
Gold-Iron Oxide Catalyst for CO Oxidation: Effect of Support Structure


Abstract
:1. Introduction
2. Results and Discussion
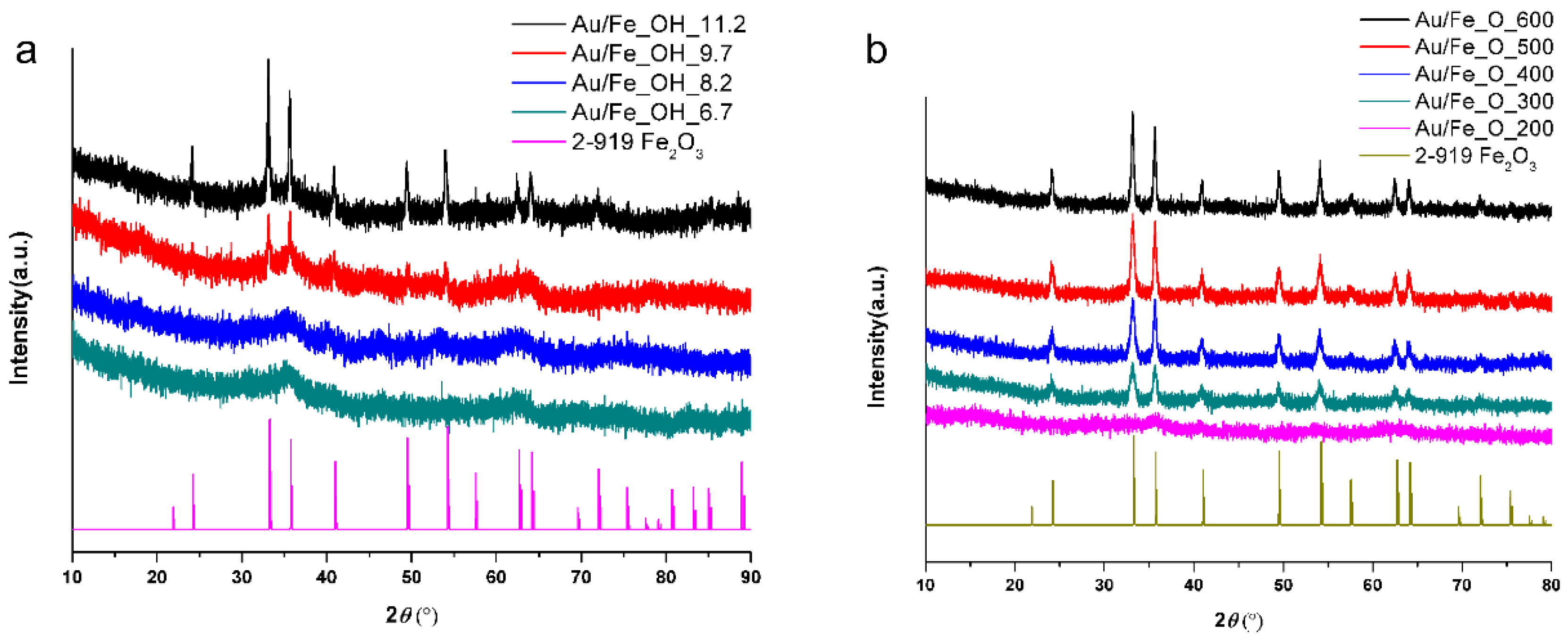
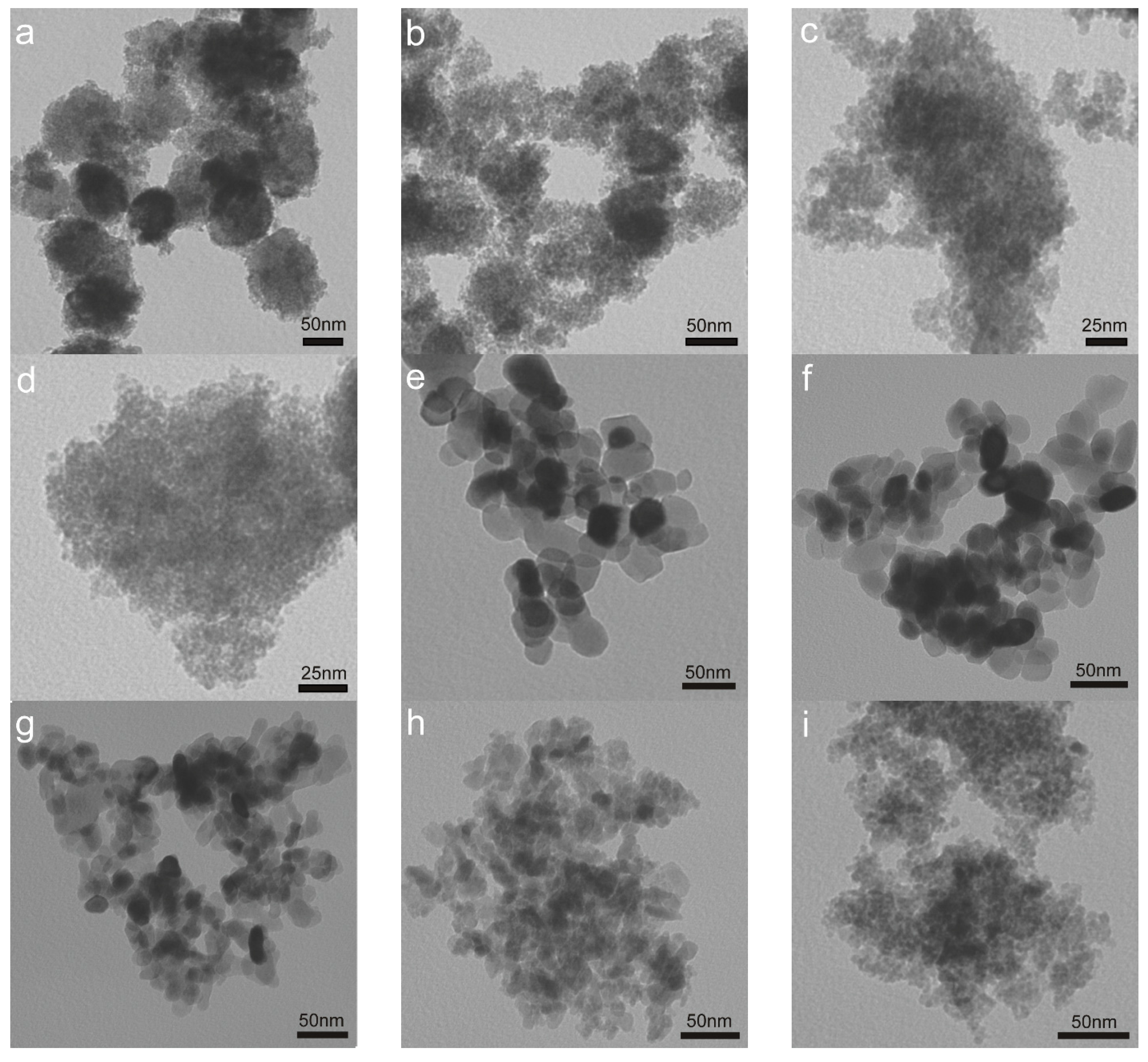
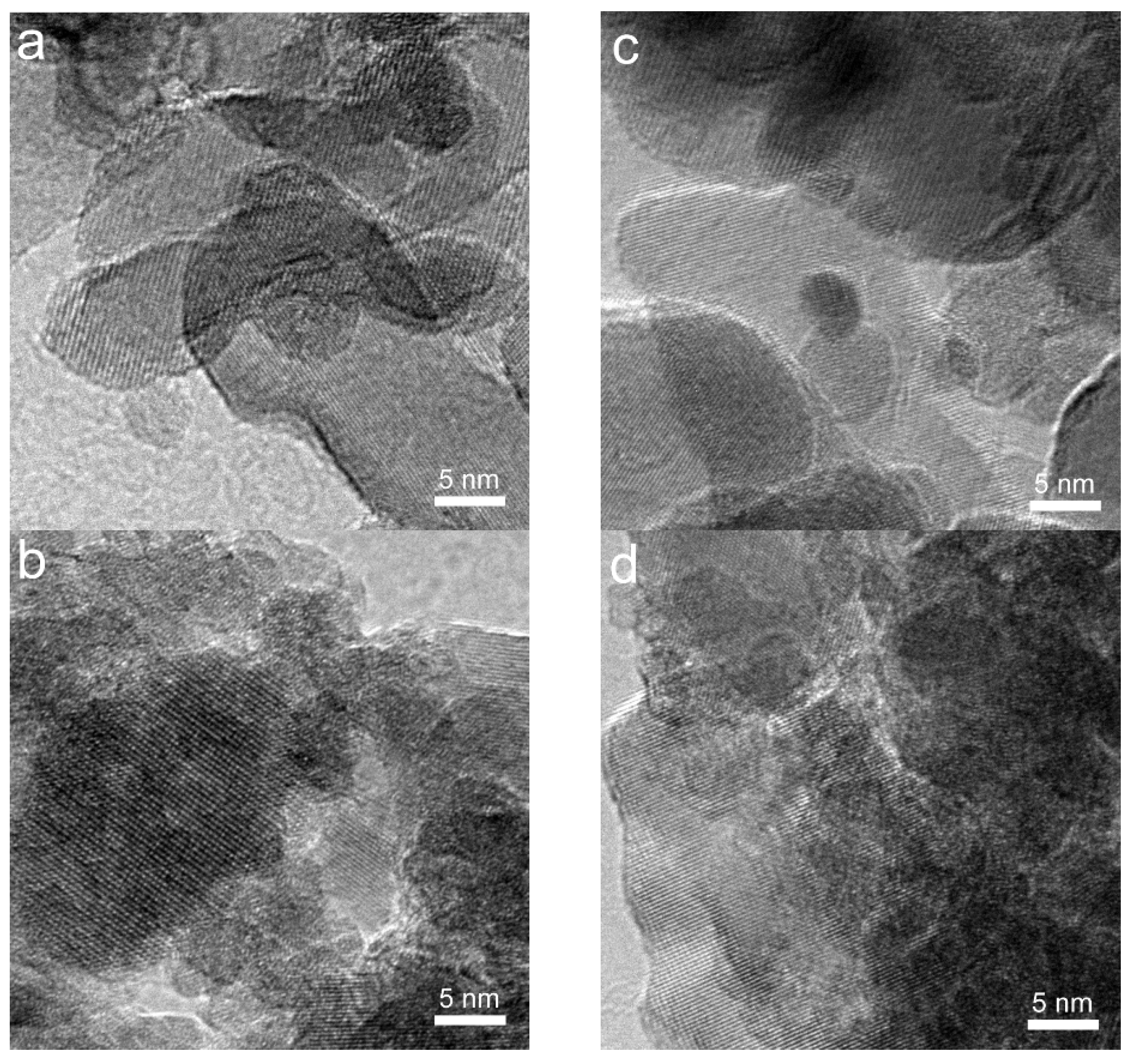
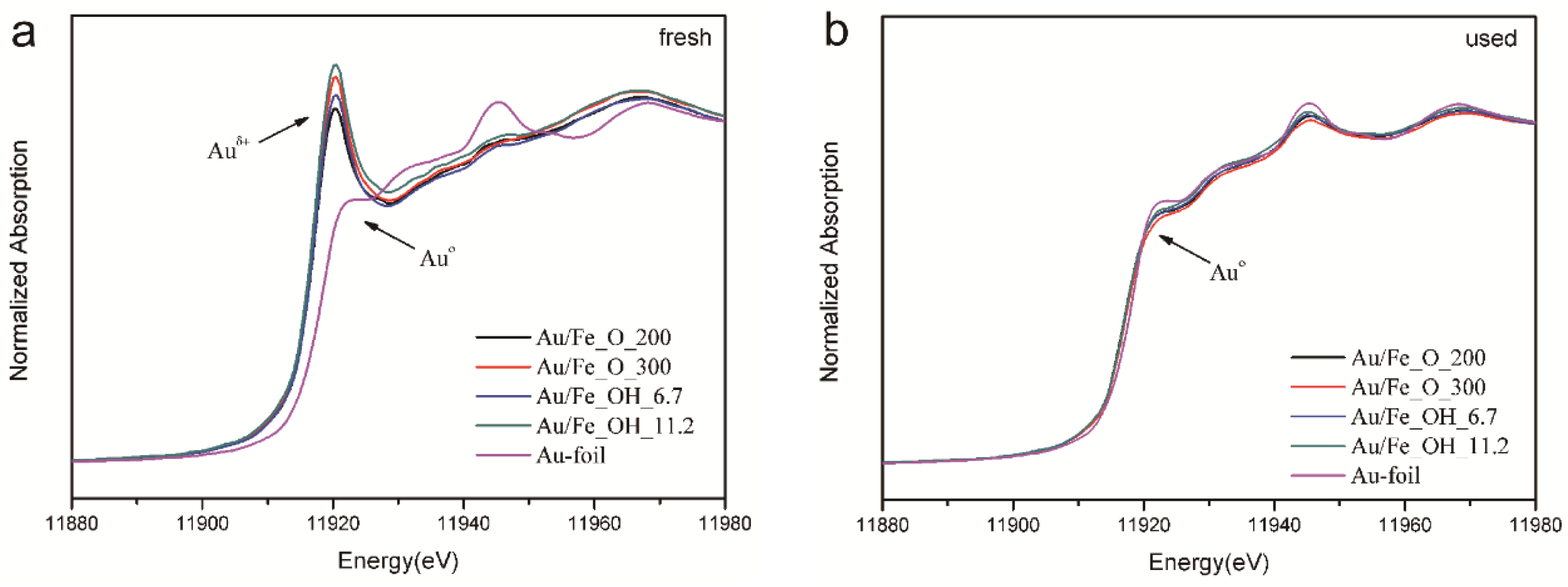
2.1. Structure and Texture of Gold-Iron Oxide Catalyst
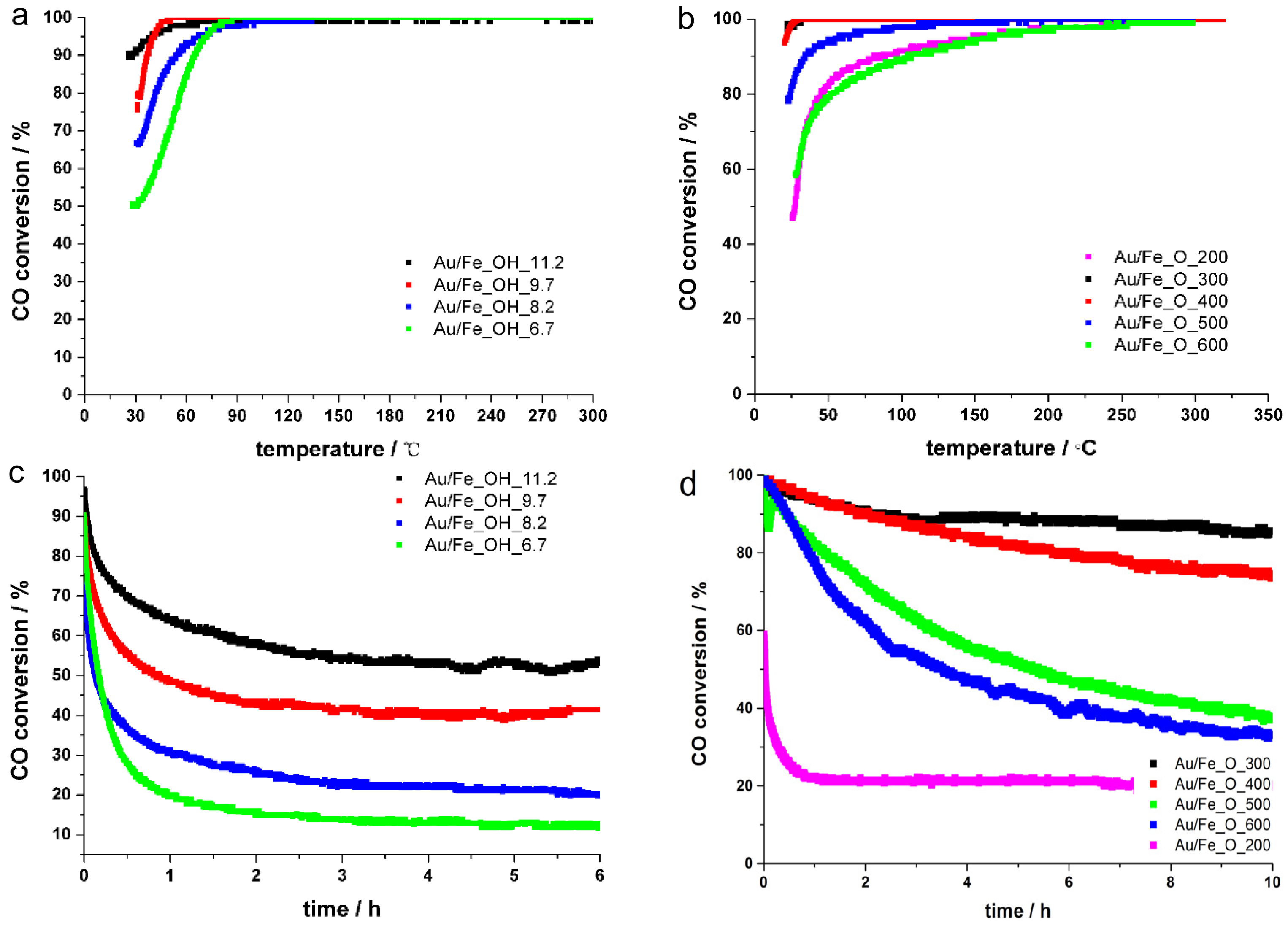
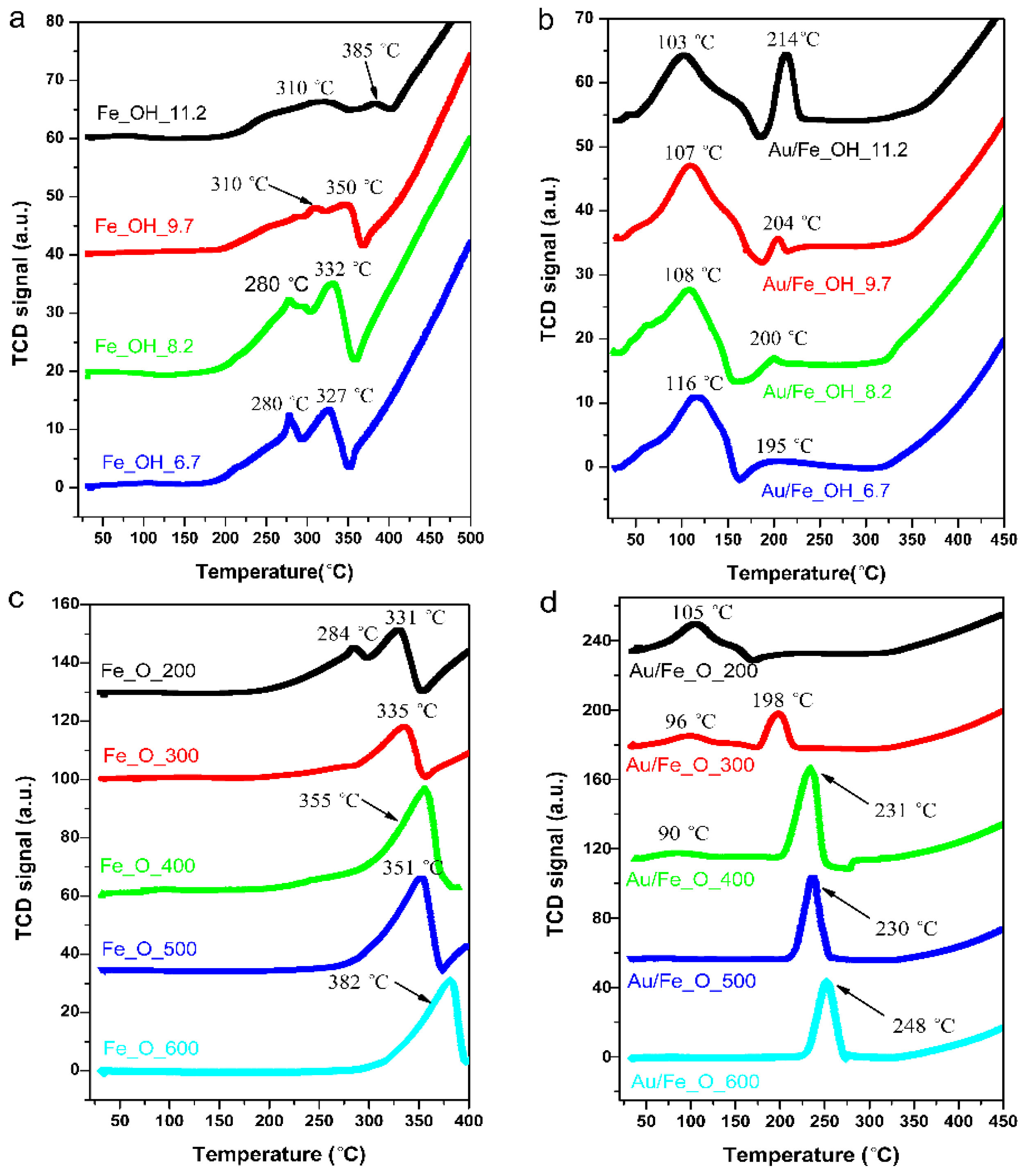
2.2. Catalytic Performance and Reducibility of Gold-Iron Oxide Catalyst
2.3. Effect of Iron Oxide Support
3. Experimental Section
3.1. Catalyst Preparation
3.1.1. Preparation of Fe_OH with Different pH Values [8]
3.1.2. Preparation of Fe_O with Different Calcination Temperatures
3.1.3. Deposition of Gold onto Fe_OH or Fe_O
3.2. Characterization
3.3. Catalytic Test
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-temperature oxidation of CO over gold supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Widmann, D.; Leppelt, R.; Behm, R.J. Activation of an Au/CeO2 catalyst for the CO oxidation reaction by surface oxygen removal/oxygen vacancy formation. J. Catal. 2007, 251, 437–442. [Google Scholar] [CrossRef]
- Camellone, M.F.; Fabris, S. Reaction mechanisms for the CO oxidation on Au/CeO2 catalysts: Activity of substitutional Au3+/Au+ cations and deactivation of supported Au+ Adatoms. J. Am. Chem. Soc. 2009, 131, 10473–10483. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Herzing, A.A.; Kiely, C.J.; Carley, A.F.; Landon, P.; Hutchings, G.J. Identification of active gold nanoclusters on iron oxide supports for CO oxidation. Science 2008, 321, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Daniells, S.T.; Overweg, A.R.; Makkee, M.; Moulijn, J.A. The mechanism of low-temperature CO oxidation with Au/Fe2O3 catalysts: A combined Mössbauer, FT-IR, and TAP reactor study. J. Catal. 2005, 230, 52–65. [Google Scholar] [CrossRef]
- Hutchings, G.J.; Hall, M.S.; Carley, A.F.; Landon, P.; Solsona, B.E.; Kiely, C.J.; Herzing, A.; Makkee, M.; Moulijn, J.A.; Overweg, A.; et al. Role of gold cations in the oxidation of carbon monoxide catalyzed by iron oxide-supported gold. J. Catal. 2006, 242, 71–81. [Google Scholar] [CrossRef]
- Li, L.; Wang, A.-Q.; Qiao, B.-T.; Lin, J.; Huang, Y.-Q.; Wang, X.-D.; Zhang, T. Origin of the high activity of Au/FeOx for low-temperature CO oxidation: Direct evidence for a redox mechanism. J. Catal. 2013, 299, 90–100. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, C.-J.; Yamasaki, J.; Terasaki, O.; Schuth, F. Highly active iron oxide supported gold catalysts for CO oxidation: How small must the gold nanoparticles be? Angew. Chem. Int. Ed. 2010, 49, 5771–5775. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.-Y.; Ho, J.; Teo, J.; Shen, S.-C.; Gedanken, A. Synthesis of porous α-Fe2O3 nanorods and deposition of very small gold particles in the pores for catalytic oxidation of CO. Chem. Mater. 2007, 19, 4776–4782. [Google Scholar] [CrossRef]
- Andreeva, D. Low temperature water gas shift over gold catalysts. Good Bull. 2002, 35, 82–88. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M.; Andreeva, D.; Tabakova, T. FTIR study of the low-temperature water-gas shift reaction on Au/Fe2O3 and Au/TiO2 catalysts. J. Catal. 1999, 188, 176–185. [Google Scholar] [CrossRef]
- Silberova, B.A.A.; Mul, G.; Makkee, M.; Moulijn, J.A. DRIFTS study of the water-gas shift reaction over Au/Fe2O3. J. Catal. 2006, 243, 171–182. [Google Scholar]
- Andreevaa, D.; Tabakovaa, T.; Idakieva, V.; Christova, P.; Giovanolib, R. Au/α-Fe2O3 catalyst for water-gas shift reaction prepared by deposition-precipitation. Appl. Catal. A 1998, 169, 9–14. [Google Scholar] [CrossRef]
- Schubert, M.M.; Venugopal, A.; Kahlich, M.J.; Plzak, V.; Behm, R.J. Influence of H2O and CO2 on the selective CO oxidation in H2-rich gases over Au/α-Fe2O3. J. Catal. 2004, 222, 32–40. [Google Scholar] [CrossRef]
- Shodiya, T.; Schmidt, O.; Peng, W.; Hotz, N. Novel nano-scale Au/α-Fe2O3 catalyst for the preferential oxidation of CO in biofuel reformate gas. J. Catal. 2013, 300, 63–69. [Google Scholar] [CrossRef]
- Landon, P.; Ferguson, J.; Solsona, B.E.; Garcia, T.; Sayari, S.A.; Carley, A.F.; Herzing, A.A.; Kiely, C.J.; Makkee, M.; Moulijn, J.A.; et al. Selective oxidation of CO in the presence of H2, H2O and CO2 utilizing Au/α-Fe2O3 catalysts for use in fuel cells. J. Mater. Chem. 2006, 16, 199–208. [Google Scholar] [CrossRef]
- Ozaki, M.; Kratohvil, S.; Matijević, E. Formation of monodispersed spindle-type hematite particles. J. Colloid Interface Sci. 1984, 102, 146–151. [Google Scholar] [CrossRef]
- Sugimoto, T.; Sakata, K. Preparation of monodisperse pseudocubic α-Fe2O3 particles from condensed ferric hydroxide gel. J. Colloid Interface Sci. 1992, 152, 587–590. [Google Scholar] [CrossRef]
- Jia, C.-J.; Sun, L.-D.; Yan, Z.-G.; You, L.-P.; Luo, F.; Han, X.-D.; Pang, Y.-C.; Zhang, Z.; Yan, C.-H. Single-crystalline iron oxide nanotubes. Angew. Chem. Int. Ed. 2005, 117, 4402–4407. [Google Scholar] [CrossRef]
- Jia, C.-J.; Sun, L.-D.; Luo, F.; Han, X.-D.; Heyderman, L.J.; Yan, Z.-G.; Yan, C.-H.; Zheng, K.; Zhang, Z.; Takano, M.; et al. Large-scale synthesis of single-crystalline iron oxide magnetic nanorings. J. Am. Chem. Soc. 2008, 130, 16968–16977. [Google Scholar] [CrossRef] [PubMed]
- Finch, R.M.; Hodge, N.A.; Hutchings, G.J.; Meagher, A.; Pankhurst, Q.A.; Siddiqui, M.R.H.; Wagnerc, F.E.; Whyman, R. Identification of active phases in Au-Fe catalysts for low-temperature CO oxidation. Phys. Chem. Chem. Phys. 1999, 1, 485–489. [Google Scholar] [CrossRef]
- Qian, K.; Zhang, W.-H.; Sun, H.-X.; Fang, J.; He, B.; Ma, Y.-S.; Jiang, Z.-Q.; Wei, S.-Q.; Yang, J.-L.; Huang, W.-X. Hydroxyls-induced oxygen activation on “inert” Au nanoparticles for low-temperature CO oxidation. J. Catal. 2011, 277, 95–103. [Google Scholar] [CrossRef]
- Hodge, N.A.; Kiely, C.J.; Whyman, R.; Siddiqui, M.R.H.; Hutchings, G.J.; Pankhurst, Q.A.; Wagner, F.E.; Rajaram, R.R.; Golunski, S.E. Microstructural comparison of calcined and uncalcined gold/iron-oxide catalysts for low-temperature CO oxidation. Catal. Today 2002, 72, 133–144. [Google Scholar] [CrossRef]
- Guo, Y.; Gu, D.; Jin, Z.; Du, P.-P.; Si, R.; Tao, J.; Xu, W.-Q.; Huang, Y.-Y.; Senanayake, S.; Song, Q.-S.; et al. Uniform 2 nm gold nanoparticles supported on iron oxides as active catalysts for CO oxidation reaction: Structure-activity relationship. Nanoscale 2015, 7, 4920–4928. [Google Scholar] [CrossRef] [PubMed]
- Venugopal1, A.; Scurrell, M.S. Low temperature reductive pretreatment of Au/Fe2O3 catalysts, TPR/TPO studies and behaviour in the water-gas shift reaction. Appl. Catal. A 2004, 258, 241–249. [Google Scholar] [CrossRef]
- Wang, D.-H.; Hao, Z.-P.; Cheng, D.-Y.; Shi, X.-C. Influence of the calcination temperature on the Au/FeOx/Al2O3 catalyst. J. Chem. Technol. Biotechnol. 2006, 81, 1246–1251. [Google Scholar] [CrossRef]
- Deng, W.-L.; Carpenter, C.; Yia, N.; Stephanopoulos, M.F. Comparison of the activity of Au/CeO2 and Au/Fe2O3 catalysts for the CO oxidation and the water-gas shift reactions. Top. Catal. 2007, 44, 199–208. [Google Scholar] [CrossRef]
- Kadkhodayan, A.; Brenner, A. Temperature-programmed reduction and oxidation of metals supported on γ-alumina. J. Catal. 1989, 117, 311–321. [Google Scholar] [CrossRef]
- Huang, J.; Dai, W.-L.; Fan, K.-N. Remarkable support crystal phase effect in Au/FeOx catalyzed oxidation of 1,4-butanediol to c-butyrolactone. J. Catal. 2009, 266, 228–235. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- Deng, W.-L.; Frenkel, A.I.; Si, R.; Flytzani-Stephanopoulos, M. Reaction-Relevant Gold Structures in the Low Temperature Water-Gas Shift Reaction on Au-CeO2. J. Phys. Chem. C 2008, 112, 12834–12840. [Google Scholar] [CrossRef]
- Zanella, R.; Giorgio, S.; Shin, C.H.; Henry, C.R.; Louis, C. Characterization and reactivity in CO oxidation of gold nanoparticles supported on TiO2 prepared by deposition-precipitation with NaOH and urea. J. Catal. 2004, 222, 357–367. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Aubulk (at. %) a | Ausurf (at. %) b | SBET (m2·g−1) c | Au 4f (eV) |
|---|---|---|---|---|
| Au/Fe_OH_11.2 | 0.53 | 0.67 (1.20) | 151 | 84.55 (84.2), 88.15 (87.9) |
| Au/Fe_OH_9.7 | 0.54 | - | 198 | - |
| Au/Fe_OH_8.2 | 0.52 | - | 203 | - |
| Au/Fe_OH_6.7 | 0.53 | 0.55 (0.42) | 212 | 84.6 (84.0), 88.15 (87.7) |
| Au/Fe_O_600 | 0.53 | - | 27 | - |
| Au/Fe_O_500 | 0.52 | - | 42 | - |
| Au/Fe_O_400 | 0.53 | - | 58 | - |
| Au/Fe_O_300 | 0.52 | 0.68 (0.74) | 97 | 84.7 (84.15), 88.4 (87.85) |
| Au/Fe_O_200 | 0.53 | 0.63 (0.54) | 136 | 84.35 (84.2), 88.05 (87.85) |
| Sample | Reduction Peak (°C) | Experimental H2-consump. (μmol·g−1) | Theoretical H2-consump. a (μmol·g−1) |
|---|---|---|---|
| DP_Au/Fe_OH_11.2 | 102 °C, 214 °C | 627 | 1300 |
| DP_Au/Fe_OH_9.7 | 107 °C, 204 °C | 549 | 1300 |
| DP_Au/Fe_OH_8.2 | 108 °C, 200 °C | 486 | 1300 |
| DP_Au/Fe_OH_6.7 | 116 °C, 195 °C | 452 | 1300 |
| DP_Au/Fe_O_200 | 105 °C | 723 | 1510 |
| DP_Au/Fe_O_300 | 96 °C, 198 °C | 778 | 1510 |
| DP_Au/Fe_O_400 | 90 °C, 231 °C | 1438 | 1510 |
| DP_Au/Fe_ O_500 | 230 °C | 861 | 1510 |
| DP_Au/Fe_ O_600 | 248 °C | 894 | 1510 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, H.-Z.; Guo, Y.; Wang, X.; Jia, C.-J.; Si, R. Gold-Iron Oxide Catalyst for CO Oxidation: Effect of Support Structure. Catalysts 2016, 6, 37. https://doi.org/10.3390/catal6030037
Cui H-Z, Guo Y, Wang X, Jia C-J, Si R. Gold-Iron Oxide Catalyst for CO Oxidation: Effect of Support Structure. Catalysts. 2016; 6(3):37. https://doi.org/10.3390/catal6030037
Chicago/Turabian StyleCui, Hui-Zhen, Yu Guo, Xu Wang, Chun-Jiang Jia, and Rui Si. 2016. "Gold-Iron Oxide Catalyst for CO Oxidation: Effect of Support Structure" Catalysts 6, no. 3: 37. https://doi.org/10.3390/catal6030037