Abstract
It is of great significance to develop sustainable processes of catalytic reaction. We report a selective procedure for the synthesis of acetylated bioactive compounds in water. The use of 1-acetylimidazole combined with Er(OTf)3 as a Lewis acid catalyst gives high regioselectivity and good yields for the acetylation of primary hydroxyl groups, as well as amino groups. The protection is achieved in short reaction times under microwave irradiation, and is successful even in the case of base-sensitive substrates.
1. Introduction
Protecting strategies are of critical importance in synthetic chemistry and represent important tools for industrial biotechnology. One of the most commonly used techniques for the protection of hydroxyl (OH) groups is acetylation. Regioselective acetylation is one of the strategies that chemists have tentatively developed, over time, in order to maximize the different reactivity of the primary hydroxyl groups in polyols and carbohydrates to be used as constituents of many biologically active compounds [1,2,3,4]. In the literature, several methods have been developed for the preparation of acetyl derivatives, using zeolites [5], various metal salts such as Mg(ClO4)2 and Zn(ClO4)2.6H2O [6,7], and some triflate catalysts or stoichiometric reagents [8,9].
Nevertheless, many of the proposed methods involve the use of non-environmentally friendly or expensive reagents, anhydrous environments and require the iterative blocking and de-blocking of all other potentially reactive OH functionalities in the molecule [10,11,12].
Earlier, carbonylimidazole derivatives were proven to be excellent acylating agents [13]. Imidazole carbamates and ureas have been successfully applied in the conversion of aliphatic and aromatic carboxylic acids into esters and amides in 70–93% yields [14]. In this endeavor, Sarpong et al. have reported the selective acetylation of indoles and oxazolidinones by using several reactive N-acyl imidazoles and/or carbamates; the reaction shows a high selectivity at the nitrogen atom of the non-nucleophilic azacycles, even in the presence of other strong nucleophiles, such as amines or hydroxyl groups [15]. The reaction, conducted at room temperature in acetonitrile for about 24 h, is catalyzed by 1,8-diazabicyclo[5.4.0]undec-5-ene (DBU) (20–50 mol %) [15]. Pei et al. reported a regioselectivebenzoylation of diols and carbohydrates using 1-benzoylimidazole and catalytic amounts of DBU in acetonitrile [16]. Nevertheless, most of these reactions were performed in conventional organic solvents because of the poor water solubility of carbonylimidazole derivatives.
The use of toxic and hazardous solvents in laboratories and in the chemical industry is considered a major risk for the environment, human health and the safety of workers. Green chemistry is a chemical philosophy encouraging the use of products and processes that reduce or eliminate the use and generation of harmful substances and solvents. Various methods, where organic synthesis can be performed without solvents, under mild conditions and with low energy consumption, have been developed. Conferences and symposia have promoted the use of alternative methods or “green” solvents [17], and water is undoubtedly the best choice to conduct chemical processes in “safe solvents”.
Only a few years ago, the use of water as solvent was considered a difficult journey because of the poor solubility of many organic compounds in aqueous solution. Nevertheless, in the last decade, the possible use of aqueous environments for chemical synthesis has directed studies towards the development of catalysts resistant to moisture. Moreover, in recent times, microwave (MW) irradiation as an alternative source of heating has been proposed in many reactions performed in water, such as Mannich reactions [18,19], substitution reactions [20,21], epoxide opening [22,23], Diels–Alder cycloadditions [24,25], heterocycle synthesis [26,27], carbonylation reactions [28,29,30] and carbon–carbon coupling reactions [31,32,33]. Microwave-assisted organic reaction offers several advantages over conventional heating, including homogeneous and rapid heating, and remarkable accelerations in reactions as a result of the heating rate producing high yields and lower quantities of side-products.
Recently, Pey et al. [34] reported the first example of acetylation of carbohydrates and diols in water using the water-soluble 1-acetylimidazole as an acetylating reagent. However, the acetylated products are only afforded after 16 h in moderate yields and in the presence of a strong base, such as tetramethylammonium hydroxide.
Over the years, the use of mild Lewis-acid catalysis has increased very quickly, and Er(OTf)3 has been proposed as an excellent catalyst in many organic transformations. Er(OTf)3is easy to handle, recyclable and is one of the cheapestcommercially available lanthanidetriflate derivatives.
Considering the stability and catalytic activity of Erbium (III) in water (that can be ascribed to their large ionic radii and an equilibrium between the Lewis acids and water [35]), and taking into account our experience in developing eco-friendly reactions [36,37,38,39,40,41] and selective procedures for the protection of natural compounds [42], we decided to test the reactivity of 1-acetylimidazole and the catalytic activity of Er(OTf)3 in water for the selective acetylation of bioactive compounds.
We propose here a catalytic procedure based upon the use of Er(OTf)3 that promotes a clean synthesis, avoiding the formation of byproducts and making the process appropriate even in the case of base-sensitive substrates.
2. Results
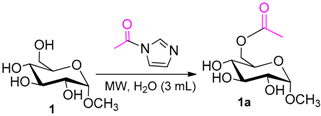
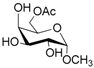
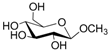
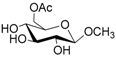
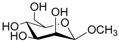
According to our ability in Er(III) catalysis in mild, non-dry reaction conditions, both in homogeneous and in heterogeneous phases [43,44,45,46,47,48,49], as well as our gained familiarity in MW/H2O synthesis [50] and “on water” reactions [42,51], we decided to test the potential use of MW in the regioselective acetylation reaction of alcohols and amino groups using 1-acetylimidazole as acetylating agent, and replacing the basic catalyst proposed by Pey et al. [35] with an eco-friendly Lewis acid catalyst like Er(OTf)3. Thus, preliminary experiments were carried out on the model reaction between methyl α-d-glucopyranoside (1 mmol) and 1-acetylimidazole (1.2 mmol) using water as the reaction solvent. Table 1 summarizes our preliminary results.

Table 1.
Optimization of regioselective acetylation of methyl α-d-glucopyranoside as the model substrate. a
In the first instance, the reaction was performed using Er(OTf)3 as catalyst, under MW irradiation. The use of 1.2 mmol of 1-acetylimidazole in the presence of 10% mmol of Er(OTf)3 provided the corresponding products in low yields, even after increasing the reaction times (Table 1, entries 1–3). Most likely, the catalytic activity of Er(III) is enhanced by the MW effect in the aqueous system. The same reaction, in fact, conducted under conventional heating, led to the formation of the acetylated product in only 14% yield (Table 1, entry 4).
Repetition of the reaction using a threefold molar excess of the acetylating agent, and extending the time of the reaction, resulted in an increase in the yield (Table 1, entries 7 and 8). Prolonged reaction times (60 min) caused lower isolated yield (Table 1, entry 9), probably due to decomposition of the starting material. No conversion of the starting substrate was obtained in the absence of catalyst also after a protracted reaction time (Table 1, entry 10). Notably, the use of an excess of catalyst did not affect the yield of the reaction (Table 1, entry 11), while reducing the amount of Er(III) resulted in poor reaction yields (Table 1, entry 12). The prolonged reaction time did not change the yield significantly (Table 1, entry 13). In any case, no by-product formation has been observed.
Thus, the optimized catalytic system (Table 1, entry 8) was chosen for the selective monoacetylation of a wide range of multifunctional natural compounds (Table 2), such as methyl d-pyranosides (Table 2, entries 1–6), phenyl d-pyranosides (Table 2, entries 7 and 8), nucleosides (Table 2, entries 9–12), phenolic antioxidant compounds (Table 2, entries 13–15) and aliphatic alcohols (Table 2, entries 16–19).
Table 2.
Selective acetylation using 1-acetylimidazole in the presence of Er(OTf)3under MWirradiation. a
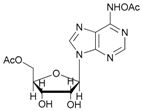
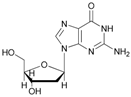
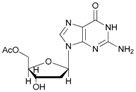
Increasing the steric hindrance of the substituents attached to the carbon atom 1, the reaction showed the same behavior as that of phenyl glucopyranoside (Table 2, entries 7 and 8). When the reagent system was used with substrates containing both primary and secondary hydroxyl groups, as well as amino groups—such as in the case of adenosine—the N,O-diacetylated derivative was obtained (Table 2, entry 9). Furthermore, the acetylation reaction of deoxyguanosine 11 led to the formation of the 5′-O-acetyl derivative as the main product, without any traces of the products acetylated on the secondary OH group and on the NH2 functionality (Table 2, entry 11).
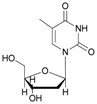
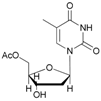
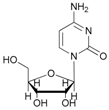
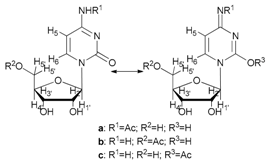
In the case of cytidine (12), the reaction deserves special attention; in fact, the presence of tautomerism in the molecule is responsible for the formation of the N-acetyl derivative as the main product (Table 2, entry 12). The 1H-NMR spectrum of the product resulting from the acetylation of cytidine showed the formation of three isomers indicated as N-acetylcytidine (A) in 40%, acetylcytidine (C) in 45% and 5′-O-acetylcytidine (B) in 15% yields, respectively (Figure 1).
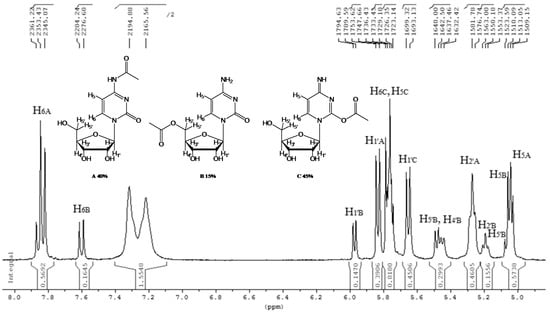
Figure 1.
1H-NMR spectrum of the crude product obtained from cytidine acetylation.
It is well known that rare-earth ions have high affinity for hard donor atoms, such as ligands containing oxygen or hybrid oxygen–nitrogen atoms [52]; various researchers have reported the extraction of lanthanides with amides, and their coordination with carbonyl groups for the synthesis of ternary 4f-element complexes [53,54].
In addition, the formation of ligands obtained by the bis-chelating mode of the pyrimidine carboxylate with lanthanides (III) is reported in crystallographic studies [55]. It can be supposed that the coordination with the carbonyl group of the pyrimidine base, assisted by an iminol–ketotautomerism of cytidine that shifts the equilibrium toward the iminol tautomer in acid conditions and at high temperatures [56], favours the formation of 2-O-acetyl-cytidine in 45% yield (Figure 2).
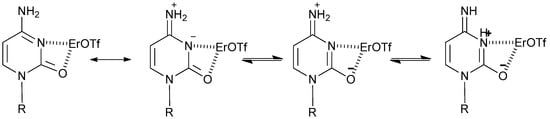
Figure 2.
Diagram of tautomeric forms of cytosine-Er(III) coordinate.
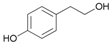
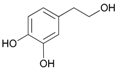
Attempted acetylation reaction of phenolic compounds, such as tyrosol and its derivatives, was particularly interesting. The reaction carried out on tyrosol led to the formation of a diacetylated product (Table 2, entry 13). On the contrary, the acetylation reaction of ortho- and meta-hydroquinone derivatives occurred only on the aliphatic OH group, probably because the hydrogenbonding to the adjacent groups hinders the acetylation reaction of the aromatic hydroxyl groups (Table 2, entries 14–16). When the method was extended to the protection of the primary hydroxyl group of glycerol, no formation of the primary acetylated product was observed. When the same reaction was carried out in 1 M aqueous NaCl, good regioselectivity and yield in the formation of glycerol 1,3-diacetate (17a) were obtained. Apparently, the presence of the salt prevents the formation of intramolecular H-bonds, favouring the acetylation reaction.
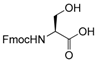
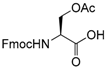
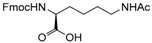
Therefore, our study proposes a new regioselective method that offers good yields, shorter reaction times and a possible extension to various substrates. With this aim, and to compare our procedure with Pei’s method [35], we applied our reaction conditions to base-sensitive substrates. The acetylation of amino groups is among the most widely used transformation in organic synthesis, and is frequently required during peptide synthesis. We attempted the acetylation of a side-chain-functionalized amino acid, such as lysine protected on the α-amino group with the base-labile [57,58,59,60,61,62] fluorenylmethyloxycarbonyl (Fmoc) protecting group (Table 2, entry 22). We observed the formation of the corresponding side chain N-acetylated N-Fmoc lysine 22a in high yields after only 10 min, without affecting the Fmoc protecting group. The procedure also works well for the acetylation of the OH group in the side-chain of N-Fmoc serine (Table 2, entry 21). Performing the acetylation reaction on the same substrates using Pei’s method, the removal of the base labile N-Fmoc protecting group occurred along with the formation of traces of by-products. In fact, thin layer chromatography (TLC) analysis of the reaction mixture after 16 h revealed the presence of dibenzofulvene, and a ninhydrin assay confirmed the formation of the free α-amino group.
3. Materials and Methods
3.1. General Methods
All chemicals and solvents were purchased from common commercial sources and were used as received without any further purification. All reactions were monitored by TLC on silica Merck 60 F254 pre-coated aluminum plates, and were developed by spraying with sulfuric acid in ethanol solution when possible. The tautomeric forms of acetyl cytosine were purified by semipreparative RP–HPLC chromatography (Phenomenex Jupiter C18, 250 × 10 mm, 10 µm, UV 272 nm, 4.0 mL/min, H2O 100%). Proton nuclear magnetic resonance (1H-NMR) spectra were recorded on a Brüker spectrometer at 300 MHz. Chemical shifts are reported in δ units (ppm) with tretramethylsilane (TMS) as reference (δ 0.00). All coupling constants (J) are reported in Hertz. Multiplicity is indicated by one or more of the following: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Carbon nuclear magnetic resonance (13C-NMR) spectra were recorded on a Brüker at 75 MHz. Chemical shifts are reported in δ units (ppm) relative to CDCl3 (δ 77.0).
MW-assisted reactions were performed on a Synthos 3000 instrument from Anton Paar (Graz, Austria), equipped with a 4 × 24 MG5 Rotor and an IR probe used for external temperature control.
LC–MS analyses were carried out using an Agilent 6540 UHD Accurate-Mass Q–TOF LC–MS (Agilent, Santa Clara, CA, USA) fitted with an electrospray ionisation source (Dual AJS ESI) operating in positive ion mode. Chromatographic separation was achieved using a C18 RP analytical column (Poroshell 120, SB-C18, 50 × 2.1 mm, 2.7 μm) at 30 °C with an elution gradient from 5% to 95% of B over 13 min, A being H2O (0.1% formic acid (FA)) and B being CH3CN (0.1% FA). Flow rate was 0.4 mL/min.
3.2. General Experimental Procedure for Microwave-Assisted Acetylation
The substrate (0.8 mmol) was dissolved in 3 mL of water in a glass vial.1-Acetylimidazole (2.4 mmol) and Er(OTf)3 (10 mol %) were then added to the solution. The mixture was reacted for 30 min in a Synthos 3000 microwave instrument, fixed on the temperature value of 60 °C (IR Limit). TLC analysis was used to monitor the progress of the reaction. After reaction completion, water was removed under reduced pressure and the resulting crude product was purified by flash chromatography (CH2Cl2/MeOH 9.5:0.5) (see Supplementary Materials).
- Methyl 6-O-acetyl α-d-glucopyranoside (1a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [63].
- Methyl 6-O-acetyl α-d-mannopyranoside (2a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [64].
- Methyl 6-O-acetyl α-d-galattopyranoside (3a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [65].
- Methyl 6-O-acetyl β-d-glucopyranoside (4a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [65].
- Methyl 6-O-acetyl β-d-mannopyranoside (5a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [64].
- Methyl 6-O-acetyl β-d-galattopyranoside (6a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [66].
- Phenyl 6-O-acetyl α-d-glucopyranoside (7a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [67].
- Phenyl 6-O-acetyl β-d-glucopyranoside (8a): 1H- and 13C-NMR (CDCl3, 300 MHz and 75.5 MHz, respectively) [67].
- N-acetyl-5′-O-acetyl adenosine (9a): 1H-NMR (CDCl3, 300 MHz): δ 2.02 (s, 3H, CH3COO), 2.18 (s, 3H, CH3CON), 3.85 (d, 1H, H5′, J = 13.15), 3.98 (d, 1H, H5′, Jgem = 13.15 Hz), 4.37 (s, 1H, H3′), 5.69 (s, 1H, H2′), 6.01–6.03 (m, 4H, H4′, H1′ 2OH), 7.84 (s, 1H, H8), 8.34 (s, 1H, H2). 13C-NMR (CDCl3, 75.5 MHz). HRMS (ESI) for [(C14H17N5O7) + Na]+: calcd 390.1026, found 390.1019 [M + Na]+.
- 5′-O-acetyl thymidine (10a): 1H-NMR (DMSO, 300 MHz): δ 1.93 (s, 3H, CH3), 2.09 (s, 1H, H2′), 2.13 (s, 3H, CH3COO), 2.18 (s, 1H, H2′), 2.46–2.44 (br, 1H, OH), 4.19 (t, 1H, H3′, J = 3.40 Hz), 4.27 (t, 1H, H4′, J = 3.29 Hz), 4.30 (dd, H5′, J = 11.84 Hz, J = 3.29 Hz), 4.36–4.40 (m, 1H, H5′), 3.98 (d, 1H, H5′, Jgem = 13.15 Hz), 6.30 (t, 1H, H1′, J = 6.47 Hz), 7.32 (s, 1H, H6), 9.78 (s, 1H, NH). 13C-NMR (DMSO, 75.5 MHz): 19.8, 21.5, 41.0, 63.9, 69.3, 82.7, 86.6, 109.8, 137.2, 150.6, 163.8, 170.3. HRMS (ESI) for [(C12H16N2O6) + Na]+: calcd 307.0906, found 307.0892 [M + Na]+.
- 5′-O-acetyl-2′-deoxyguanosine (11a): 1H-NMR (DMSO, 300 MHz): δ 2.07 (s, 3H, CH3COO), 2.36 (m, 1H, H2′), 2.77 (m, 1H, H2′), 3.57–3.58 (m br, 2H, H3′ OH), 3.99–4.02 (m, 2H, H5′), 5.28 (m, 1H, H4′), 6.10 (m, 1H, H1′), 6.72 (s, 2H, NH2), 7.94 (s, 1H, H8). 13C-NMR (DMSO, 75.5 MHz): 21.0, 40.7, 61.8, 75.3, 82.9, 85.2, 116.9, 135.4, 150.1, 154.2, 157.1, 170.3. HRMS (ESI) for [(C12H15N5O2) + Na]+: calcd 332.0971, found 332.0965 [M + Na]+.
- 2′-deoxy-3′,5′-di-O-acetylguanosine (11b): 1H-NMR (DMSO, 300 MHz): δ 2.03 (s, 3H, CH3COO), 2.08 (s, 3H, CH3COO), 2.41 (1H, ddd, 2J = 14.26 Hz, 3J1 = 5.47 Hz, 3J2 = 2.1 Hz, H-2b′) (m, 1H, H2′), 2.92 (1H, ddd, 2J = 14.27 Hz, 3J1 = 8.7 Hz, 3J2 = 6.4 Hz, H-2a′), 4.15-4.30 (m, 3H, H5′, H3′), 5.28 (d, 1H, H4′, J = 6.83), 6.08–6.15 (m, 1H, H1′), 6.52 (2H, s, NH2), 7.92 (1H, s, H8), 10.68 (1H, s, NH, 13C-NMR (DMSO, 75.5 MHz): 20.4, 20.7, 35.4, 63.6, 74.4, 81.4, 82.9, 116.7, 151.0, 153.7, 156.9, 169.9, 170.1.45. HRMS (ESI) for [(C14H17N5O6) + Na]+: calcd 374.1077, found 374.1071 [M + Na]+.
- N-acetyl-cytidine (12a), 5′-O-acetyl-cytidine (12b), 6-O-acetyl-cytidine (12c): 1H-NMR (DMSO, 300 MHz): δ 2.05 (s, 3H, CH3A), 2.08 (s, 6H, CH3B + CH3C), 3.51–3.71 (m, 6H, H5′A, H5′C, H3′C, H2′C), 3.82–4.01 (m, 3H, H5′A, H5′C, H4′A), 4.11–4.29 (m, 4H, H3′A, H4′A, H4′C, H3′B), 5.03–5.07 (m, 3H, H5A, H5B, H5′B), 5.19 (t, 1H, H2′B, J = 4.81 Hz), 5.27 (t, 1H, H2′A, J = 5.04 Hz), 5.45 (d, H4′B, J = 5.04 Hz), 5.48 (d, 1H, H5′B), 5.65 (d, 1H, H1′C, J = 6.19 Hz), 5.76 (m, 2H, H6C, H5C), 5.83 (d, 1H, H1′A, J = 5.96), 5.97 (d, 1H, H1′B), 7.60 (d, 1H, H6B, J = 7.56 Hz), 7.83 (d, 1H, H6A, J = 7.56 Hz). 13C-NMR (DMSO, 75.5 MHz): 60.5, 60.7, 68.1, 69.6, 71.8, 72.4, 73.2, 75.3, 79.1, 80.4, 81.9, 84.7, 86.7, 88.7, 90.0, 94.2, 94.3, 94.4, 141.3, 155.2, 155.4, 155.4, 165.5, 165.5, 169.6, 169.7, 178.3. HRMS (ESI) for [(C11H18N3O6) + Na]+: calcd 311.1093, found 311.1093 [M + Na]+, 311.1095 [M + Na]+, 311.1086 [M + Na]+.
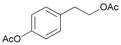
- 4-[2-(Acetyloxy)ethyl]phenyl acetate (13a): 1H-NMR (DMSO, 300 MHz): δ 1.98 (s, 3H, CH3), 2.25 (s, 3H, CH3), 2.88 (t, 2H, CH2, J = 7.50 Hz), 4.20 (t, 2H, CH2, J = 7.11 Hz), 7.05 (d, 2H, Har, J = 8.49 Hz), 7.28 (d, 2H, Har, J = 8.46 Hz), 13C-NMR (DMSO, 75.5 MHz): Spectroscopic data compared to those reported in the literature [68].
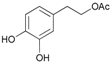
- 3,4-dihydroxyphenethyl acetate (14a): 1H-NMR (DMSO, 300 MHz): δ 1.97 (s, 3H, CH3), 2.68 (t, 2H, CH2, J = 6.87 Hz), 4.10 (t, 2H, CH2, J = 7.11 Hz), 6.45–6.48 (m, 1H, Har), 6.48–6.62 (m, 2H, Har). 13C-NMR (DMSO, 75.5 MHz): 20.4, 34.3, 65.2, 115.0, 116.9, 121.1, 128.9, 144.2, 145.6, 170.6. HRMS (ESI) for [(C10H12O4) + H]+: calcd 197.0814, found 197.0818, 219.0635 [M + Na]+.
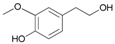
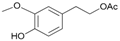
- 4-hydroxy-3-methoxyphenethyl acetate (15a): 1H-NMR (CDCl3, 300 MHz): δ 1.99 (s, 3H, CH3), 2.25 (s, 3H, CH3), 2.85 (t, 2H, CH2, J = 7.50 Hz), 3.73 (s, 3H, CH3), 4.33 (t, 2H, CH2, J = 7.11 Hz), 7.03 (d, 2H, Har, J = 8.43 Hz), 7.25 (d, 2H, Har, J = 8.43 Hz). 13C-NMR (CDCl3, 75.5 MHz): 20.4, 34.8, 56.9, 64.8, 113.7, 151.8, 121.5, 133.1, 142.9, 151.1, 170.0. HRMS (ESI) for [(C11H14O4) + H]+: calcd 211.0970, found 211.0978, 233.0781 [M + Na]+.
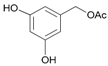
- Acetyl-3,5-dihydroxybenzyl alcohol (16a): 1H-NMR (DMSO, 300 MHz): 2.05 (s, 3H, CH3), 4.88 (s, 2H, CH2), 6.14–6.18 (m, 3H, Har). 13C-NMR (DMSO, 75.5 MHz): 20.6, 35.1, 64.8, 121.5, 128.3, 134.1, 143.2, 169.1, 170.0. 13C-NMR (DMSO, 75.5 MHz): 20.6, 65.3, 101.9, 105.6, 105.6, 137.9, 158.3, 170.0. HRMS (ESI) for [(C9H11O4) + H]+: calcd 183.0657, found 183.0655, 205.0480 [M + Na]+.
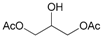
- Glycerol 1,3-diacetate (17a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those reported in the literature [69].
- n-Butyl acetate (18a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz) [70].
- 1,4-Butanediol, diacetate (19a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those of the pure product. GC-MS (EI): 114.0 (11), 73.0 (20), 71.0 (30), 54.0 (45), 43.0 (100) [71].
- n-Octyl acetate (20a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those of the pure product. GC-MS (EI): 112.0 (10), 84.0 (30), 70.0 (36), 56.0 (30), 43.0 (100) [71].
- Fmoc-Ser(Ac)-OH (21a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those of the pure product.
- Fmoc-Lys(Ac)-OH (22a): 1H- and 13C-NMR (CDCl3, 300 MHz, and 75.5 MHz). Spectroscopic data compared to those of the pure product.
4. Conclusions
An effective procedure for the acetylation of various natural compounds has been developed. The reaction showed high selectivity for the acetylation of the primary hydroxyl groups in carbohydrates and diols; in the case of nucleosides, the acetylation reaction led to the formation of the 5′-O-acetyl derivative.
The procedure, unlike most other acetylation methods reported in the literature, allows the use of water as the solvent, thus avoiding the use of organic solvents. Moreover, other advantages of this method are the use of a catalytic amount of Er(OTf)3, short reaction time and high regioselectivity. The catalyst is inexpensive and can be easily and consistently recovered [44]. It was reused for two consecutive cycles without any significant loss in catalytic activity in the synthesis of 1a (56% and 45%), with negligible release into the environment.
The use of Lewis-acid catalysis is a valuable method with respect to the previously reported procedure [25]: the reaction yields are increased, the reaction times are very short and the acetylating agent is activated, allowing a greater selectivity in the acetylation process, thus avoiding the formation of by-products. Furthermore, the reagent system is also applicable to base-labile substrates such as N-Fmoc amino acids, with the formation of the desired side-chain-acetylated products without the removal of the α-amino protecting group. Thus, our reaction conditions can be considered orthogonal to the basic conditions reported by Pey et al. [25].
Supplementary Materials
The following are available online at www.mdpi.com/2073-4344/7/9/269/s1, NMR spectra for all new products.
Author Contributions
M.N. and A.P. conceived and designed the experiments; F.O. and P.C. performed the experiments; A.D.N. and L.M. analyzed the data; M.O. contributed reagents/materials/analysis tools; M.N. and M.L.D.G. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lu, K.; Hsieh, S.; Patkar, L.N.; Chen, C.; Lin, C. Simple and efficient Per-O-acetylation of Carbohydrates by lithium perchlorate catalyst. Tetrahedron 2004, 60, 8967–8973. [Google Scholar] [CrossRef]
- Kasiganesan, H.; Wright, G.L.; Chiacchio, M.A.; Gumina, G. Novel l-adenosine analogs as cardioprotective agents. Bioorg. Med. Chem. 2009, 17, 5347–5352. [Google Scholar] [CrossRef] [PubMed]
- Ferry, A.; Guinchard, X.; Retailleau, P.; David, C. Synthesis, characterization, and coupling reactions of six-membered cyclic p-chiral ammonium phosphonite–boranes; reactive H-phosphinateequivalents for the stereoselective synthesis of glycomimetics. J. Am. Chem. Soc. 2012, 134, 12289–12301. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.X.; Rahm, M.; Wu, B.; Dong, H. H-bonding activation in highly regioselective acetylation of diols. J. Org. Chem. 2013, 78, 11618–11622. [Google Scholar] [CrossRef] [PubMed]
- Ballini, R.; Bosica, G.; Carloni, L.; Maggi, R.; Sartori, G. Zeolite HSZ-360 as a new reusable catalyst for the direct acetylation of alcohols and phenols under solventless conditions. Tetrahedron Lett. 1998, 39, 6049–6052. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Dalpozzo, R.; Marcantoni, E.; Massaccesi, M.; Rinaldi, S.; Sambri, L. Mg(ClO4) as a powerful catalyst for the acylation of alcohols under solvent-free conditions. Synlett 2003, 2003, 39–42. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Dalpozzo, R.; Marcantoni, E.; Massaccesi, M.; Sambri, L. Zn(ClO4) 6H2O as a powerful catalyst for a practical acylation of alcohols with acid anhydrides. Eur. J. Org. Chem. 2003, 23, 4611–4617. [Google Scholar] [CrossRef]
- Chandra, K.L.; Saravanan, P.; Singh, R.K.; Singh, V.K. Lewis acid catalyzed acylation reactions: Scope and limitations. Tetrahedron 2002, 58, 1369–1374. [Google Scholar] [CrossRef]
- Dalpozzo, R.; Nino, A.D.; Maiuolo, L.; Procopio, A.; Nardi, M.; Bartoli, G.; Romeo, R. Highly efficient and versatile acetylation of alcohols catalyzed by cerium (III) triflate. Tetrahedron Lett. 2003, 44, 5621–5624. [Google Scholar] [CrossRef]
- Danieli, B.; Luisetti, M.; Sampognato, G.; Carrea, G.; Riva, S. Regioselective acylation of polyhydroxylated natural compounds catalyzed by Candida Antarctica lipase B (Novozym 435) in organic solvents. J. Mol. Catal. B Enzym. 1997, 3, 193–201. [Google Scholar] [CrossRef]
- Sugahara, K.; Satake, N.; Kamata, K.; Nakajima, T.; Mizuno, N. A basic germanodecatungstate with a −7 charge: Efficient chemoselective acylation of primary alcohols. Angew. Chem. Int. Ed. 2014, 53, 13248–13252. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, W.; William, J.M.; Onomura, O. Selective monobenzoylation of 1,2- and 1,3-diols catalyzed by Me2SnCl2in water (organic solvent free) under mild conditions. J. Org. Chem. 2012, 77, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, R.U.; Driguez, H. Chemical synthesis of 2-acetamido-2-deoxy-4-O-(alpha-l-fucopyranosyl)-3-O-(beta-d-galactopyranosyl)-d-glucose. Lewis a blood-group antigenic determinant. J. Am. Chem. Soc. 1975, 97, 4063–4069. [Google Scholar] [CrossRef]
- Heller, S.T.; Sarpong, R. Chemoselectiveesterification and amidation of carboxylic acids with imidazole carbamates and ureas. Org. Lett. 2010, 12, 4572–4575. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.T.; Schultz, E.E.; Sarpong, R. Chemoselective N-acylation of indoles and oxazolidinones with carbonylazoles. Angew. Chem. Int. Ed. 2012, 51, 8304–8308. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hou, C.; Ren, J.; Xin, X.; Xu, H.; Pei, Y.; Dong, H.; Pei, Z. Regioselectivebenzoylation of diols and carbohydrates by catalytic amounts of organobase. Molecules 2016, 21, 641. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.; Ali, M.A.; Clifford, A.A. Reactions in unusual media. Curr. Top. Med. Chem. 2004, 4, 729–771. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Dou, R.; Song, G.; Jiang, J. Dramatically accelerated synthesis of aminoketones via aqueous mannich reaction under combined microwave and ultrasound irradiation. Synlett 2005, 14, 2245–2247. [Google Scholar] [CrossRef]
- Shi, L.; Wang, M.; Zhang, F.M.; Fan, C.A. Microwave-promoted three-component coupling of aldehyde, alkyne, and amine via C-H activation catalyzed by copper in water. Org. Lett. 2004, 6, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Cherng, Y.J. Efficient nucleophilic substitution reaction of aryl halides with amino acids under focused microwave irradiation. Tetrahedron 2000, 56, 8287–8289. [Google Scholar] [CrossRef]
- Kaval, N.; Dehaen, W.; Matyus, P.; Van der Eycken, E. Convenient and rapid microwave-assisted synthesis of pyrido-fused ring systems applying the tert-amino effect. Green Chem. 2004, 6, 125–127. [Google Scholar] [CrossRef]
- Pironti, V.; Colonna, S. Microwave-promoted synthesis of β-hydroxy sulfides and β-hydroxysulfoxides in water. Green Chem. 2005, 7, 43–45. [Google Scholar] [CrossRef]
- Lindsay, K.B.; Pyne, S.G. Studies on the synthesis of croomine: Synthesis of the tricyclic B,C,D-ring core structure. Synlett 2004, 5, 779–782. [Google Scholar]
- Chen, I.H.; Young, J.N.; Yu, S.J. Recyclable organotungsten Lewis acid and microwave assisted Diels-Alder reactions in water and in ionic liquids. Tetrahedron 2004, 60, 11903–11909. [Google Scholar] [CrossRef]
- Kranjc, K.; Kocevar, M.; Iosif, F.; Coman, S.M.; Parvulescu, V.I.; Genin, E.; Genet, J.P.; Michelet, V. Efficient and green access to functionalized and highly constrained heteropolycyclic derivatives via a microwave accelerated Diels-Alder cycloaddition and hetereogeneous hydrogenation sequence. Synlett 2006, 2006, 1075–1079. [Google Scholar]
- Molteni, V.; Hamilton, M.M.; Mao, L.; Crane, C.M.; Termin, A.P.; Wilson, D.M. Aqueous one-pot synthesis of pyrazoles, pyrimidines and isoxazoles promoted by microwave irradiation. Synthesis 2002, 12, 1669–1674. [Google Scholar] [CrossRef]
- Bryson, T.A.; Stewart, J.J.; Gibson, J.M.; Thomas, P.S.; Berch, J.K. Green heterocycle synthesis, isochromenones and artemidin. Green Chem. 2003, 5, 174–176. [Google Scholar] [CrossRef]
- Kaiser, N.F.K.; Hallberg, A.; Larhed, M. In situ generation of carbon monoxide from solid molybdenum hexacarbonyl. A convenient and fast route to palladium-catalyzed carbonylation reactions. J. Comb. Chem. 2002, 4, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Georgsson, J.; Hallberg, A.; Larhed, M. Rapid palladium-catalyzed synthesis of esters from aryl halides utilizing Mo(CO)6as a solid carbon monoxide source. J. Comb. Chem. 2003, 5, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Wannberg, J.; Larhed, M. Increasing rates and scope of reactions: Sluggish amines in microwave-heated aminocarbonylation reactions under air. J. Comb. Chem. 2003, 68, 5750–5753. [Google Scholar]
- Leadbeater, N.E.; Marco, M. Rapid and amenable suzuki coupling reaction in water using microwave and conventional heating. J. Org. Chem. 2003, 68, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Capek, P.; Pohl, R.; Hocek, M. Cross-coupling reactions of unprotected halopurine bases, nucleosides, nucleotidesand nucleoside triphosphates with 4-boronophenylalanineinwater. Synthesis of (purin-8-yl)- and (purin-6-yl)phenylalanines. Org. Biomol. Chem. 2006, 4, 2278–2284. [Google Scholar] [CrossRef] [PubMed]
- Arvela, R.K.; Leadbeater, N.E. Microwave-promoted heck coupling using ultralow metal catalyst concentrations. J. Org. Chem. 2005, 70, 1786–1790. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wei, P.; Pei, Y.; Xu, H.; Xina, X.; Pei, Z. Regioselective acetylation of carbohydrates and diols catalyzed by tetramethyl-ammonium hydroxide in water. Green Chem. 2014, 16, 4510–4514. [Google Scholar] [CrossRef]
- Kobayashi, S.; Nagayama, S.; Busujima, T. Lewis acid catalysts stable in water. Correlation between catalytic activity in water and hydrolysis constants and exchange rate constants for substitution of inner-sphere water ligands. J. Am. Chem. Soc. 1998, 120, 8287–8288. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Barattucci, A.; Bonaccorsi, P.; Leggio, A.; Minuti, L.; Romio, E.; Temperini, A.; Siciliano, C. Deprotection/reprotection of the amino group in α-amino acids and peptides. A one-pot procedure in [Bmim][BF4] ionic liquid. RSC Adv. 2014, 4, 2678–2686. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Gagliardi, A.; Leggio, A.; Leotta, V.; Romio, E.; Liguori, A. N-Urethane protection of amines and amino acids in an ionic liquid. RSC Adv. 2015, 5, 63407–63420. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Costanzo, P.; De Nino, A.; Maiuolo, L.; Nardi, M.; Olivito, F.; Procopio, A. Simple and efficient Fmoc removal in ionic liquid. RSC Adv. 2017, 7, 36482–36491. [Google Scholar] [CrossRef]
- Procopio, A.; Dalpozzo, R.; De Nino, A.; Maiuolo, L.; Nardi, M.; Russo, B. Synthesis of acetonides from epoxides catalyzed by erbium(III) triflate. Adv. Synth. Catal. 2005, 347, 1447–1450. [Google Scholar] [CrossRef]
- Dalpozzo, R.; De Nino, A.; Nardi, M.; Russo, B.; Procopio, A. 1,2-Diacetates by epoxide ring opening promoted by erbium(III) triflate. Arkivoc 2006, VI, 67–73. [Google Scholar]
- Dalpozzo, R.; Nardi, M.; Oliverio, M.; Paonessa, R.; Procopio, A. Erbium(III) triflate is a highly efficient catalyst for the synthesis of β-alkoxy alcohols, 1,2-diols and β-hydroxysulfides by ring opening of epoxides. Synthesis 2009, 2009, 3433–3438. [Google Scholar]
- Oliverio, M.; Nardi, M.; Cariati, L.; Vitale, E.; Bonacci, S.; Procopio, A. “On Water” MW-Assisted Synthesis of Hydroxytyrosol Fatty Esters. ACS Sustain. Chem. Eng. 2016, 4, 4661–4665. [Google Scholar] [CrossRef]
- Procopio, A.; Das, G.; Nardi, M.; Oliverio, M.; Pasqua, L. A mesoporous ErIII-MCM-41 catalyst for the cyanosilylation of aldehydes and ketones under solvent-free conditions. ChemSusChem 2008, 1, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Nardi, M.; Cozza, A.; De Nino, A.; Oliverio, M.; Procopio, A. One-pot synthesis of dibenzo [b,e] [1,4] diazepin-1-ones. Synthesis 2012, 44, 800–804. [Google Scholar] [CrossRef]
- Nardi, M.; Cozza, A.; Maiuolo, L.; Oliverio, M.; Procopio, A. 1,5-Benzoheteroazepines through eco-friendly general condensation reactions. Tetrahedron Lett. 2011, 52, 4827–4834. [Google Scholar] [CrossRef]
- Procopio, A.; Costanzo, P.; Curini, M.; Nardi, M.; Oliverio, M.; Sindona, G. Erbium(III) chloride in ethyl lactate as a smart ecofriendly system for efficient and rapid stereoselective synthesis of trans-4,5-diaminocyclopent-2-enones. ACS Sustain. Chem. Eng. 2013, 1, 541–544. [Google Scholar] [CrossRef]
- Nardi, M.; Oliverio, M.; Costanzo, P.; Sindona, G.; Procopio, A. Eco-friendly stereoselective reduction of α,β-unsaturated carbonyl compounds by Er(OTf)3/NaBH4in 2-MeTHF. Tetrahedron 2015, 71, 1132–1135. [Google Scholar] [CrossRef]
- Oliverio, M.; Nardi, M.; Cariati, L.; Vitale, E.; Bonacci, S.; Procopio, A. Facile ecofriendly synthesis of monastrol and its structural isomers via biginelli reaction. ACS Sustain. Chem. Eng. 2016, 2, 1228–1233. [Google Scholar] [CrossRef]
- De Nino, A.; Maiuolo, L.; Merino, P.; Nardi, M.; Procopio, A.; Roca-López, D.; Russo, B.; Algieri, V. New efficient organocatalyst supported on simple ionic liquid as recoverable system for asymmetric diels-alder reaction in presence of water. ChemCatChem 2015, 7, 830–835. [Google Scholar] [CrossRef]
- Nardi, M.; Herrera Cano, N.; Costanzo, P.; Oliverio, M.; Sindona, G.; Procopio, A. Aqueous MW eco-friendly protocol for amino group protection. RSC Adv. 2015, 5, 18751–18760. [Google Scholar] [CrossRef]
- Procopio, A.; Cravotto, G.; Oliverio, M.; Costanzo, P.; Nardi, M.; Paonessa, R. An eco-sustainable erbium(III)-catalyzed method for formation/cleavage of O-tert-butoxy carbonates. Green Chem. 2011, 13, 436–443. [Google Scholar] [CrossRef]
- Musikas, C. Solvent extraction for the chemical separation of the 5f elements. Inorg. Chem. Acta 1987, 140, 197–206. [Google Scholar] [CrossRef]
- Musikas, C.; Hubert, H. Extraction by N,N′-tetraalkylmalonamides II. Sol. Extr. Ion Exch. 1987, 5, 877–893. [Google Scholar] [CrossRef]
- Narita, H.; Yaita, T.; Tachimori, S. Solvent extraction of trivalent lanthanoid ions with N,N’-Dimethyl-N,N′-Diphenyl-3-Oxapentanediamide. Radiochim. Acta 1998, 81, 223–226. [Google Scholar] [CrossRef]
- Narita, H.; Yaita, T.; Tachimori, S. Study on the extraction of trivalent lanthanide ions with N,N′-dimethyl-N,N′-diphenyl-malonamide and diglycolamide. Radioanal. J. Nucl. Chem. 1999, 239, 381–384. [Google Scholar] [CrossRef]
- Cepeda, J.; Balda, R.; Beobide, G.; Castillo, O.; Fernández, J.; Luque, A.; Pérez-Yáñez, S.; Román, P.; Sánchez, D.V. Lanthanide(III)/Pyrimidine-4,6-dicarboxylate/Oxalate extended frameworks: A detailed study based on the lanthanide contraction and temperature effects. Inorg. Chem. 2011, 50, 8437–8451. [Google Scholar] [CrossRef] [PubMed]
- Kocienski, P.J. Protecting Groups; Georg ThiemeVerlag: Stuttgart, Germany, 2005. [Google Scholar]
- Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A preparation of N-Fmoc-N-methyl-alpha-amino acids and N-nosyl-N-methyl-alpha-amino acids. Amino Acids 2010, 38, 133–143. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A new non-natural arginine-like amino acid derivative with a sulfamoyl group in the side-chain. Amino Acids 2010, 38, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, M.L.; Leggio, A.; Malagrinò, F.; Romio, E.; Siciliano, C.; Liguori, A. N-Methylated α-Amino acids and peptides: Synthesis and biological activity. Mini Rev. Med. Chem. 2016, 16, 683–690. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Di Gioia, M.L.; Liguori, A.; Perri, F.; Siciliano, C.; Spinella, M. N-alkylation of N-aryl-α-amino acid methyl esters by trialkyloxonium tetrafluoroborates. Tetrahedron 2011, 67, 9708–9714. [Google Scholar] [CrossRef]
- Siciliano, C.; Barattucci, A.; Bonaccorsi, P.; Di Gioia, M.L.; Leggio, A.; Minuti, L.; Romio, E.; Temperini, A. Synthesis of d-erythro-sphinganine through serine-derived α-amino epoxides. J. Org. Chem. 2014, 79, 5320–5326. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Brufani, M.; Melchioni, C.; Romagnoli, P. Protection of primary alcoholic function with rare-earths salts. Tetrahedron Lett. 1997, 38, 651–652. [Google Scholar] [CrossRef]
- Jansson, P.E.; Kenne, L.; Schweda, E. Nuclear magnetic resonance and conformational studies on monoacetylated methyl d-gluco- and d-galacto-pyranosides. J. Chem. Soc. Perkin Trans. 1987, 1, 377–383. [Google Scholar] [CrossRef]
- Osada, M.; Kikuta, K.; Yoshida, K.; Oqata, M.; Usui, T. Non-catalytic synthesis of Chromogen I and III from N-acetyl-d-glucosamine in high-temperature water. Green Chem. 2013, 15, 2960–2966. [Google Scholar] [CrossRef]
- McClure, M.S.; Berry, M.B.; Caine, D.; Crawford, C.; Crump, B.C.; Glover, B.N.; Kedia, S.B.; Millar, A.; Mitchell, M.B.; Nichols, C.J.; et al. Highly selective primary alkoxycarboxylation and esterification of unprotected pyranose derivatives mediated by scandium(III) triflate catalysis. Eur. J. Org. Chem. 2012, 19, 3561–3565. [Google Scholar] [CrossRef]
- Ciuffreda, P.; Casati, S.; Manzocchi, A. Complete (1)H and (13)C NMR spectral assignment of alpha- and beta-adenosine, 2’-deoxyadenosine and their acetate derivatives. Magn. Reson. Chem. 2007, 45, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, B.; Phukan, P. A new ferrocene-based bulky pyridine as an efficient reusable homogeneous catalyst. RSC Adv. 2013, 3, 15327–15336. [Google Scholar] [CrossRef]
- Lange, K.; Koenig, A.; Roegler, C.; Seeling, A.; Lehmann, J. NO donors—Part 18: Bioactive metabolites of GTN and PETN—Synthesis and vasorelaxant properties. Bioorg. Med. Chem. Lett. 2009, 19, 3141–3144. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.; Zhang, G.; Ren, X.; Xu, X.; Yang, R.; Luo, S.; Yin, S. Air-stable titanocenebis(perfluorooctanesulfonate) as a new catalyst for acylation of alcohols, phenols, thiols, and amines under solvent-free condition. J. Organomet. Chem. 2010, 695, 1182–1188. [Google Scholar] [CrossRef]
- Ruijie, Z.; Hongting, S.; Yongcang, Z.; Yan, F.; Zhi, C.; Junfeng, W.; Man, C.; Manzhou, Z.; Qingxiang, G. Heterobimetallicdinuclear lanthanide alkoxide complexes as acid-basedifunctional catalysts for transesterification. J. Org. Chem. 2014, 79, 9246–9252. [Google Scholar]




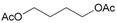



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).