Evolution of Water Diffusion in a Sorption-Enhanced Methanation Catalyst
by
 , , ,
, , ,
Renaud Delmelle
1,* ,
,
Jasmin Terreni
2,
Arndt Remhof
3 ,
,
Andre Heel
1,
Joris Proost
4 and
Andreas Borgschulte
2 1
Institute of Materials and Process Engineering (IMPE), Zurich University of Applied Sciences (ZHAW), Technikumstrasse 9, CH-8401 Winterthur, Switzerland
2
Laboratory for Advanced Analytical Technologies, Swiss Federal Laboratories for Materials Science and Technology (Empa), Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
3
Materials for Energy Conversion, Swiss Federal Laboratories for Materials Science and Technology (Empa), Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
4
Institute of Mechanics, Materials and Civil Engineering (iMMC), Université catholique de Louvain, Place Sainte-Barbe 2, B-1348 Louvain-la-Neuve, Belgium
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(9), 341; https://doi.org/10.3390/catal8090341
Submission received: 28 May 2018
/
Revised: 18 August 2018
/
Accepted: 18 August 2018
/
Published: 21 August 2018
(This article belongs to the Special Issue Catalytic, Photocatalytic and Electrocatalytic Processes for the Valorisation of CO2)
Abstract
:Sorption-enhanced methanation has consequent advantages compared to conventional methanation approaches; namely, the production of pure methane and enhanced kinetics thanks to the application of Le Châtelier’s principle. In this paper, we address the question of the long-term stability of a sorption-enhanced methanation catalyst-support couple: Ni nanoparticles on zeolite 5A. Compared to most conventional methanation processes the operational conditions of sorption-enhanced methanation are relatively mild, which allow for stable catalyst activity on the long term. Indeed, we show here that neither coking nor thermal degradation come into play under such conditions. However, a degradation mechanism specific to the sorption catalysis was observed under cyclic methanation/drying periods. This severely affects water diffusion kinetics in the zeolite support, as shown here by a decrease of the water-diffusion coefficient during multiple cycling. Water diffusion is a central mechanism in the sorption-enhanced methanation process, since it is rate-limiting for both methanation and drying.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The key issues of intermittency and dispersion of primary renewable electricity sources find an answer in power-to-gas (P2G) strategies, where the excess of renewable electricity is converted into synthetic gas fuels, using hydrogen produced by water electrolysis as a primary reactant [1]. Renewable methane is produced from renewable hydrogen and carbon dioxide with a high efficiency (Sabatier reaction). This process can be implemented on a large scale [2], namely because renewable methane plants are based on relatively simple chemical reactors using earth-abundant Ni-based catalysts located near areas of renewable electricity production, abundant CO2 sources such as biogas production and access to the natural gas grid.
Established methanation processes [3,4] involve fixed-bed, fluidized-bed and three-phase reactors with classic metal-support catalyst systems. Although other elements are studied at a fundamental level (e.g., Ru [5], Co [6], Mo [7] and Fe [8]), nickel remains the most suitable active metal (and by far the most widely used in commercial applications) when considering activity, selectivity and price [3,9]. However, the reaction temperature is above 250 °C, resulting in a thermodynamically limited conversion yield of less than 96% [10], which is further reduced by finite kinetics. Although the latter may be improved by appropriate catalysts, the thermodynamic limit can only be overcome by modifying thermodynamic conditions. A straightforward possibility is to increase the reaction pressure, which comes with an additional energy cost. The thermodynamic equilibrium is also shifted by the active removal of the water product from the catalyst reaction centers by adsorbing it in the water affine catalyst support such as zeolites in order to improve the reaction yield and kinetics (i.e., making use of the well-known Le Châtelier’s principle) [11,12]. We have shown that this process runs optimally with specific parameters, notably with gas hourly space velocities (GHSV) which are lower by orders of magnitude than processes used in classic catalyzed methanation (i.e., on the order of 100 h−1 [13]). The reason for this is that, under certain gas flow conditions, moisture evolves in the reactor as a stable water front. As long as this front does not reach the reactor outlet (sorption-enhanced mode), pure methane is produced. When the zeolite support is saturated with water, a drying step is required. Specific criteria also come into play in terms of reactor temperature: an optimum needs to be found between the zeolite support water sorption capacity and the metal catalytic activity. In the case of Ni nanoparticles on zeolite 5A, the optimum is 300 °C at atmospheric pressure [13]. The need for a fine optimization of sorption-enhanced processes was also recently shown by numerical simulations [14].
Similar to normal methanation conditions, nickel is facing durability issues when used as a sorption catalyst that simultaneously affect the process performance, cost and environmental impact. Depending on the process considered and on the methanation conditions—GHSV, temperature, pressure, stoichiometry, and reactant purity—different deactivation mechanisms can potentially occur [3,15]: poisoning [16], fouling (coking) [17], thermal degradation [18], mechanical degradation (attrition, crushing) [19] and corrosion (leaching) [16,20]. Despite their differences, these mechanisms all affect the concentration of active sites on the catalyst surface, in turn lowering the apparent rate constant for methanation. The development of effective solutions to these deactivation issues is a crucial research topic for future applications [9,15]. Another emerging field is the synergy between the catalyst and its support [9,21], which can affect the system performance in many ways. There was concern that the Ni sorption catalyst is particularly sensitive to coking due to the low water concentration at the catalytically active centers [10,16,19]. We show in this publication that the degradation of the catalytic activity relevant during the reaction phase of a sorption catalyst is negligible at optimized conditions. We attribute this to the encapsulation of the Ni-particle in the inner of the zeolite structure, preventing irreversible carbon growth, but allowing exchange of reactants and products to and from the active surface, respectively. However, we observe a diminution of the water diffusion, a process relevant only in sorption catalysts during the regeneration (drying) phase.
In this paper, a focus is made on the long-term performance of the catalyst-support couple under optimum conditions for sorption-enhanced methanation, the latter of which were determined elsewhere [13], using nickel nanoparticles as the active metal and zeolite 5A as the support. The choice of zeolite 5A is justified elsewhere [22,23]. In short, the most important parameters for the choice of a sorption enhanced support are (i) the pore size, which allows manipulating the reaction path towards the desired reaction intermediates and products—because of the differences in product kinetic diameters, pore sizes ≥5 Å favor CH4 formation while pore sizes ≤3 Å favor CO formation—and (ii) the water sorption capacity at the catalyst operation temperature, which determines the extent of Le Châtelier’s effect.
The performance of this system was studied by means of a thermogravimetric method; i.e., with specimens under the gas stream in a magnetic suspension balance. This approach allows for both equilibrium and kinetic analyses through real-time monitoring of the specimen mass change. This reflects the evolution of the Sabatier reaction, because water is one of its products and is entirely adsorbed on the zeolite support as long as the reaction is sorption-enhanced. We also used this experimental approach to measure the equilibrium CO2 and H2O uptake capacity at conditions relevant for methanation. Methanation and drying have been studied over long periods of time, both in steady-state and multiple-cycling conditions. The evolution of the performance was then linked to the catalyst surface chemistry, crystal structure and water desorption kinetics.
2. Results and Discussion
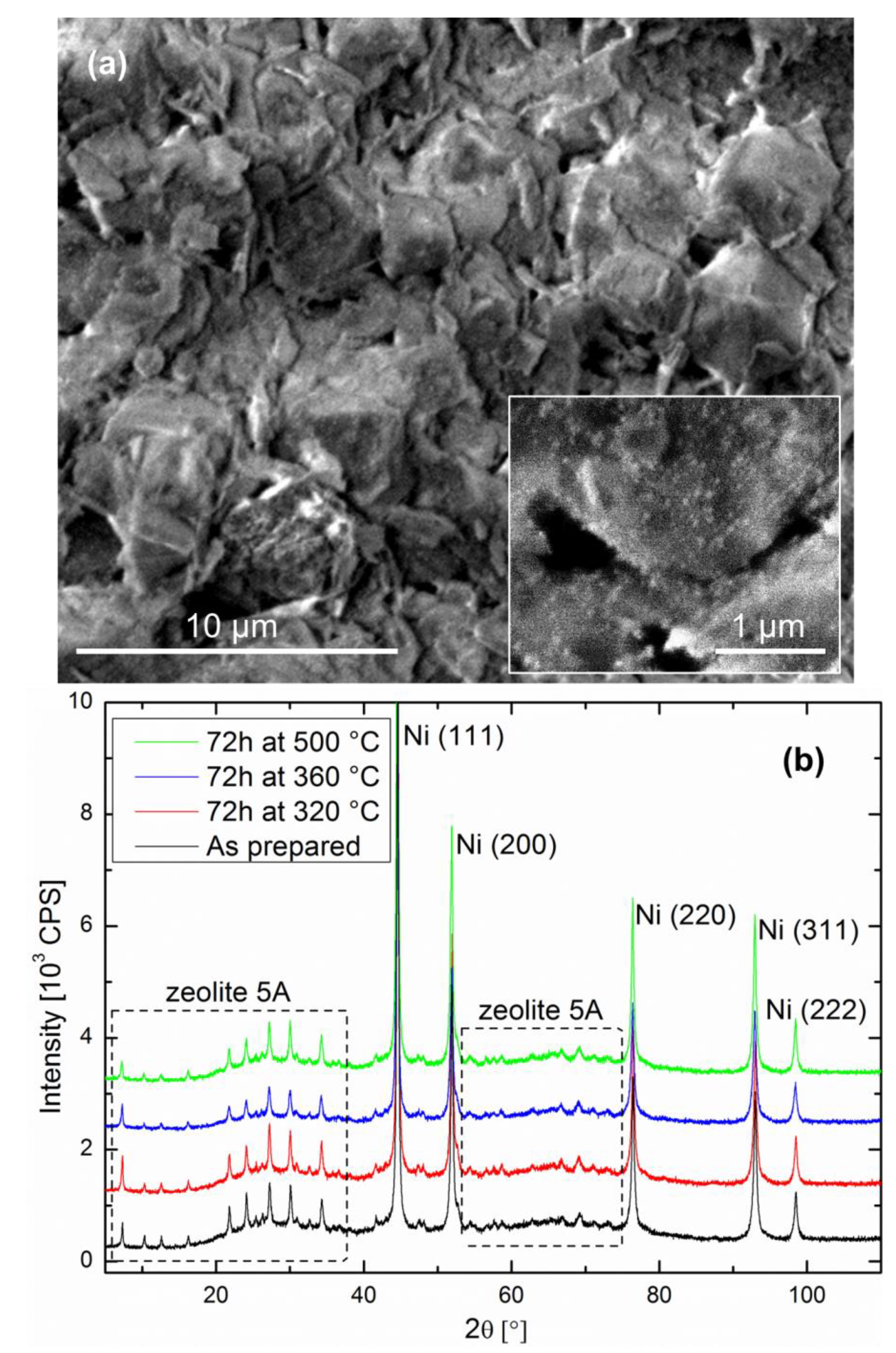
The catalyst-support couple after reduction is as follows: Ni particles with sizes in the 20–30 nm range are present on the zeolite surface as well as in the bulk. The cube-like zeolite crystallites are typically between 2 and 5 µm in size, exhibiting flat facets over which the Ni particles are homogeneously distributed (see inset of Figure 1a). The fracture surface shown in Figure 1a is representative of the pellet bulk, as well as the surface, on which no noticeable difference was observed. The XRD data of the as-prepared specimen shown in Figure 1b confirms the Ni and zeolite crystallite sizes, as well as crystal structures.
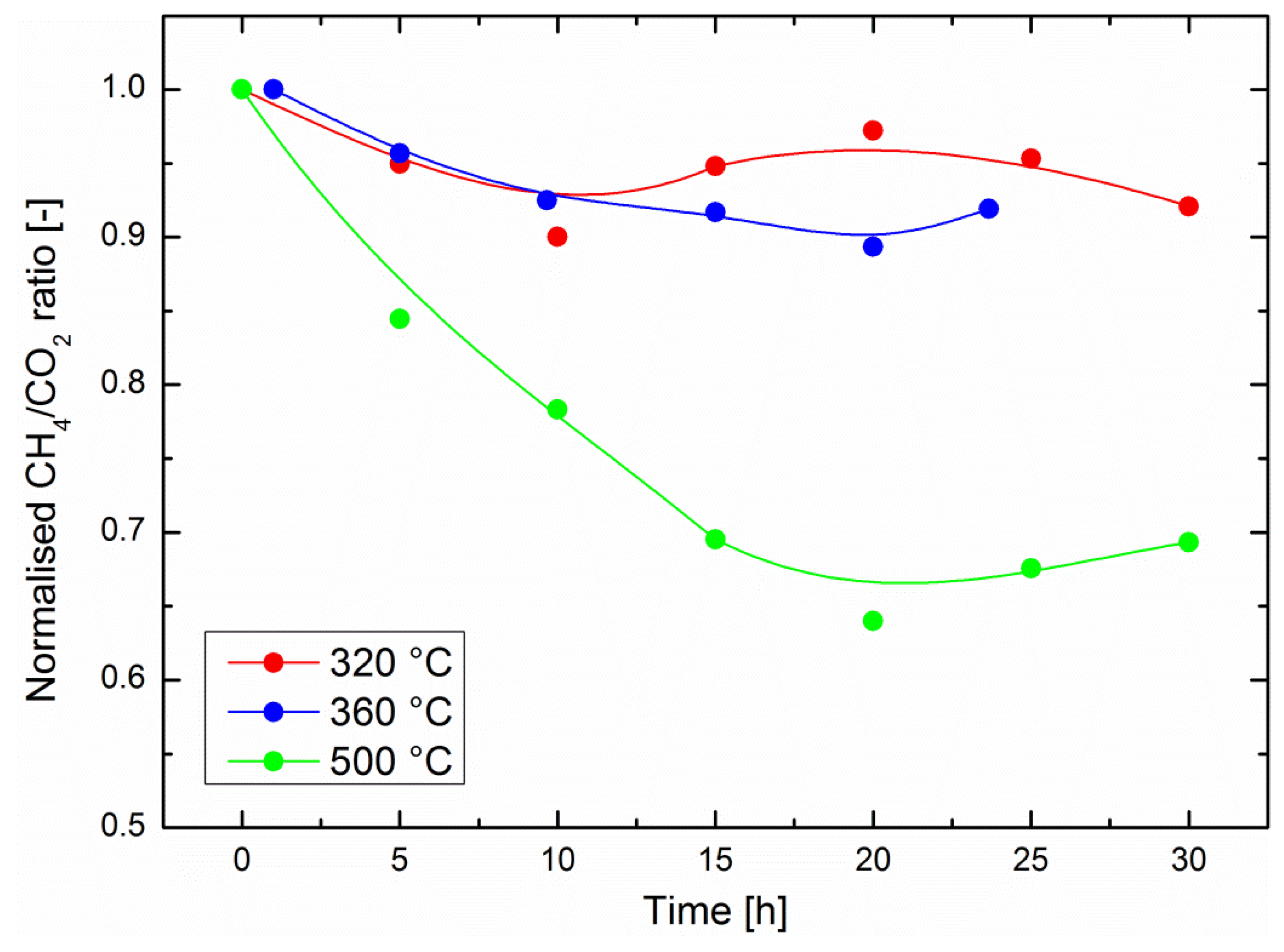
Continuous methanation experiments were carried out at different temperatures. In such experiments, the catalyst was simply subjected to a stoichiometric H2/CO2 ratio for a given time period. We will first focus on the catalyst performance, illustrated by the normalized CH4/CO2 ratio in Figure 2. One should note here that this ratio is only a relative estimation of the catalyst performance because part of the gas stream is bypassing the catalyst (see experimental section). It cannot be used to quantitatively assess the catalyst performance, e.g., by deducing the process methane yield. Although experiments up to 75 h were performed with in-situ gravimetric measurements, it should be noted that the operation time with in-situ IR measurements was currently limited to about 30 h. As explained above, the outlet gas line was heated to avoid water condensation, but still some water condensed in the optical cell. After about 30 h, the IR background was affected by the water signal and did not allow for a quantitative evaluation of the data. Alternative solutions are currently being studied in order to improve the setup.
Relatively small performance drops were observed at 320 °C and at 360 °C, with a normalized CH4/CO2 ratio losing about 10% of its original value (i.e., at the start of the experiment, immediately after introduction of CO2 into the system) in 30 h of operation in the optimum region for sorption-enhanced methanation. The performance drop was significantly higher at 500 °C, where a decrease of the CH4/CO2 ratio of 30% was observed after 30 h operation. This is an important result, which encourages the development of the still recent sorption-enhanced methanation strategy, because the latter exhibits mild operation conditions compared to most classic methanation catalysts [3,4]. One might indeed expect either Ni crystallite growth, catalyst poisoning or catalyst coking to be favored at higher operation temperatures. In the present study, poisoning can be ruled out because only pure commercial gases were used. Nickel crystallite growth is a possible scenario, especially at 500 °C. However, the XRD diffractograms in Figure 1 indicate there was no reduction in the peak width after 72 h of catalyst operation at any of the three temperatures considered here. This does not exclude fine changes in the microstructure of the nanoparticles. Studies on thermal degradation of classic methanation catalysts indicate that temperatures above 500 °C should generally be avoided [3]. The excellent stability of the catalyst activity in the optimal region for sorption-enhanced methanation (around 300 °C [13]) is encouraging. However, such materials are operated in alternating methanation/drying phases. The main challenge of the current study is the evaluation of the catalyst stability under such phases, which imply a focus on reversible water evolution in the zeolite support.
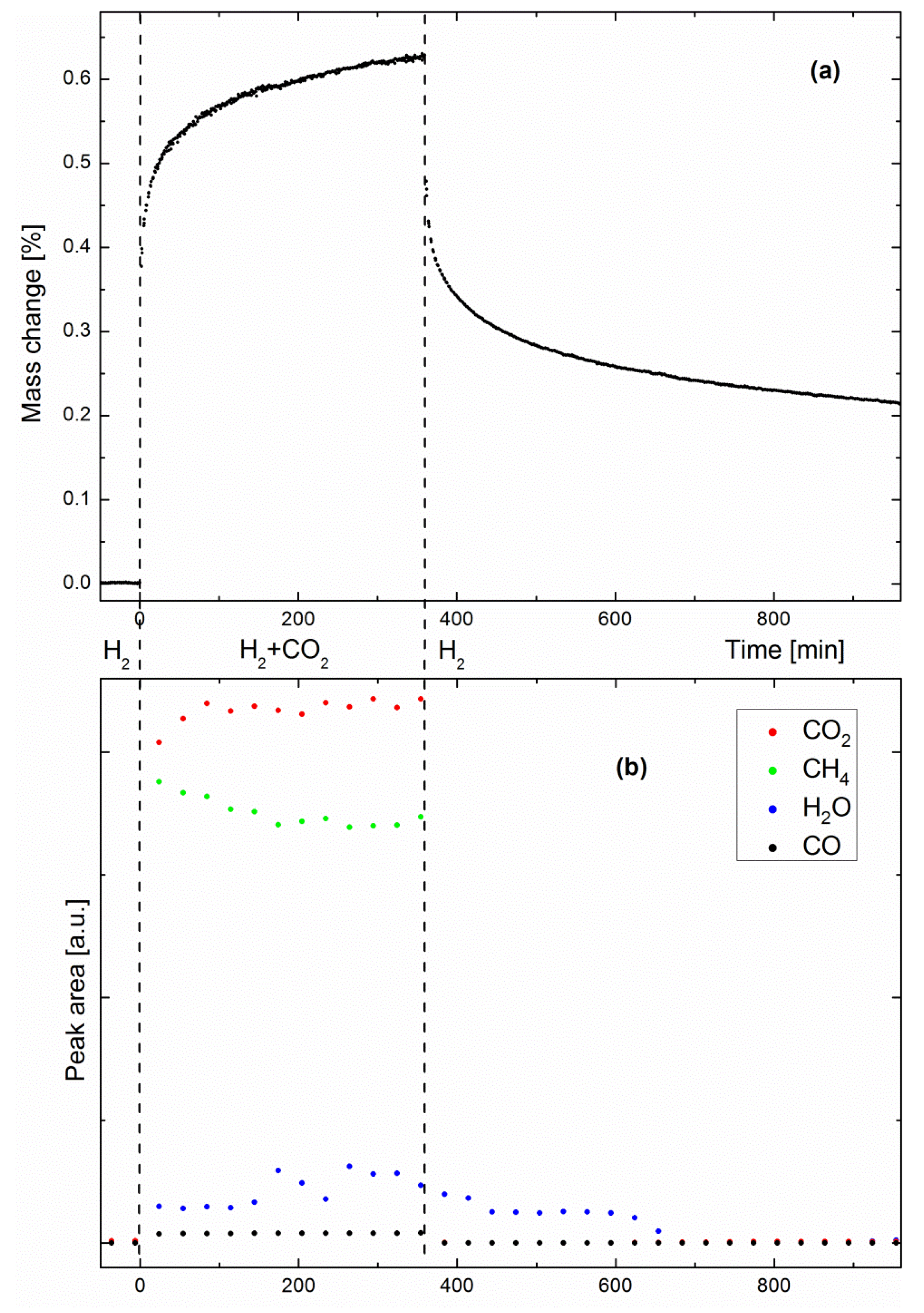
Figure 3a shows the evolution of the specimen mass as a function of time in a methanation/drying experiment performed at 360 °C. The steep early-stage kinetics is due to the sorption-enhanced effect, which results in fast methane production, and simultaneously, fast water uptake by the zeolite support. As water uptake approaches the zeolite saturation point, the sorption-enhanced effect becomes weaker, as shown by the slower late-stage kinetics. When the zeolite is saturated with water, the catalyst still produces methane, behaving like a classic methanation catalyst, where the system reflects the sole activity of the Ni nanoparticles. One must note here that the mass change shown in Figure 3a could not be exclusively the result of water adsorption by the zeolite support. As will be discussed later, the possibilities of specimen coking and CO2 adsorption will be considered as well. Such phenomena would explain why the specimen mass was still significantly higher than the baseline acquired before methanation, even after 10 h drying.
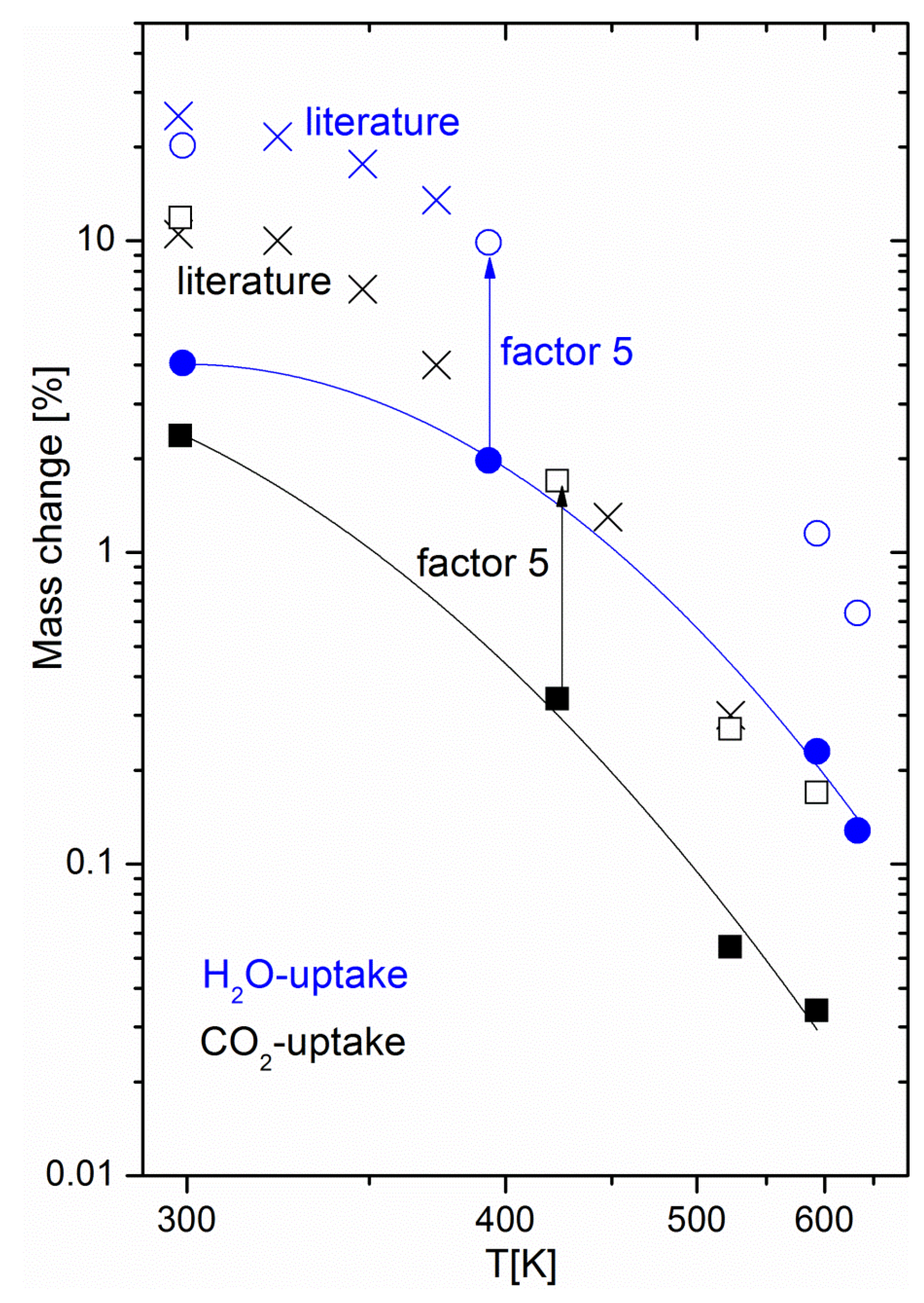
Figure 3b shows the peak areas of CO2, CH4, H2O and CO as measured by IR spectrometry (one spectrum was recorded every 30 min). No IR peaks were observed before CO2 introduction, as expected, since H2 is not IR-active. As soon as the second reactant came into play, both CH4 and H2O products were observed, together with small amounts of CO, a well-known intermediate of the Sabatier reaction [22]. Small water peaks are readily observed because the system is not designed as a catalyst bed. It has previously been shown that pure methane can be produced only as long as the catalyst bed at the outlet consists of dry zeolite [13]. When the so-called water front reaches the outlet, a dynamic equilibrium state is reached between water on the zeolite surface and water in the gas stream. Here, water could escape the specimen at any time, even when the Sabatier reaction was still sorption-enhanced. Although no clear trends can be distinguished from the evolutions of the H2O and CO peak areas, the trend observed in Figure 2 (i.e., a decreasing CH4/CO2 ratio) is already visible in a shorter experiment, such as in Figure 3b. When drying was initiated, water progressively desorbed from the zeolite, while the other gas species immediately dropped to zero. To corroborate the hypothesis of water being the origin of the mass change observed by gravimetry, we performed adsorption equilibrium measurements. Figure 4 shows the equilibrium uptake of CO2 and water at fixed partial pressures and various temperatures, including the temperature range relevant for methanation. The CO2 partial pressure of 200 mbar corresponded to that used in the methanation experiments, while the partial water pressure used for adsorption experiments was significantly lower than that expected to occur during methanation: at 50% CO2 conversion, a water partial pressure of 200 mbar was reached, while the adsorption experiments were limited to a water partial pressure of 15.8 mbar for technical reasons (humidification of the carrier gas had to take place at room temperature). Still, the equilibrium water uptake exceeded that of CO2 by a factor of five; the mass change observed during drying can thus be attributed to water. Apart from confirming the hypothesis of water being the origin of the mass change, the experiments yielded important information for the development of materials for sorption-enhanced methanation. The uptake curves match literature data [24] of the pure 5A zeolite over the full investigated temperature range, if scaled by a factor of around five (see Figure 4). This means that when anticipating performance data of sorption catalysts, it is possible to refer to reference data for the zeolite host with a “correction factor”. This factor is due to the increased weight from the additional catalyst added to the sorption material (about 6 wt %), and partial blocking of adsorption sites due to the impregnation process (in a previous study on the same materials, we showed the effect of Ni loading on the catalyst BET surface area [22], e.g., the addition of 6 wt % Ni decreases the latter from 460 m2g−1 to 330 m2g−1). The maximum uptake of the host sorption material determines the performance of the sorption catalyst. As pure zeolite 13X has a higher water uptake capacity than 5A catalysts, a better performance by it as the host material for the sorption catalyst is expected [13] and was found (the duration of the sorption enhanced mode is multiplied roughly by a factor of three when switching from 5A to 13X [23]). Furthermore, thermodynamic parameters, such as the heat of adsorption, relevant for the methanation and drying process remain unchanged. For energy balance calculations of the process, one may thus rely on reference data of the pure zeolites. As the energetics of the water-zeolite system remains unchanged, we can thus safely assume that the kinetic properties, such as the diffusion of the sorption catalyst, also reflect that of the pure host material.
Crank developed a model based on Fick’s second law of diffusion for the evolution of water in porous solids [25], describing it as a water vapor diffusion process. The following expression accounts for the evolution of the average water content in a porous solid under the hypothesis of a uniform surface water concentration [26]:
where mt and meq, are the masses adsorbed at time t and at equilibrium, R is the average zeolite crystallite radius and D is the diffusion coefficient. Equation (1) is valid for both water adsorption and desorption [26] and can therefore be used to fit a water uptake mechanism such as sorption-enhanced methanation (Figure 3a) but also for zeolite drying [27], i.e., the two fundamental steps of the sorption-enhanced methanation process [13]. This means that in both cases, meq is the water uptake capacity of the zeolite support at the reaction temperature. In the case of methanation, it is the mass of water taken up by the zeolite; in the case of drying, it is the mass of water desorbed from the zeolite. A simplified expression of Equation (1) accounts for late-stage kinetics (long term asymptote, mt/meq > 0.75) [26]:
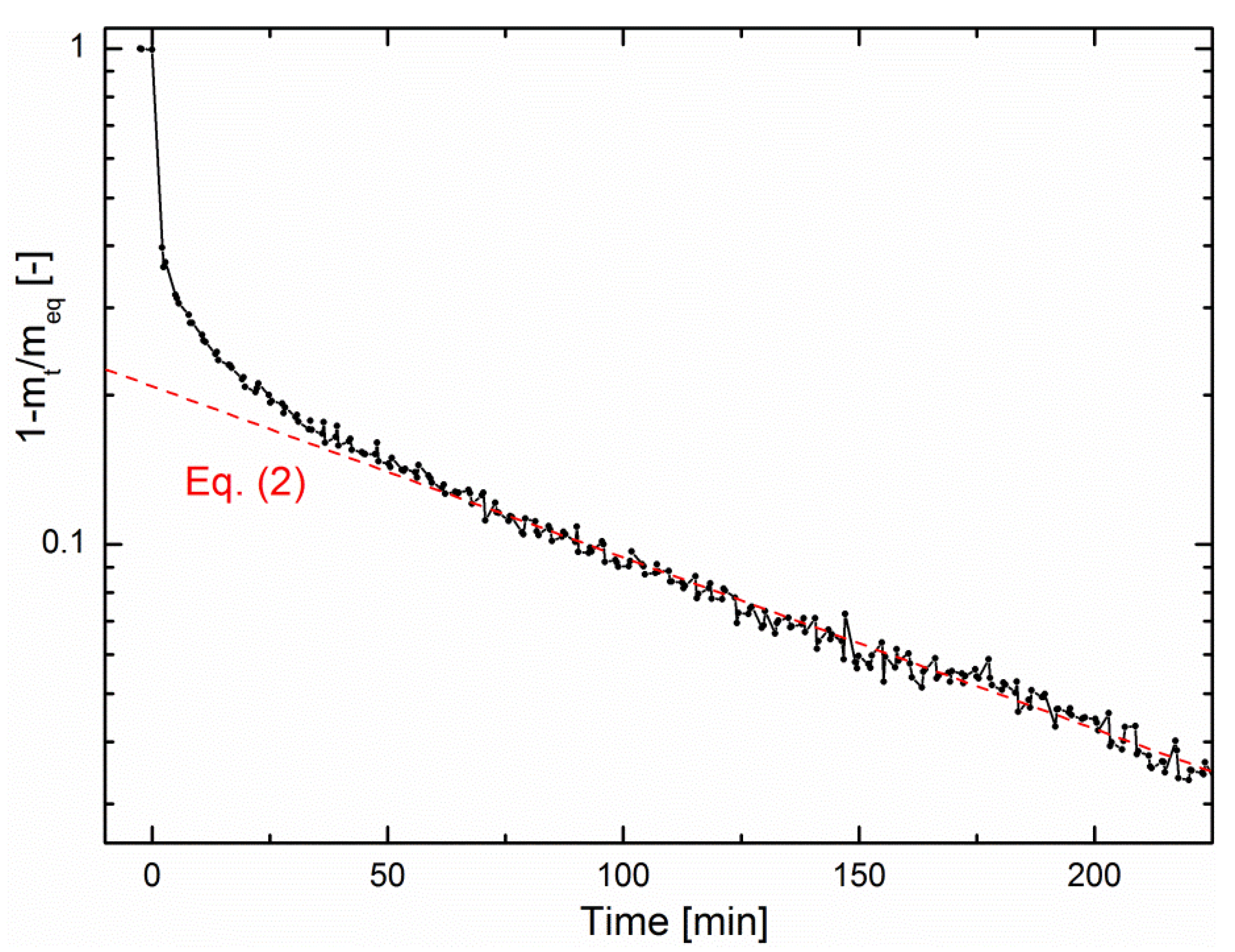
According to Equation (2), Figure 5 shows a logarithmic plot of the fractional mass uptake as a function of time from the data of Figure 3a. One can conclude from this plot that the methanation reaction is limited by water diffusion. However, on the one hand, choosing R as the zeolite crystallite radius does not result in realistic values of D. On the other hand, choosing R as the zeolite pellet size (R = 0.075 cm) results in D = 7.6 ± 0.1 × 10−8 cm2/s, in good agreement with literature [28]. This indicates that the rate-limiting step of sorption-enhanced methanation is water diffusion through the pellet bed rather than intracrystalline diffusion. The same analysis could be performed in the early-stage kinetics, for which a simplified expression of Equation (1) also exists [26], but this stage is too fast compared to the minimum time resolution of the magnetic suspension balance, so that no reliable quantitative analysis could be performed.
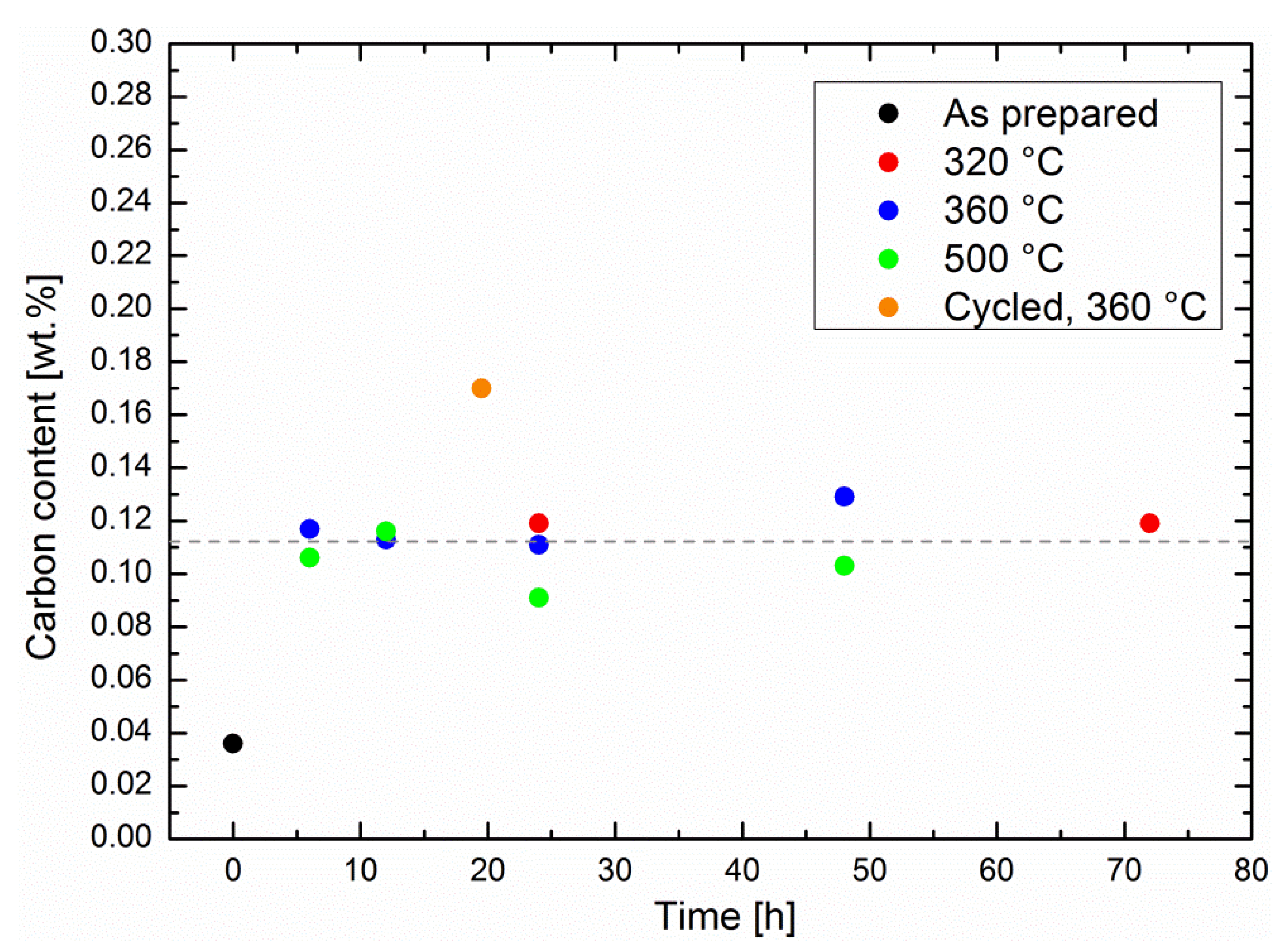
The possibility of coking was studied by means of specimen melting in pure oxygen and subsequent quantitative infrared analysis (see experimental details). The methanation experiments described in Figure 1 were repeated with different operation times in order to evaluate the carbon concentration in the related specimens (shown in Figure 6). First, the carbon content increased roughly by a factor of four between the as-prepared specimens and the specimens that had been utilized for catalytic screening. Classical coking on the surface of the catalytic Ni particles is expected to increase with time as well as temperature [19]. The carbon content was influenced neither by the methanation time, nor by the reaction temperature, although the catalytic performance decreased at very high temperatures. It remained at an average of 0.11 ± 0.01 wt %; i.e., there was no significant carbon uptake by the sorption catalysts during catalysis. Therefore, the results from the performance tests, as well as the carbon analysis, demonstrate a very robust behavior of the catalysts, in particular at typical operation temperatures. This observation is in line with studies on coking of classic methanation catalysts [3], which indicate that coking is not an issue with operation temperatures under 500 °C.
Apart from water, zeolites adsorb CO2 [29,30], CO, and various other carbon-containing side products, such as carbonates and higher hydrocarbons. For example, adsorbed CO2 could remain in the zeolite pores even at zero CO2 partial pressure [29], i.e., also during specimen transport in air between the methanation experiments and melting in the tubular oven, but this amount would not increase with reaction time. Since diffusion of CO2 in the pores of zeolite 5A is relatively fast [30], other adsorbed carbon-containing intermediates may contribute to the total carbon content. Again, these compounds would not accumulate during the course of the reaction. With concentrations as low as 0.1 wt %, the chemical analysis of these compounds is a challenge. Diffusive reflective infrared Fourier transform spectroscopy (DRIFTS) can identify most of possible adsorbates on a catalyst surface. These DRIFTS measurements on Ni-zeolite showed the presence of three well-known reaction intermediates in the Sabatier process: CO as well as the formate and carbonate ions [22]. As the latter ones may also be formed on the zeolite surface, their diffusion to the catalytic Ni surfaces may be slightly slower than their formation, and thus cause accumulation in the zeolites. This hypothesis is underlined by the measurement of the carbon uptake of a cycled sorption catalyst. The sequence, consisting of a methanation step of 30 min and a drying step of 1 h, was repeated 39 times, for a total methanation time of 19.5 h. Here, the carbon content was higher than in the specimens that had undergone continuous methanation experiments, even with longer methanation times at the same temperature (see Figure 6). During drying, any carbonaceous species was no longer adsorbed in competition with water, which eased its adsorption in the zeolite, and may have blocked some pores. We believe that the special degradation phenomenon described above was taking place preferentially on a dry, rather than a wet, surface.
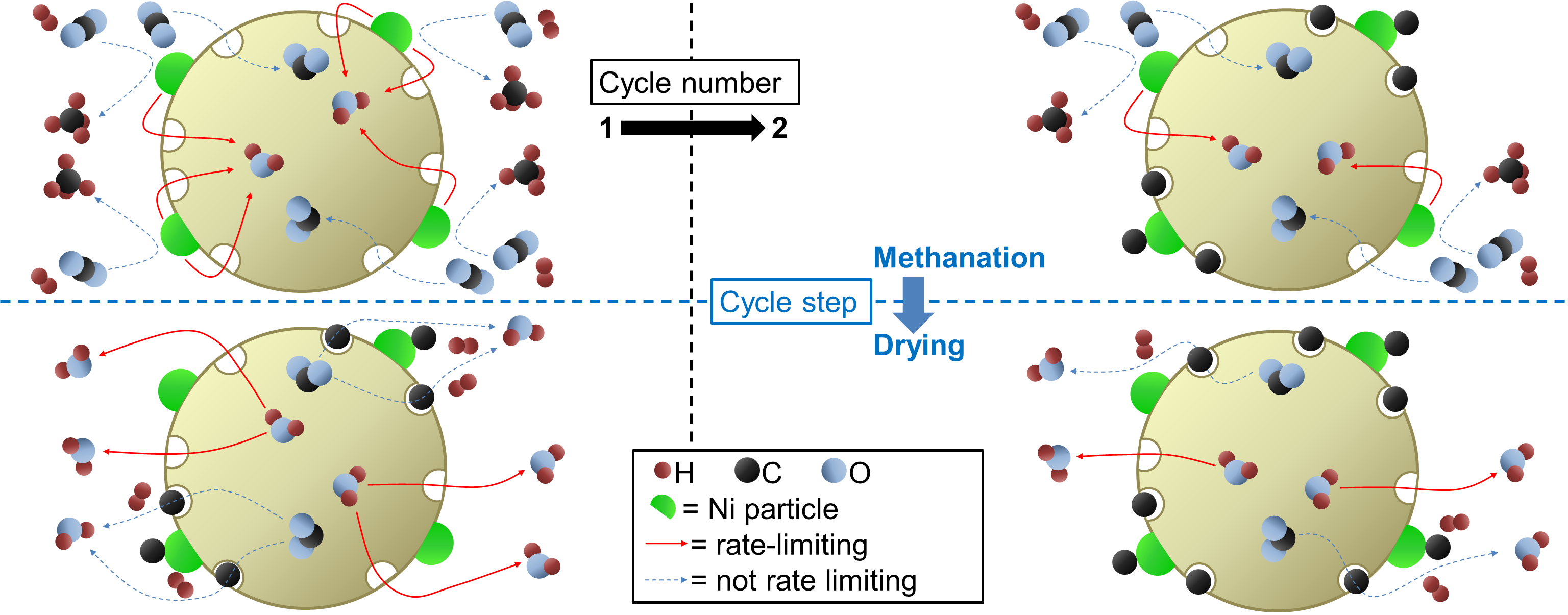
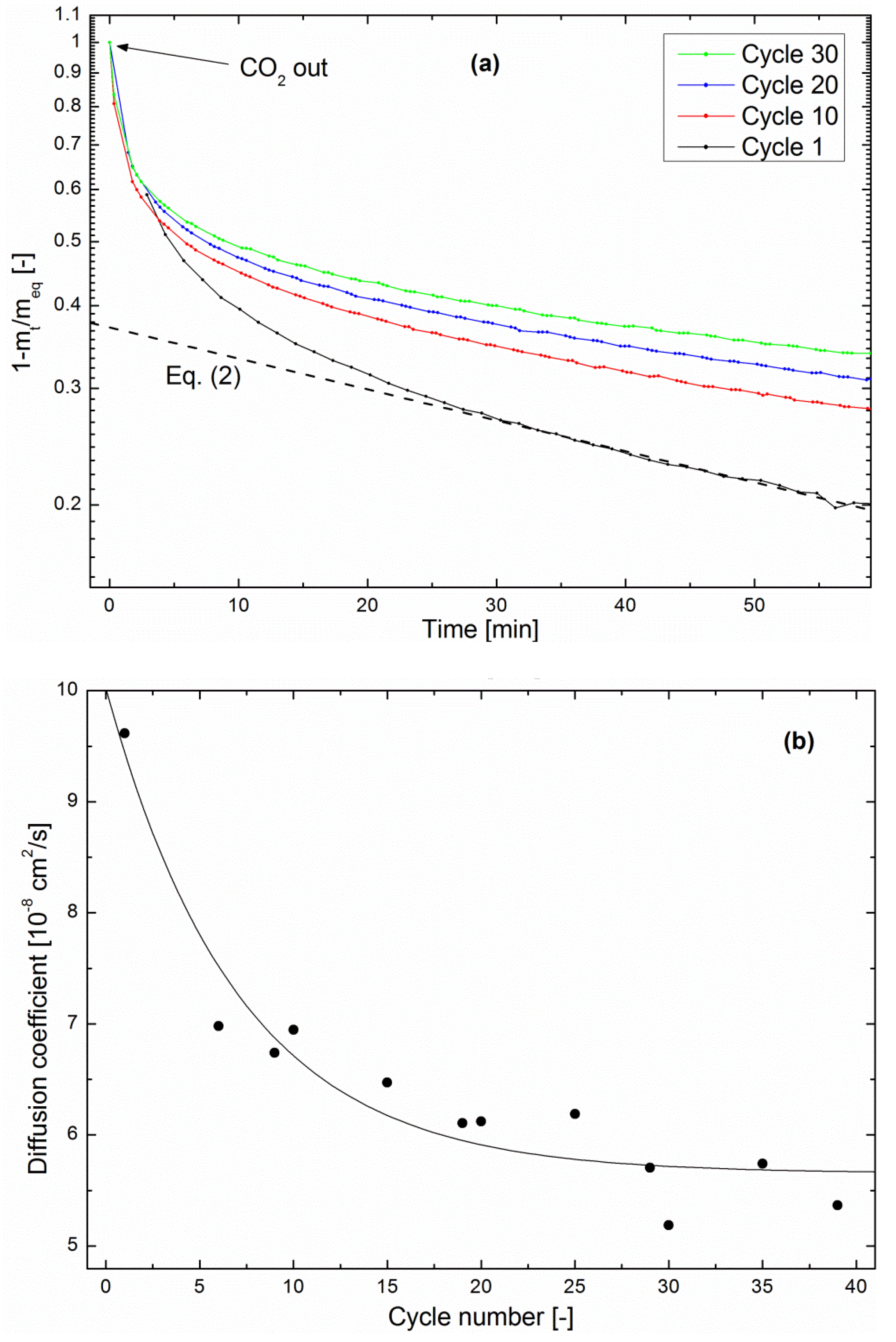
This phenomenon is quantitatively assessed here through a study of the zeolite drying kinetics of each methanation/drying cycle in the multiple cycling experiment described above. Figure 7a shows that the drying kinetics is clearly slowed from cycle to cycle. As in the methanation kinetics shown in Figure 5, the fractional mass loss can be fitted with Equation (2) in the late stage [27]. Here again, realistic values of the diffusion coefficient were obtained by using R as the pellet size, confirming the above hypothesis that water evolution in the impregnated zeolite is limited by diffusion in the pellets. Figure 7b shows the evolution of D as a function of the cycle number. The order of magnitude is in agreement with the above-calculated value of D in the methanation regime. Moreover, it decreased by about 40% after 39 cycles, indicating a deterioration of the water-diffusion properties. Such a trend was not visible in the continuous methanation experiments, which points again towards a degradation mechanism taking place preferentially during the drying process.
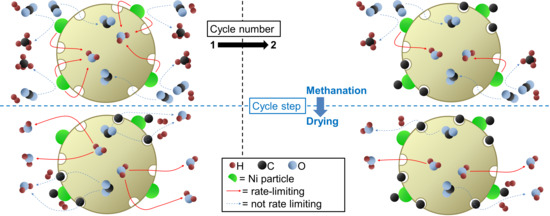
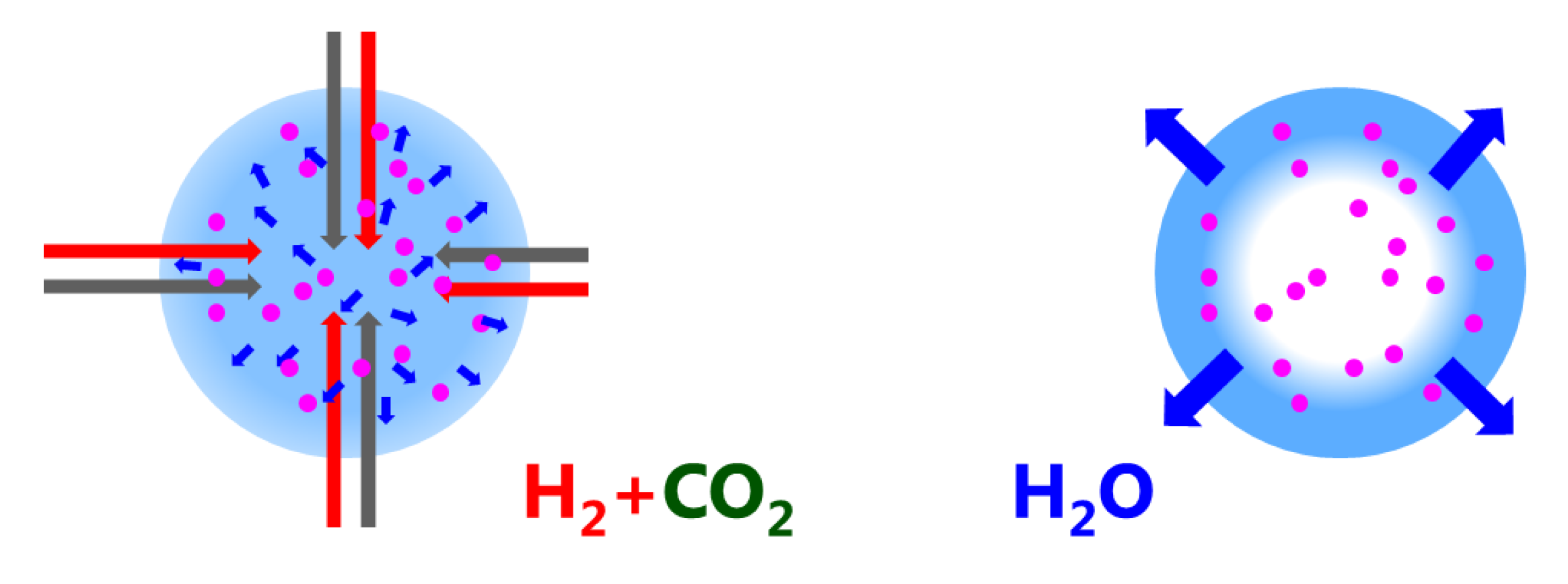
The degradation phenomenon investigated here is specific to sorption-enhanced methanation. It takes place during the drying phases, where the CO2 adsorbed on the zeolite surface during the methanation phases is converted to intermediates, such as carbon monoxide, formates, and carbonates [22]. There is an indication that this adsorption takes place preferentially in dry conditions. This means that here, catalyst degradation is not associated to a decrease of the catalyst activity (i.e., catalyst deactivation). Despite the fact that the Ni nanoparticles maintain their activity throughout long sequences of methanation and drying, the diffusion properties of the water adsorbing zeolite host matrix are significantly affected, which in turns affects the process performance rather than the catalyst performance itself. Carbon containing adsorbates will affect the water adsorption kinetics and equilibrium [31] by a partial blocking of pores in the zeolite and hinder the Le Châtelier’s effect in the sorption-enhanced methanation process. At higher temperatures, additional phenomena cannot be excluded: namely, the performance drop observed at 500 °C (see Figure 2) could be the result of hydriding of the Ni nanoparticles. Such particles embedded in a support could undergo important microstructural [32] and stress [33] effects, which in turn could strongly affect the distribution and mobility of hydrogen on the Ni surface. These possibilities will also be investigated in the future. An important outcome of the paper is the finding that the water diffusion path length is of the order of mm (Equation (2)). This is relevant for regeneration (drying) only: during methanation water diffuses from the catalytic reaction centers to the neighboring zeolite crystallites (i.e., of the order of m); for regeneration, water has to leave the catalyst pellet, i.e., diffusion path lengths of m (see Figure 8). This explains why the catalyst degradation affects the regeneration mode, while the catalytic performance during methanation is nearly unaffected.
Sorption-enhanced methanation enables the production of pure methane from CO2 and hydrogen based on the active removal of water from the catalytically active (Ni-) centers. In a previous study, we showed that sorption-enhanced methanation catalysts indeed show better performances than commercial methanation catalysts with similar nanostructures operated in the same experimental conditions [11]. The reaction steps, which are enhanced, involve the formation of water, i.e., the detachment of oxygen from the intermediates (see, e.g., Shi et al. [34]). The subsequent reaction steps responsible for the hydrogenation of the carbons remain untouched. An unwanted side reaction is the formation of carbon deposits on the active catalyst, which will be enhanced if the hydrogenation of carbon is the rate-limiting step [19]. Thus, there are methanation concepts using a high water partial pressure preventing the accumulation of carbon on the catalyst [10]. The lower methanation yield of the single reaction is overcome by repeating the reaction in serial reactors [35]. Our reaction concept, based on a low water partial pressure, may thus be vulnerable to catalyst coking. We demonstrated in this paper that classical coking, i.e., carbon deposition on Ni, does not take place. However, due to a more complex materials system (sorption catalyst) and the specific operation conditions, an additional degradation mechanism was found to lead to reduced water diffusion in the sorbent. However, this problem can find solutions in the zeolite drying step, i.e., by making it a catalyst regeneration step as well. As a matter of fact, it was shown that drying in oxidizing atmosphere leads to better catalytic activity in the sorption-enhanced mode, even if it oxidizes the active metal at its surface (an increase of the duration of the sorption enhanced mode of 26% was observed when using air as a drying gas instead of hydrogen [23]). Oxidizing atmospheres can also be used in order to clean carbon compounds from the catalyst surface. Additionally, the simple fact that drying takes place in oxidizing conditions hinders the reduction of carbon oxides on the surface, and thereby the formation of coke. Of course, such oxidizing drying steps need to be balanced with reducing environments in order to limit oxidation of the active Ni surface.
3. Experimental Details
As stated above (see introduction), this study starts from a fully optimized process [13] with known catalyst microstructure and properties [11,23], from which the question of the long-term stability of the related catalyst arises. The metal-support system was prepared as follows: nickel nitrate hexahydrate (Ni(NO3)2, Sigma-Aldrich, St. Louis, MO, USA) was dissolved in deionized water. Pellets of zeolite 5A (Linde Type A, cylindrical shape, 1.5 mm diameter, 3 mm length) were immersed in this solution for 24 h at room temperature. Ni partly ion-exchanged with alkali/alkaline earth elements from the zeolite structure (zeolite 5A formula: CanNa12−2n[(AlO2)12(SiO2)12]·xH2O), while non-impregnated Ni could also remain in the zeolite pores after immersion. Therefore, the present synthesis is a combination of liquid ion exchange and impregnation. Washing of the pellets was omitted in order to avoid producing toxic Ni waste. The pellets were then dried at 100 °C for another 24 h, and reduced in hydrogen flow for 2 h at 650 °C. In these conditions, Ni atoms incorporated in the zeolite structure cluster and form nanoparticles on the surface as well as in the bulk, for a Ni loading of about 6 wt % [13].
This catalyst was characterized by the following methods: scanning electron microscopy (SEM) images of the zeolite surface were acquired using a Nova NanoSEM 230 FEI (Thermo Fischer Scientific, Waltham, MA, USA) with a 20 kV acceleration voltage. SEM observations were conducted at pellet surfaces and fracture surfaces without noticeable difference. The crystal structure of the specimens was investigated by X-ray powder diffraction (XRD) with a Bruker D8 diffractometer (Bruker, Billerica, MA, USA) with Cu-Kα radiation in a 2θ range of 5°–90° and a step size of 0.017° (pattern PDF codes: 01-077-1335 (zeolite 5A) and 01-071-4740 4-850 (Ni)). Topas software (v. 5.0, Bruker AXS, Karlsruhe, Germany, 2014) was used for determination of crystallite sizes (Scherrer equation). Carbon concentrations were determined by melting the specimens in pure oxygen (with a slight overpressure compared to atmosphere) in a tubular oven at 1350 °C (IRF 1600 from SYLAB, Metz, France) and measuring the CO2 emitted by carbon combustion in real time with nondispersive infrared spectrometry (Infrared Analyzer CSbox from SYLAB, with a resolution of 0.1 ppm).
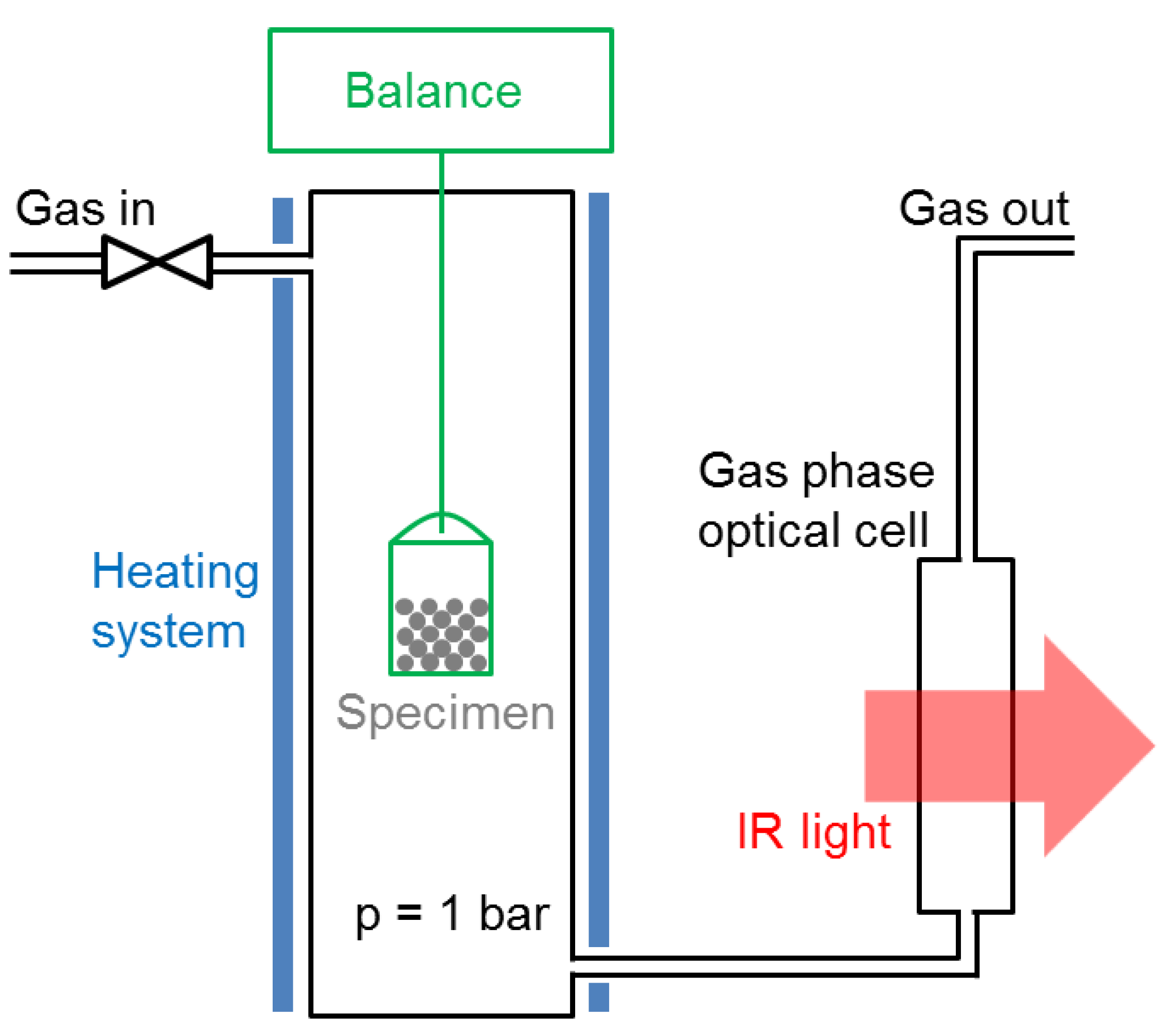
The catalyst specimens were studied in-situ in a magnetic suspension balance (Rubotherm, Bochum, Germany) modified in order to encapsulate the specimen holder in a gas stream. This system is shown schematically in Figure 9. The outlet gas stream was connected to an infrared spectrometer (Alpha from Bruker, Billerica, MA, USA, equipped with an 8-cm gas cell, resolution: 0.9 cm−1). The specimen could be heated up to 500 °C. The bucket-shaped specimen holder was micrometrer-sized meshed—any gas species could penetrate easily without undesirable flow effects. The gas connections were made with Swagelock tubing (Swagelock, Solon, OH, USA), and the gas flows controlled by thermal-based mass flow controllers (EL-FLOW Select series from Bronkhorst, Ruurlo, The Netherlands). The latter could be programmed with a LabVIEW interface. The outlet gas line was heated to 100 °C with a heating band in order to avoid water condensation in the optical cell. The main advantage of this system is the robustness of the magnetic suspension balance measurements. Reliable data could be acquired for days, either by continuous methane production or by repeated methanation/drying cycles. Apart from kinetic measurements, the system was used to acquire equilibrium data for the uptake of water and CO2 as a function of temperature. For these experiments, the catalyst was exposed either to a 4:1 He/CO2 gas mixture at 1 bar, or to humidified He gas corresponding to a water partial pressure of 15.8 mbar (50% humidity at 25 °C).
The specimen mass change during sorption-enhanced methanation and drying was monitored in this system, providing quantitative insight into the kinetics and equilibrium behaviors of the catalyst-support couple. The specimen mass was in the range 2.5–3 g. Both methanation and drying were performed at atmospheric pressure with a total flow of 250 mL/min and a stoichiometric H2/CO2 ratio. It should be noted that this setup is not a catalyst screening setup. No gas hourly space velocity, and thereby no catalyst turnover frequency, could be deduced from the total gas flow because the specimen holder was hanging in empty space, and therefore could not be considered as a catalyst bed. A significant CO2 signal was always observed because the inlet gas species could easily bypass the specimen. Additionally, water could escape the specimen at any time and from any region. Consequently, the long-term performance of the catalyst will be discussed in terms of in-situ IR signal ratio between gas species, rather than absolute gas composition.
In the methanation experiments considered here, the specimens were permanently subjected to a H2 flow; the CO2 flow was then switched on to trigger methanation and simply switched off to trigger drying. In order to discuss the long-term catalyst performance in both classic and sorption-enhanced methanation, temperatures between 320 and 500 °C were considered; i.e., by screening the catalyst activity from the sorption-enhanced methanation optimum of 300 °C described by Borgschulte et al. [13] up to more commonly encountered temperatures for classic catalytic methanation [3,4].
4. Conclusions
Sorption-enhanced methanation catalysts allow for relatively mild operational conditions compared to conventional methanation; namely, in the case of Ni nanoparticles distributed on 5A zeolite, the active nanoparticles under operational conditions shows practically no long-term deactivation close to the sorption-enhanced methanation optimum, as coking and thermal degradation are excluded in the present study. However, sorption-enhanced methanation requires alternating methanation/drying periods. Water diffusion in the zeolite pellet bed is the rate-limiting step in both cases. A comparison of the equilibrium adsorption data of CO2 and H2O suggests that thermodynamic properties such as diffusion may be estimated from that of the pure zeolite, for which much more data exist. For equivalent methanation times, specimens screened under such cyclic treatments exhibited carbon contents about 55% higher than specimens subjected to continuous methanation conditions. This suggests a degradation phenomenon specific to sorption-enhanced methanation, which does not directly affect the catalytic activity of the active metal. In this sense, this degradation phenomenon is not a deactivation phenomenon. Reaction intermediates and products in the zeolite formed during methanation phases block pores in the sorbent during drying phases. The consequence of this was a decrease in the water diffusion coefficient of 40% observed after 39 cycles. The decrease in diffusion hardly affects the catalytic performance but has a considerable impact on the regeneration due to the much longer water diffusion path lengths. Further investigations are necessary in order to determine the mechanism of the reduction of diffusion, as well as the factors that influence the adsorption of reaction intermediates and products in the zeolite under dry conditions. Catalyst drying in oxidizing conditions is a good solution to this degradation phenomenon. More generally, we believe that the sorption-enhanced methanation strategy could be adapted to other power-to-fuel processes in the future (e.g., methanol synthesis, Fischer–Tropsch process).
Author Contributions
R.D. and A.B. conceived and designed the experiments, R.D. and J.T. performed the experiments, R.D. and A.B. analyzed the data, A.R. and J.P. performed materials analysis. R.D., A.H. and A.B. wrote and revised the paper.
Funding
This research was funded by the Swiss Federal Office of Energy (SFOE) and the ‘Forschungs-, Entwicklungs- und Förderungsfonds der Schweizer Gaswirtschaft’ (FOGA) through the SMARTCAT project (grant number SI/501130-01 and 0268) and by the Swiss National Science Foundation (SNSF) in the NRP70 programme ‘Energy Turnaround’.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Borgschulte, A. The Hydrogen Grand Challenge. Front. Energy Res. 2016, 4. [Google Scholar] [CrossRef]
- Meylan, F.D.; Moreau, V.; Erkman, S. Material constraints related to storage of future European renewable electricity surpluses with CO2 methanation. Energy Policy 2016, 94, 366–376. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Götz, M.; Lefebvre, J.; Mörs, F.; Koch, A.M.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable power-to-gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. CO2 reduction on supported Ru/Al2O3 catalysts: Cluster size dependence of product selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- Janlamool, J.; Praserthdam, P.; Jongsomjit, B. Ti-Si composite oxide-supported cobalt catalysts for CO2 hydrogenation. J. Nat. Gas Chem. 2011, 20, 558–564. [Google Scholar] [CrossRef]
- Aksoylu, A.E.; Misirli, Z.; Önsan, Z.I. Interaction between nickel and molybdenum in Ni-Mo/Al2O3 catalysts: I: CO2 methanation and SEM-TEM studies. Appl. Catal. A 1998, 168, 385–397. [Google Scholar] [CrossRef]
- Merkache, J.; Fechete, I.; Maamache, M.; Bernard, M.; Turek, P.; Al-Dalama, K.; Garin, F. 3D ordered mesoporous Fe-KIT-6 catalysts for methylcyclopentane (MCP) conversion and carbon dioxide (CO2) hydrogenation for energy and environmental applications. Appl. Catal. A 2015, 504, 672–681. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Borgschulte, A.; Gallandat, N.; Probst, B.; Suter, R.; Callini, E.; Ferri, D.; Arroyo, Y.; Erni, R.; Geerlings, H.; Züttel, A. Sorption enhanced CO2 methanation. Phys. Chem. Chem. Phys. 2013, 15, 9620–9625. [Google Scholar] [CrossRef] [PubMed]
- Walspurger, S.; Elzinga, G.D.; Dijkstra, J.W.; Sarić, M.; Haije, W.G. Sorption enhanced methanation for substitute natural gas production: Experimental results and thermodynamic considerations. Chem. Eng. J. 2014, 242, 379–386. [Google Scholar] [CrossRef]
- Borgschulte, A.; Delmelle, R.; Duarte, R.B.; Heel, A.; Boillat, P.; Lehmann, E. Water distribution in a sorption enhanced methanation reactor by time resolved neutron imaging. Phys. Chem. Chem. Phys. 2016, 18, 17217–17223. [Google Scholar] [CrossRef] [PubMed]
- Im, S.I.; Lee, K.B. Novel sorption-enhanced methanation with simultaneous CO2 removal for the production of synthetic natural gas. Ind. Eng. Chem. Res. 2016, 55, 9244–9255. [Google Scholar] [CrossRef]
- Moulijn, J.A.; van Diepen, A.E.; Katepijn, F. Catalyst deactivation: Is it predictable? What to do? Appl. Catal. A 2001, 212, 3–16. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Bibby, D.M.; Howe, R.F.; McLellan, G.D. Coke formation in high-silica zeolites. Appl. Catal. A 1992, 93, 1–34. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, T.; Peng, J.; Wang, S.; Wang, S. A comparison of Ni/SiC and Ni/Al2O3 catalysed total methanation for production of synthetic natural gas. Appl. Catal. A 2013, 462–463, 75–81. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Szabo, S.; Bakos, I. Corrosion accelerating surface catalysts. Corros. Rev. 2002, 20, 95–104. [Google Scholar] [CrossRef]
- Yang, N.; Wang, R. Sustainable technologies for the reclamation of greenhouse gas CO2. J. Clean. Prod. 2015, 103, 784–792. [Google Scholar] [CrossRef]
- Borgschulte, A.; Callini, E.; Stadie, N.; Arroyo, Y.; Rossell, M.D.; Erni, R.; Geerlings, H.; Züttel, A.; Ferri, D. Manipulating the reaction path of the CO2 hydrogenation reaction in molecular sieves. Catal. Sci. Technol. 2015, 5, 4613–4621. [Google Scholar] [CrossRef]
- Delmelle, R.; Duarte, R.B.; Franken, T.; Burnat, D.; Holzer, L.; Borgschulte, A.; Heel, A. Development of improved nickel catalysts for sorption enhanced CO2 methanation. Int. J. Hydrog. Energy 2016, 41, 20185–20191. [Google Scholar] [CrossRef]
- Wang, Y.; LeVan, M.D. Adsorption equilibrium of carbon dioxide and water vapor on zeolites 5A and 13X and silica gel: Pure components. J. Chem. Eng. Data 2009, 54, 2839–2844. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion; Clarendon Press: Oxford, UK, 1970. [Google Scholar]
- Ruthven, D.M. Diffusion in type A zeolites: New insights from old data. Micropor. Mesopor. Mater. 2012, 162, 69–79. [Google Scholar] [CrossRef]
- Keey, R. Drying of Loose and Particulate Materials; Hemisphere Publishing Corporation: New York, NY, USA, 1992. [Google Scholar]
- Turgman-Cohen, S.; Araque, J.C.; Hoek, E.M.V.; Escobedo, F.A. Molecular dynamics of equilibrium and pressure-driven transport properties of water through LTA-type zeolites. Langmuir 2013, 29, 12389–12399. [Google Scholar] [CrossRef] [PubMed]
- Triebe, R.W.; Tezel, F.H. Adsorption of nitrogen, carbon monoxide, carbon dioxide and nitric oxide on molecular sieves. Gas Sep. Purif. 1995, 9, 223–230. [Google Scholar] [CrossRef]
- Ruthven, D.M.; Lee, L.K.; Yucel, Y. Kinetics of non-isothermal sorption in molecular sieve crystals. AIChE J. 1980, 26, 16–23. [Google Scholar] [CrossRef]
- Silva, J.A.C.; Mata, V.G.; Dias, M.M.; Lopes, J.C.B.; Rodrigues, A.E. Effect of coke in the equilibrium and kinetics of sorption on 5A molecular sieve zeolites. Ind. Eng. Chem. Res. 2000, 39, 1030–1034. [Google Scholar] [CrossRef]
- Delmelle, R.; Amin-Ahmadi, B.; Sinnaeve, M.; Idrissi, H.; Pardoen, T.; Schryvers, D.; Proost, J. Effect of structural defects on the hydriding kinetics of nanocrystalline Pd thin films. Int. J. Hydrog. Energy 2015, 40, 7335–7347. [Google Scholar] [CrossRef] [Green Version]
- Delmelle, R.; Michotte, S.; Sinnaeve, M.; Proost, J. Effect of internal stress on the hydriding kinetics of nanocrystalline Pd thin films. Acta Mater. 2013, 61, 2320–2329. [Google Scholar] [CrossRef]
- Shi, C.; O’Grady, C.P.; Peterson, A.A.; Hansen, H.A.; Norskov, J. Modeling CO2 reduction on Pt(111). Phys. Chem. Chem. Phys. 2013, 15, 7114–7122. [Google Scholar] [CrossRef] [PubMed]
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S.M.A. Production of synthetic natural gas (SNG) from coal and dry biomass—A technology review from 1950 to 2009. Fuel 2010, 89, 1763–1783. [Google Scholar] [CrossRef]
Figure 1.
(a) Representative SEM image of the fracture surface of a Ni-impregnated 5A zeolite pellet after reduction. (b) X-ray powder diffraction patterns of the catalyst as prepared and after methanation under different conditions.
Figure 1.
(a) Representative SEM image of the fracture surface of a Ni-impregnated 5A zeolite pellet after reduction. (b) X-ray powder diffraction patterns of the catalyst as prepared and after methanation under different conditions.

Figure 2.
Evolution of the normalized CH4/CO2 ratio calculated from in-situ IR data during long-term methanation experiments at different temperatures. The solid lines are guides for the eye.
Figure 2.
Evolution of the normalized CH4/CO2 ratio calculated from in-situ IR data during long-term methanation experiments at different temperatures. The solid lines are guides for the eye.

Figure 3.
(a) Evolution of the mass of a Ni-impregnated 5A zeolite catalyst measured by a magnetic suspension balance during methanation (6 h) and drying (10 h) at 360 °C with stoichiometric H2/CO2 feed ratio. H2 is always present in the inlet gas stream, and t = 0 s corresponds to CO2 introduction into the system. (b) Evolution of the IR peak areas of CH4, CO2, H2O, and CO in the outlet gas stream.
Figure 3.
(a) Evolution of the mass of a Ni-impregnated 5A zeolite catalyst measured by a magnetic suspension balance during methanation (6 h) and drying (10 h) at 360 °C with stoichiometric H2/CO2 feed ratio. H2 is always present in the inlet gas stream, and t = 0 s corresponds to CO2 introduction into the system. (b) Evolution of the IR peak areas of CH4, CO2, H2O, and CO in the outlet gas stream.

Figure 4.
Equilibrium uptake of CO2 and water at fixed partial pressures for various temperatures. The partial pressures are 200 mbar CO2 and 15.8 mbar H2O, respectively, in the carrier gas He. Literature data on pure zeolite 5A24 is included for comparison. The literature data matches that for the sorption catalyst if scaled by a factor of five.
Figure 4.
Equilibrium uptake of CO2 and water at fixed partial pressures for various temperatures. The partial pressures are 200 mbar CO2 and 15.8 mbar H2O, respectively, in the carrier gas He. Literature data on pure zeolite 5A24 is included for comparison. The literature data matches that for the sorption catalyst if scaled by a factor of five.

Figure 5.
Evolution of the fractional mass uptake of a Ni-impregnated 5A zeolite catalyst during methanation at 360 °C with stoichiometric H2/CO2 feed ratio. The red line represents Equation (2).
Figure 5.
Evolution of the fractional mass uptake of a Ni-impregnated 5A zeolite catalyst during methanation at 360 °C with stoichiometric H2/CO2 feed ratio. The red line represents Equation (2).

Figure 6.
Gravimetric carbon content in Ni-impregnated 5A zeolite specimens subjected to continuous methanation experiments (red, blue and green dots, average of the latter shown by the grey line) and multiple methanation/drying cycles (orange).
Figure 6.
Gravimetric carbon content in Ni-impregnated 5A zeolite specimens subjected to continuous methanation experiments (red, blue and green dots, average of the latter shown by the grey line) and multiple methanation/drying cycles (orange).

Figure 7.
(a) Evolution of the fractional mass loss of a Ni-impregnated 5A zeolite catalyst during drying at 360 °C in a multiple cycling experiment. t = 0 s corresponds to CO2 removal from the system. (b) Diffusion coefficient as a function of cycle number, calculated from fitting Equation (2) to the late-stage region of drying curves. The solid line is a guide to the eye.
Figure 7.
(a) Evolution of the fractional mass loss of a Ni-impregnated 5A zeolite catalyst during drying at 360 °C in a multiple cycling experiment. t = 0 s corresponds to CO2 removal from the system. (b) Diffusion coefficient as a function of cycle number, calculated from fitting Equation (2) to the late-stage region of drying curves. The solid line is a guide to the eye.

Figure 8.
Sketch to illustrate the different length scale of diffusion: water diffusion is a local phenomenon during methanation (from the catalytic reaction centers to the neighboring zeolite crystallites, i.e., of the order of m); for regeneration, water has to leave the catalyst pellet, i.e., diffusion path lengths of m.
Figure 8.
Sketch to illustrate the different length scale of diffusion: water diffusion is a local phenomenon during methanation (from the catalytic reaction centers to the neighboring zeolite crystallites, i.e., of the order of m); for regeneration, water has to leave the catalyst pellet, i.e., diffusion path lengths of m.

Figure 9.
Schematic representation of the experimental setup used in this study for sorption-enhanced methanation and drying.
Figure 9.
Schematic representation of the experimental setup used in this study for sorption-enhanced methanation and drying.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Delmelle, R.; Terreni, J.; Remhof, A.; Heel, A.; Proost, J.; Borgschulte, A. Evolution of Water Diffusion in a Sorption-Enhanced Methanation Catalyst. Catalysts 2018, 8, 341. https://doi.org/10.3390/catal8090341
AMA Style
Delmelle R, Terreni J, Remhof A, Heel A, Proost J, Borgschulte A. Evolution of Water Diffusion in a Sorption-Enhanced Methanation Catalyst. Catalysts. 2018; 8(9):341. https://doi.org/10.3390/catal8090341
Chicago/Turabian StyleDelmelle, Renaud, Jasmin Terreni, Arndt Remhof, Andre Heel, Joris Proost, and Andreas Borgschulte. 2018. "Evolution of Water Diffusion in a Sorption-Enhanced Methanation Catalyst" Catalysts 8, no. 9: 341. https://doi.org/10.3390/catal8090341
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.