with increasing Pt crystallite size, and that activation energy for oxygen chemisorption also decreased with increasing Pt particle size, which resulted in an increased reaction rate. Relatively weak Pt–O bonds were reported to form on larger Pt particles. No data on Pt oxidation state was reported, but the authors believed that the Pt was metallic since their experiments were performed at temperatures where stable Pt oxide formation was not favoured. Garetto and Apesteguía observed similar behaviour for cyclopentane oxidation [
20]. They reported that the turnover frequency values for cyclopentane increased with increasing Pt particle size. Pt existed predominantly as metallic particles and larger Pt crystallite sizes increased the density of active Pt–O species. A redox mechanism was postulated, the first step of which was oxidation of metallic Pt to Pt–O. The dissociative adsorption of oxygen was reported to be the rate determining step. In a time on stream experiment cyclopentane conversion increased with time. This was reported to be due to agglomeration of the Pt particles to larger ones, and hence more active Pt particles. The observations of larger Pt particles being more active, and the importance of metallic Pt are consistent with our observations for the total oxidation of naphthalene. It can be envisaged that for total oxidation adsorption of the naphthalene molecule flat on the surface would be favourable, whereas adsorption end on would more likely result in partial oxidation to products like phthalic anhydride. Hence, larger Pt particles will facilitate such preferential naphthalene adsorption, as well as facilitating O
2 chemisorption and increasing the density of more weakly bound Pt–O species [
19,
20].
The application of catalytic oxidation for VOC control requires catalysts with stable activity. The stability of the most active catalyst containing 2.5 wt.% Pt and calcined at 550 °C was investigated. Firstly the stability of the catalyst activity was studied by cycling through a series of light-off curves, during the cycle the catalyst temperature was reduced to ambient before commencing the next cycle (
Figure 4). After the first cycle the catalyst activity increased, with the temperature required to achieve equivalent conversion shifting to approximately 15 °C lower. The second and third cycles showed very similar activity, at each temperature the measured activity was greater for the third cycle, but with the exception of one data point the conversion for each cycle was within experimental error. The reason for the increase of activity after the first cycle is not clear, as it was not possible to recover sufficient catalyst to perform meaningful characterisation. However, speculatively an increase in the Pt particle size, aided by the reaction temperature and the exothermic nature of naphthalene oxidation, may be important as the same effect has been shown to increase the activity of a supported Pt catalyst for cyclopentane oxidation [
20].
Figure 4.
Conversion of naphthalene to CO2 as a function of temperature for a 2.5 wt.% Pt/SiO2 catalyst calcined at 550 °C used in repeat cycles: ▲ First cycle; ♦ Second cycle; ■ Third cycle.
Figure 4.
Conversion of naphthalene to CO2 as a function of temperature for a 2.5 wt.% Pt/SiO2 catalyst calcined at 550 °C used in repeat cycles: ▲ First cycle; ♦ Second cycle; ■ Third cycle.
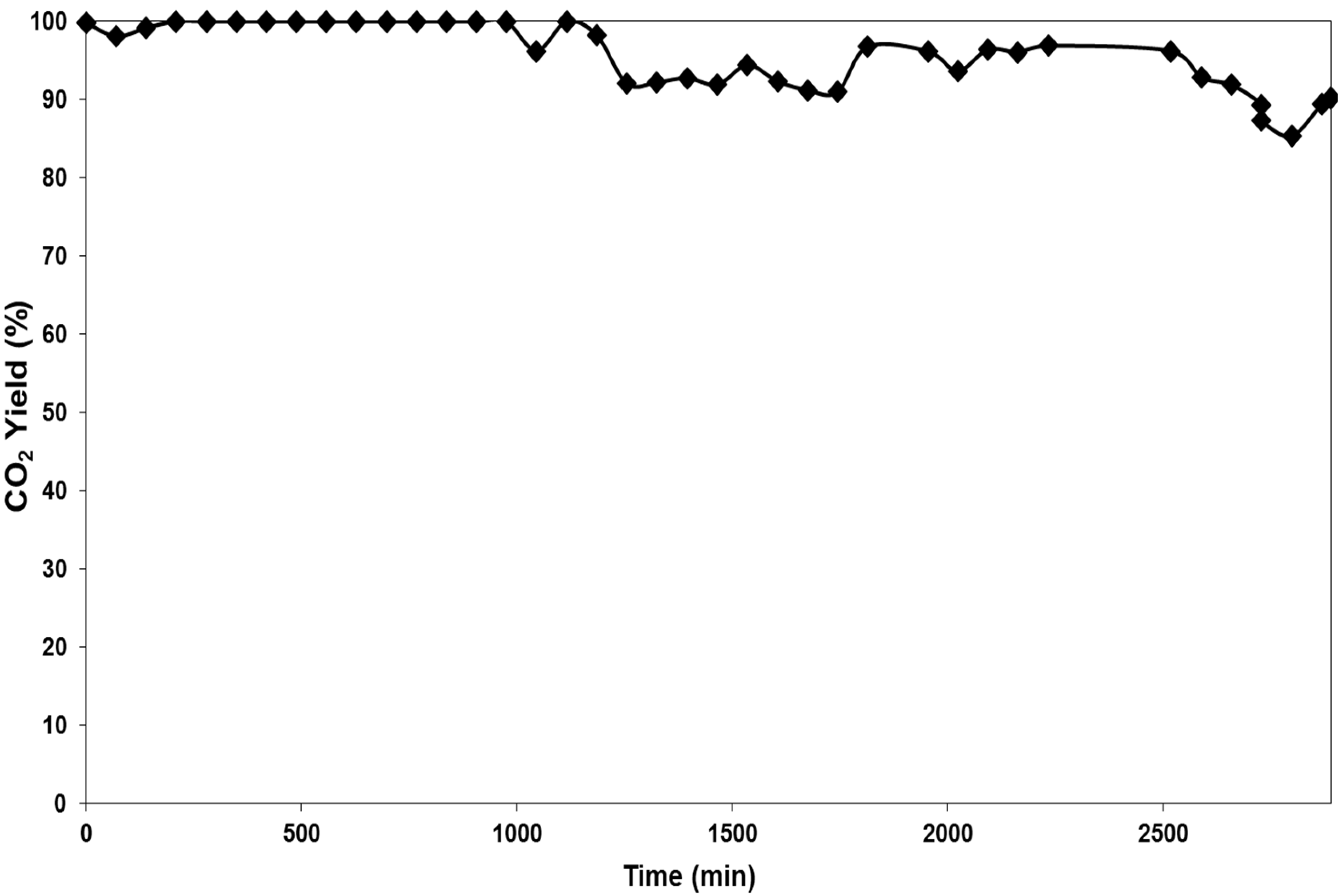
Catalyst stability was also probed by measuring activity as a function of time-on-stream (
Figure 5). Total naphthalene conversion was observed for approximately the first 1000 minutes on-stream. After this initial period there was a decrease of activity with conversion to CO
2 remaining above 90%. This result is in contrast to the increase of activity after cycling. Again it was not possible to recover sufficient catalyst after use for characterisation, and these deactivation processes are areas for further potential study.
Figure 5.
Conversion of naphthalene to CO2 at 300 °C as a function of time for a 2.5 wt.% Pt/SiO2 catalyst calcined at 550 °C.
Figure 5.
Conversion of naphthalene to CO2 at 300 °C as a function of time for a 2.5 wt.% Pt/SiO2 catalyst calcined at 550 °C.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



