
Pyrazole Supported Zinc(II) Benzoates as Catalysts for the Ring Opening Copolymerization of Cyclohexene Oxide and Carbon Dioxide

 and
and
Abstract
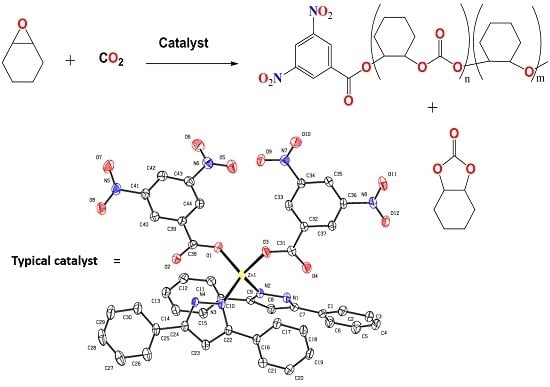
:
1. Introduction
2. Results and Discussion
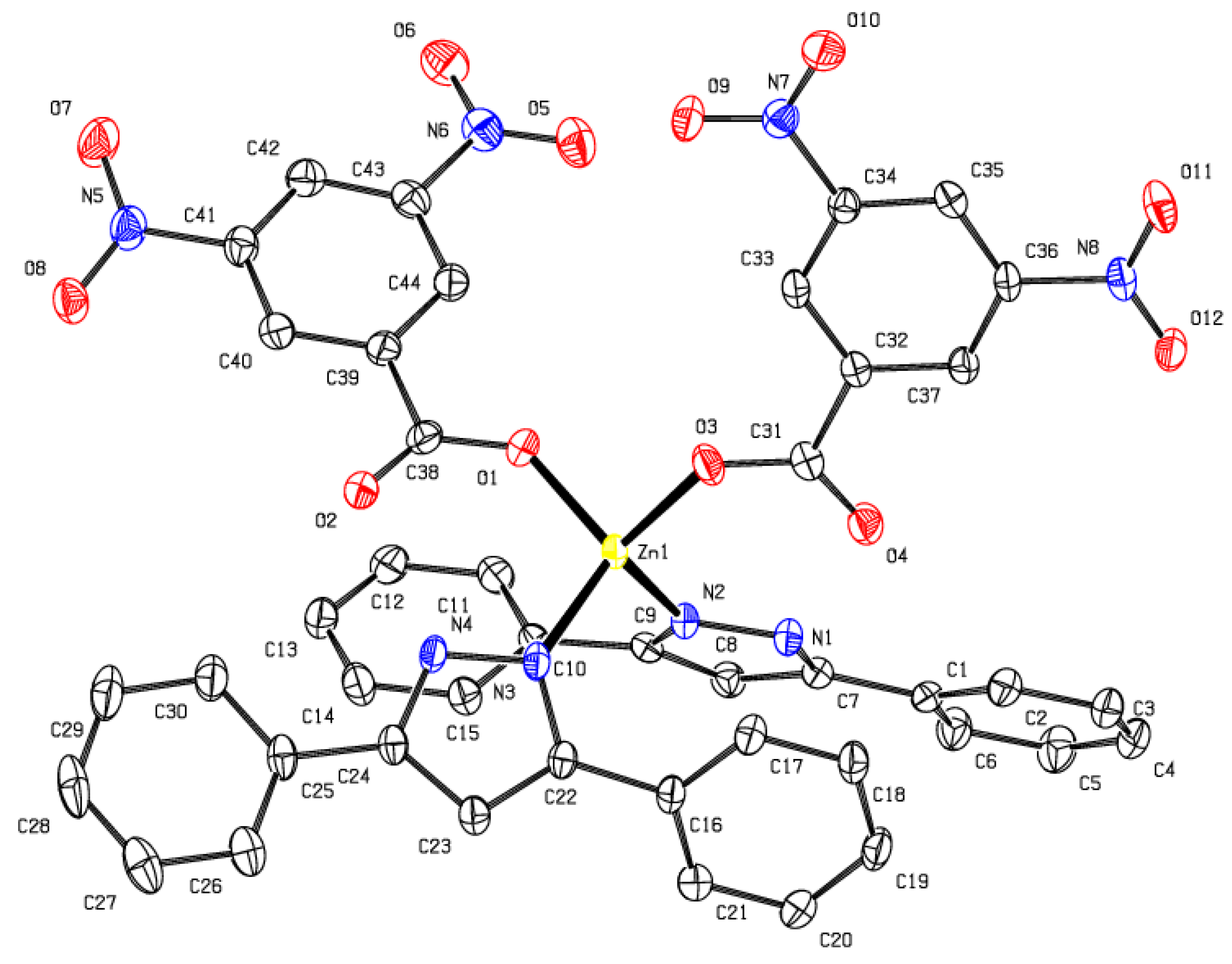
2.1. Synthesis and Characterization of Zinc(II) Complexes


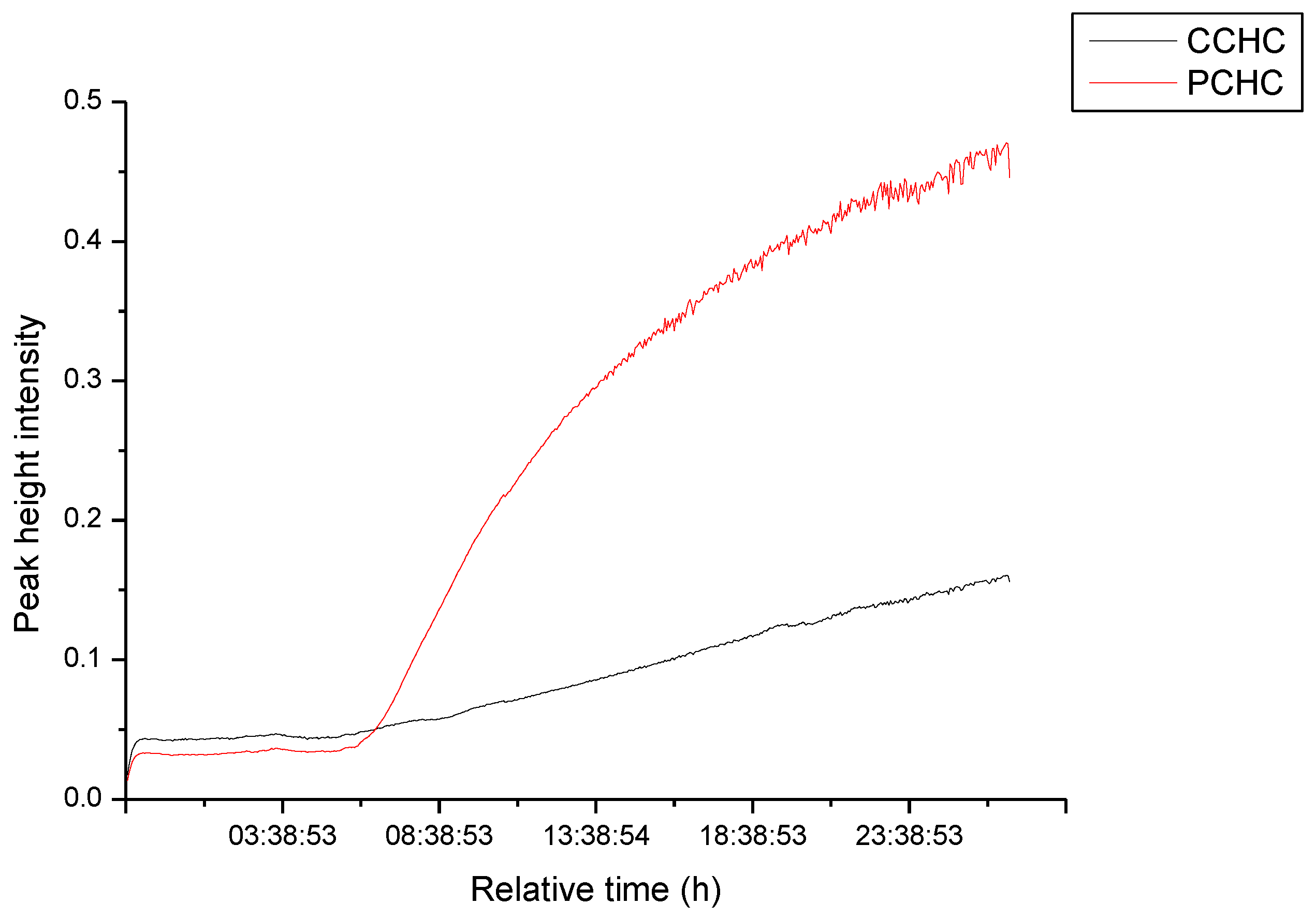
2.2. Ring-Opening Copolymerization of CO2 and CHO with Complexes 1–4

{kind=link}
{kind=link}
{kind=link}
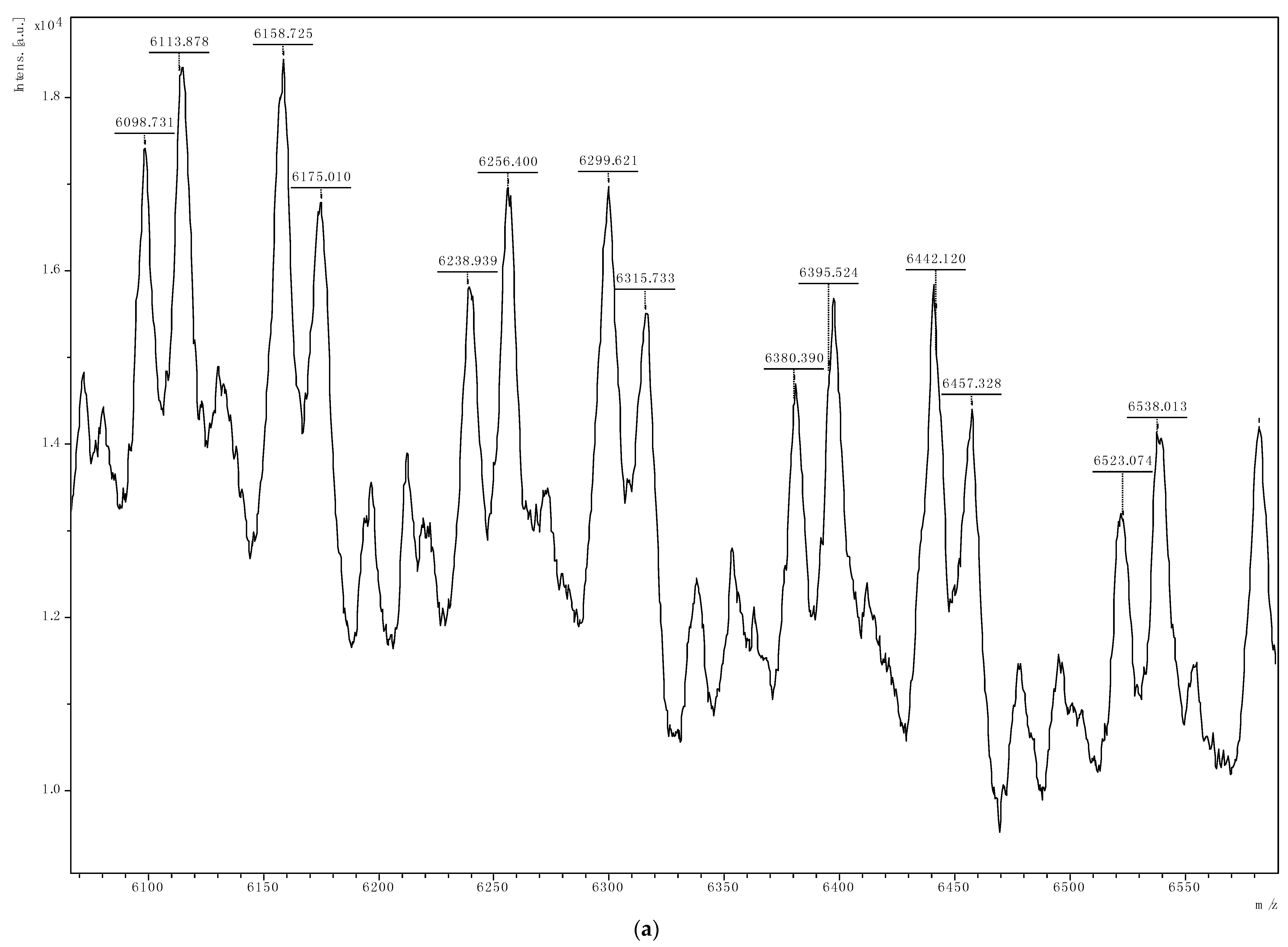{kind=link}
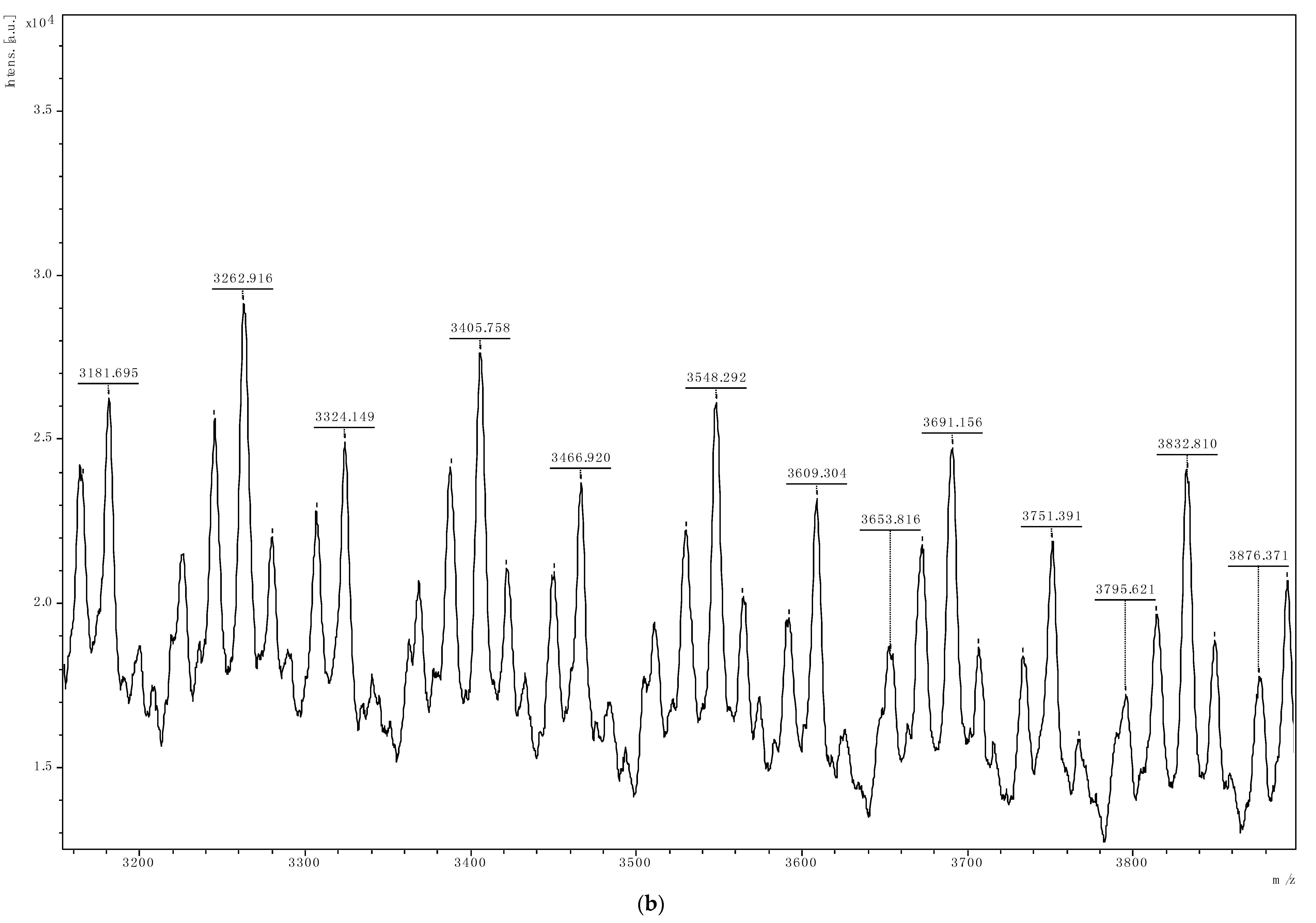{kind=link}
{kind=link}
| Entry | Complex | [M]/[C] | Poly(cyclohexene carbonate) (PCHC) (%) b | Cyclic cyclohexene carbonate (CCHC) (%) b | Selectivity | TOF (h−1) c |
|---|---|---|---|---|---|---|
| 1 | 1 | 1000:1 | 4.4 | 2.1 | 68 | 4.1 |
| 2 | 1 | 500:1 | 18 | 5.6 | 76 | 7.7 |
| 3 | 1 | 250:1 | 19.3 | 6.1 | 76 | 7.7 |
| 4 | 2 | 1000:1 | 0 | 0 | - | - |
| 5 | 2 | 500:1 | 26.5 | 3.9 | 87 | 9.9 |
| 6 | 2 | 250:1 | 28 | 5.7 | 83 | 10 |
| 7 | 3 | 1000:1 | 0 | 0 | - | - |
| 8 | 3 | 500:1 | 8.2 | 2.6 | 76 | 3.5 |
| 9 | 3 | 250:1 | 11.2 | 4.5 | 71 | 2.5 |
| 10 | 4 | 1000:1 | 0 | 0 | - | - |
| 11 | 4 | 500:1 | 0 | 0 | - | - |
| 12 | 4 | 250:1 | 50.1 | 33.2 | 64 | 15 |

| Entry | Catalyst | CO2 Pressure (MPa) | Poly(cyclohexene carbonate) (PCHC) (%) b | Cyclic cyclohexene carbonate (CCHC) (%) b | Selectivity for PCHC (%) | Carbonate content in PCHC (%) c | TOF (h−1) d | Mn (g/mol) e | PDI (Mw/Mn) | Tg (°C) f |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 18.0 | 5.6 | 76 | 40 | 7.7 | 1600 | 1.64 | - |
| 2 | 2 | 1 | 26.5 | 3.9 | 87 | 15 | 9.9 | 5400 | 1.64 | - |
| 3 | 3 | 1 | 11.2 | 4.5 | 71 | 66 | 2.5 | - | - | - |
| 4 | 4 | 1 | 50.1 | 33.2 | 60 | 85 | 15 | 6300 | 1.61 | 109.4 |
| 5 | 1 | 2 | 25.0 | 5.8 | 81 | 81 | 10.1 | 9300 | 1.34 | - |
| 6 | 2 | 2 | 33.0 | 3.9 | 89 | 44 | 18 | 6500 | 2.05 | 116.7 |
| 7 | 3 | 2 | 11.6 | 4.3 | 73 | 76 | 2.5 | - | - | - |
| 8 | 4 | 2 | 55.6 | 41.5 | 57 | 89 | 17 | 6900 | 1.57 | 105.4 |
| 9 | 1 | 3 | 43.0 | 3.9 | 92 | 88 | 15.2 | 10,500 | 1.78 | 104.4 |
| 10 | 2 | 3 | 48.0 | 6.2 | 83 | 60 | 31 | 7700 | 2.19 | 114.4 |
| 11 | 3 | 3 | 31.0 | 7.0 | 82 | 89 | 6 | 6300 | 2.25 | - |
| 12 | 4 | 3 | 57.9 | 39.1 | 60 | 92 | 17 | 6300 | 1.44 | 107.8 |
| 13 | 1 | 4 | 58.0 | 10.9 | 84 | 98 | 22.3 | 12,300 | 1.19 | - |
| 14 | 2 | 4 | 55.0 | 6.5 | 89 | 77 | 32 | 5200 | 2.50 | 103.5 |
| 15 | 3 | 4 | 53.8 | 9.7 | 85 | 89 | 10 | - | - | - |
| 16 | 4 | 4 | 67.7 | 29.9 | 69 | 95 | 18 | 7600 | 1.52 | 112.9 |


3. Experimental Section
3.1. Materials
3.2. Instrumentation
3.3. Representative Procedure for CO2 and CHO Copolymerization Reaction
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Coates, G.W.; Moore, D.R. Discrete metal-based catalysts for the copolymerization of CO2 and epoxides: Discovery, reactivity, optimization, and mechanism. Angew. Chem. Int. Ed. 2004, 43, 6618–6639. [Google Scholar] [CrossRef]
- Koning, C.; Wildeson, J.; Parton, R.; Plum, B.; Steeman, P.; Darensbourg, D. Synthesis and characterization of poly(cyclohexane carbonate), synthesis from CO2 and cyclohexene oxide. J. Polym. 2001, 42, 3995–4004. [Google Scholar] [CrossRef]
- Harnett, C.K.; Coates, G.W.; Craighead, H.G. Heat-depolymerizable polycarbonates as electron beam patternable sacrificial layers for nanofluidics. J. Vac. Sci. Technol. 2001, B19, 2842–2845. [Google Scholar] [CrossRef]
- Harnett, C.K.; Satyalakshmi, K.M.; Coates, G.W.; Craighead, H.G. Direct electron-beam patterning of surface coating and sacrificial layers for micro-total analysis systems. J. Photopolym. Sci. Technol. 2002, 15, 493–496. [Google Scholar] [CrossRef]
- Wu, G.-P.; Jiang, S.-D.; Lu, X.-B.; Ren, W.-M.; Yan, S.-K. Stereoregular poly(cyclohexene carbonate)s: Unique crystallization behaviour. Chin. J. Polym. Sci. 2012, 30, 487–492. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making plastics from carbon dioxide: salen metal complexes for the production of polycarbonates from expoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef]
- Coates, G.W.; Jeske, R.C. Handbook of Green Chemistry; Crabtree, R.H., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; p. 343. [Google Scholar]
- Nakano, K.; Hashimoto, S.; Nozaki, K. Bimetallic mechanism operating in the copolymerization of proplyene oxide and carbon dioxide catalyzed by cobalt-salen complexes. Chem. Sci. 2010, 1, 369–373. [Google Scholar] [CrossRef]
- Nakano, K.; Nakamura, M.; Nozaki, K. Alternating copolymerization of cyclohexene oxide and carbon dioxide catalyzed by (salalen)CrCl complexes. Macromolecules 2009, 42, 6972–6980. [Google Scholar] [CrossRef]
- Nakano, K.; Kamada, T.; Nozaki, K. Selective formation of polycarbonate over cyclic carbonate: copolymerization of epoxides and carbon dioxide catalyzed by a cobalt(III) with piperidinium end-capping arm. Angew. Chem. Int. Ed. 2006, 45, 7274–7277. [Google Scholar] [CrossRef]
- Ren, W.-M.; Zhang, X.; Liu, Y.; Li, J.-F.; Wang, H.; Lu, X.-B. Highly active, bifunctional Co(III)salen catalyst for alternating copolymerization of CO2 with cyclohexene oxide and terpolymerization with aliphatic epoxides. Macromolecules 2010, 43, 1396–1402. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, W.-M.; Liu, J.; Lu, X.-B. Asymmetric copolymerization of CO2 with meso-epoxides mediated by dinuclear cobalt(III) complexes: unprecedented enantioselectivity and activity. Angew. Chem. Int. Ed. 2013, 52, 11594–11598. [Google Scholar] [CrossRef]
- Super, M.; Berluche, E.; Costello, C.; Beckman, E. Copolymerization of 1,2-epoxicyclohexane and carbon dioxide using carbon dioxide as both reactant and solvent. Macromolecules 1997, 30, 368–372. [Google Scholar]
- Super, M.; Beckman, E. Co polymerization of CO2 and cyclohexene oxide. J. Macromol. Symp. 1998, 127, 89–108. [Google Scholar]
- Darensbourg, D.J.; Holtcamp, M. Catalytic activity of zinc(II) phenoxides which possess readily accessible coordination sites. Copolymerization and terpolymerization of epoxides and carbon dioxide. Macromolecules 1995, 28, 7577–7579. [Google Scholar]
- Cheng, M.; Lobkovsky, E.B.; Coates, G.W. Catalytic reactions involving C1 feedstocks: new high-activity zinc(II)-based catalysts for the alternating copolymerization of carbon dioxide and epoxides. J. Am. Chem. Soc. 1998, 120, 11018–11019. [Google Scholar]
- Chapman, A.M.; Keyworth, C.; Kembert, M.R.; Lennox, A.J.J.; Williams, C.K. Adding value to power station captured CO2: tolerant Zn and Mg homogeneous catalysts for polycarbonate polyol production. ACS Catal. 2015, 5, 1581–1588. [Google Scholar] [CrossRef]
- Kembert, M.R.; Knight, P.D.; Reung, P.T.; Williams, C.K. Highly active dizinc catalyst for the copolymerization of carbon dioxide and cyclohexene oxide at one atmosphere pressure. Angew. Chem. Int. Ed. 2009, 48, 931–933. [Google Scholar] [CrossRef]
- Elmas, S.; Subhani, M.A.; Leitner, W.; Muller, T.E. Anion effect controlling the selectivity in the zinc catalysed copolymerisation of CO2 and cyclohexene oxide. Beilstein J. Org. Chem. 2015, 11, 42–49. [Google Scholar] [CrossRef]
- CCDC 1000656 Contains the Supplementary Crystallographic Data for this Paper. This Data Can Be Obtained Free from the Cambridge Crystallographic Data Centre. Available online www.ccdc.cam.ac.uk/data_request/cif (accessed on 18 January 2016).
- Sarma, R.; Kalita, D.; Baruah, B. Solvent induced reactivity of 3,5-dimethylpyrazole towards zinc(II) carboxylates. Dalton Trans. 2009, 7428–7436. [Google Scholar] [CrossRef]
- Klaus, S. Development of efficient catalysts for the CO2/epoxide copolymerization reaction. Ph.D. Thesis, Technical University of Munich, Munich, Germany, 2011. [Google Scholar]
- Darensbourg, D.J.; Wildeson, J.R.; Yarbrough, J.C. Coplymerization and terpolymerization of CO2 and epoxides using soluble zinc crotonate catalyst precursor. Inorg. Chem. 2002, 41, 973–980. [Google Scholar] [CrossRef]
- Cheng, M.; Moore, D.R.; Reczek, J.J.; Chamberlain, B.M.; Lobkovsky, E.B.; Coates, G.W. Single-site β-diimine zinc catalysts for the alternating copolymerization of CO2 and expoxides: catalyst synthesis and unprecedented activity. J. Am. Chem. Soc. 2001, 123, 8738–8749. [Google Scholar] [CrossRef]
- Moore, D.R.; Cheng, M.; Lobkovsky, E.B.; Coates, G.W. Electronic and steric effects on CO2/epoxide polymerization: subtle modification resulting in superior activities. Angew. Chem. Int. Ed. 2002, 41, 2599–2602. [Google Scholar] [CrossRef]
- Cheng, M.; Moore, D.R.; Lobkovsky, E.B.; Coates, G.W. Mechanism for the polymerization of epoxides and CO2 using β-diiminate zinc catalysts: evidence for bimetallic epoxide enchainment. J. Am. Chem. Soc. 2003, 125, 11911–11924. [Google Scholar]
- Darensbourg, D.J.; Mackiewicz, R.M.; Phelps, A.L.; Billodeaux, D.R. Copolymerization of epoxides catalyzed by metal salen complexes. Acc. Chem. Res. 2004, 37, 836–844. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Yarbrough, J.C.; Ortiz, C.; Fang, C.C. Comparative kinetics studies of the copolymerization of cyclohexene oxide and propylene oxide with carbon dioxide in the presence of chromium salen derivatives. In situ FTIR measurements of copolymers vs cyclic carbonate production. J. Am. Chem. Soc. 2003, 125, 7586–7591. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Yarbrough, J.C.; Wildeson, J.R.; Reibenspiea, J.H. Bis 2,6-difluorophenoxide dimeric complexes of zinc and cadmium and their phosphine adducts: lessons learnt relative to carbon dioxide/cyclohexene oxide alternation copolymerization processes catalyzed by zinc phenoxides. J. Am. Chem. Soc. 2000, 122, 12487–12496. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Mackiewicz, R.M.; Phelps, A.L.; Rodgers, J.L. The coupling of epoxides and carbon dioxide in the presence of homogeneous transition-metal catalysts. Production of polycarbonates vs cyclic carbonates. Prepr. Pap-Am. Chem. Soc. Div. Fuel Chem. 2004, 49, 5–6. [Google Scholar]
- Kitajima, N.; Fujisawa, K.; Fujimoto, C.; Morooka, Y.; Hashimoto, S.; Kitagawa, T.; Toriumi, K.; Tatsumi, K.; Nakamura, A. A new model for dioxygen binding in hemocyanin. Synthesis, characterization and molecular structure of the.mu.-.eta.2:.eta.2 peroxo dinuclear copper(II) complexes, [Cu(HB(3,5-R2pz)3]2(O2) (R = isopropyl and Ph). J. Am. Chem. Soc. 1992, 114, 1277–1291. [Google Scholar] [CrossRef]
- Appavoo, D. Pyrazole and pyrazolyl copper and zinc complexes in ring opening polymerization of ε-caprolactone and d,L-lactide. Master’s Thesis, University of Johannesburg, Johannesburg, South Africa, September 2011. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lephoto, M.L.; Nakano, K.; Appavoo, D.; Owaga, B.O.; Nozaki, K.; Darkwa, J. Pyrazole Supported Zinc(II) Benzoates as Catalysts for the Ring Opening Copolymerization of Cyclohexene Oxide and Carbon Dioxide. Catalysts 2016, 6, 17. https://doi.org/10.3390/catal6010017
Lephoto ML, Nakano K, Appavoo D, Owaga BO, Nozaki K, Darkwa J. Pyrazole Supported Zinc(II) Benzoates as Catalysts for the Ring Opening Copolymerization of Cyclohexene Oxide and Carbon Dioxide. Catalysts. 2016; 6(1):17. https://doi.org/10.3390/catal6010017
Chicago/Turabian StyleLephoto, Mapudumo L., Koji Nakano, Divambal Appavoo, Bernard O. Owaga, Kyoko Nozaki, and James Darkwa. 2016. "Pyrazole Supported Zinc(II) Benzoates as Catalysts for the Ring Opening Copolymerization of Cyclohexene Oxide and Carbon Dioxide" Catalysts 6, no. 1: 17. https://doi.org/10.3390/catal6010017
APA StyleLephoto, M. L., Nakano, K., Appavoo, D., Owaga, B. O., Nozaki, K., & Darkwa, J. (2016). Pyrazole Supported Zinc(II) Benzoates as Catalysts for the Ring Opening Copolymerization of Cyclohexene Oxide and Carbon Dioxide. Catalysts, 6(1), 17. https://doi.org/10.3390/catal6010017