1. Introduction
Biomass is an abundant and easy to harvest resource, but faces difficulties associated with distribution logistics (collection, transportation, and distribution). Furthermore, biomass, especially wood, has lower energy density (2–3 GJ/m
3) than other common fuels such as ethanol (23 GJ/m
3) and gasoline (35 GJ/m
3) [
1,
2]. One of the most traditional and promising technologies for converting solid biomass into energy, fuels and commodities is gasification [
3], but obtaining the desired products can be challenging. The composition of gasification gases (CO
2, H
2, CO, CH
4) can vary depending on gasifying agent (i.e., steam, air, oxygen), gasification temperature, reactors design, as well as feedstock composition. For example, Molino et al. [
4] have reported that Pine sawdust gasification result in a syngas composition with high content of CO (35–43%) and H
2 (21–39%), while CH
4 (6–10%) and CO
2 (18–20%) appear in lower concentrations. However, changing biomass type by α-cellulose the balance is quite different with less CO (6.5–11.2%), H
2 (13.5–18.5%), and CH
4 (2.2–3.7%) and increasing CO
2 (26.3–27.7%). Regardless of the gasification conditions, the syngas is contaminated with traces of undesirable species such as ammonia (NH
3) and sulphur compounds (H
2S, SO
x), with condensable polyaromatics known as tar and with solid particles [
5]. These contaminants are not allowed for most technologies used downstream of the gasifier such as IC engine, turbines, FT-synthesis reactors, etc. Tar elimination is the higher investment and operations cost in gasification; thus, the removal of tar is a major means to reduce the overall cost of biomass gasification [
5]. However, ammonia, even at low concentrations, is inadmissible for F-T synthesis processes and may limit the use of combustion gases due to the generation of NO
x [
6]. Physical cleaning pathways eliminate these compounds by means of filters, water traps, rotary separators, electrostatic precipitators, etc. [
7]. All of these technologies miss the potential energy present in the contaminants (especially tar) [
8] and transfer the contaminants to another effluent (liquid or solid), which also requires treatment before disposition, thus elevating the operation costs [
9]. A promising way to eliminate tar and ammonia is via thermal or catalytic cracking/reforming, which could also contribute to increase the concentration of CO
x and/or H
2 in the syngas [
10,
11,
12]. Thermal cracking depends on temperature and begins to be effective over 1000 °C, so it requires special equipment, which elevates the installation costs [
11]. Above this temperature, each 100 °C increase implies a caloric power loss of 3.5% [
10]. Wet and dry reforming processes do not require high temperatures but depending on gasification conditions they could require an auxiliary service to supply steam or CO
2, respectively. Furthermore, both technologies require catalysts to reduce the activation energies of specific reactions [
8,
13,
14,
15]. Catalytic cracking works at the same temperature as reforming, but without the need of an auxiliary service, making it the most economically attractive technology. Oxides of alkaline and alkaline-earth metals, dolomites, and olivine have been reported as active for cleaning gasification gases, nevertheless, all of them are deactivated by agglomeration, coke deposition, and/or sintering at the reaction conditions [
8,
16]. According to [
17,
18], nickel is the most effective catalyst for catalytic upgrading of gasification gases. It has high activity for tar cracking, with high selectivity to H
2, and it is also capable of decomposing ammonia [
19,
20]. Iron is also used in this kind of reaction because it is considered the metal with highest activity for breaking carbon-carbon bonds [
21]. It is also active for ammonia decomposition [
22], and it is more abundant and environmentally manageable than nickel [
23]. One of the main drawbacks of using metal catalysts to clean gasification gases is the coke formation, which is influenced by the operational conditions, and the catalyst and support properties [
8].
The most used supports (e.g., alumina) act as reaction promoters, but they have a strong interaction with hydrocarbons so they are affected by coke formation, mostly on acid sites [
24]. On the other hand, carbonaceous materials have proven to have a high affinity and adsorption selectivity to hydrocarbon compounds [
25] such as tar. Apart from an easily tailorable porous structure and surface chemistry, carbon presents other advantages as a catalyst support: metals at the surface can be easily reduced; the structure is resistant to acids and bases and is stable at high temperatures if a proper treatment is used; the active phase can be easily recovered; conventional carbon supports usually have lower costs than other conventional supports, such as silica [
26,
27]; and higher resistance to surface coke formation [
24]. Nevertheless, carbon supports also present some disadvantages, such as their low reproducibility, since different carbonization batches of the same raw material can contain varying ash amounts.
In particular, carbon aerogels (CAG) solve the problem of reproducibility because they are obtained from standardized raw materials, mostly by carbonization of cellulose microfibers (MFCs) [
28] or aerogels obtained via organic synthesis [
29]. CAGs can be synthesized using a sol-gel process, generating an organic gel that is dried via freeze drying. The dry gel is pyrolyzed (carbonized) to obtain the corresponding CAG. Cellulose-based organic gels are of particular interest, as cellulose has high carbon content and it is renewable, chemically stable, and globally abundant [
30]. Alternative synthesis methods, using different raw materials, include: sol–gel systems based on the crosslinking of cellulose acetate with polyfunctional isocyanate in acetone [
31]; sol-gel polymerization of linear phenolic resin and hexamethylenetetramine in alcoholic solutions [
32]; sol–gel polymerization reaction of resorcinol and formaldehyde in water [
33]. All of them correspond to organic synthesis with hazardous substances, which make more attractive the use of cellulose-based raw materials. Regardless of the synthesis method, CAGs present several advantages as catalyst supports, compared to common carbon materials. They allow a uniform distribution of metal particles at the surface, and the metal dispersion is stable upon heating treatments [
34]; because of their nanostructure, they show higher thermal resistance [
35] and mechanical strength [
36]. In addition to catalyst supports, CAGs can be used as adsorbents, electrodes, and supercapacitors for secondary batteries, hydrogen storage, and desalination [
37,
38,
39].
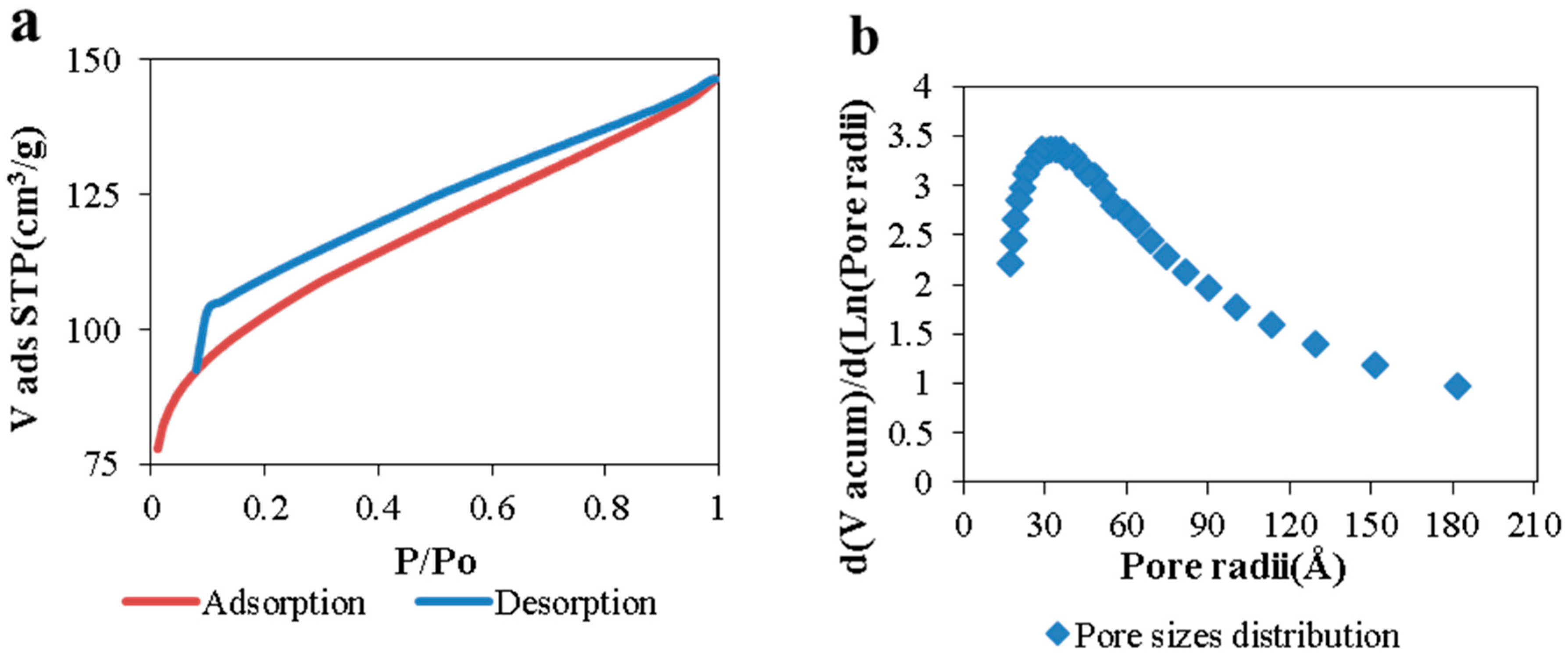
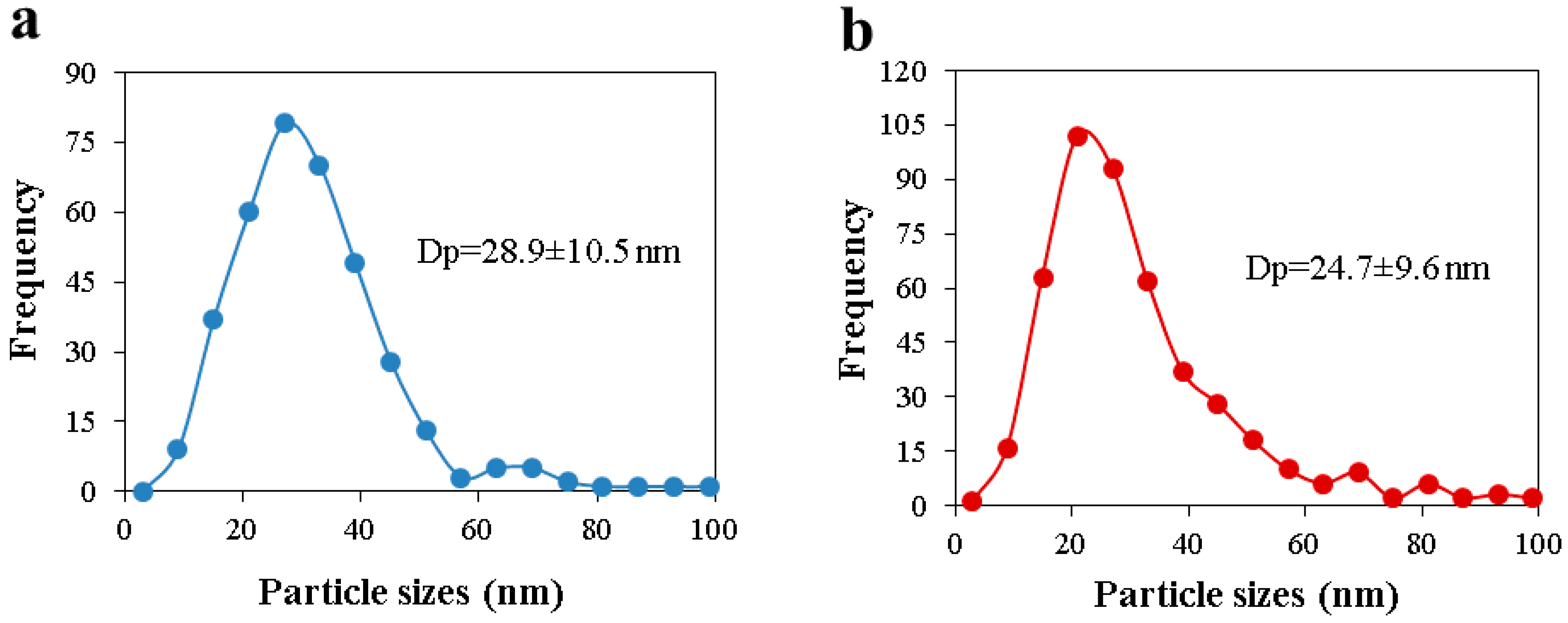
Morphological and structural properties of CAGs from cellulose can be easily manipulated by controlling the carbonization process (
Table 1). According to Jazaeri et al., temperature and heating rate are key parameters in the carbonization [
30]. Their experiments indicated that long holding times (above 17 h) and slow heating rates (below 2 °C/min) help to preserve desired fibrous morphology and acceptable mass yield, although reported mass yields are far from the theoretical 44.4 % of carbon in cellulose (about 15% [
40]). Different strategies have been reported to increase mass yield: Impregnation of raw material with ammonium salts [
41], carbonization under acid atmosphere (HCl) to promote a fast dehydration and avoid carbon losses [
42], and modification of the carbonization conditions [
43,
44]. In particular, the impregnation with ammonium salts (i.e., ammonium sulphate) modifies the pyrolysis mechanism that is normally observed for biomass. It favours dehydration at lower temperatures and stabilizes the carbonaceous structure [
45], which results in a lower mass loss.
Usually, higher carbonization temperatures lead to carbons with higher specific surface area, up to the point (different for each raw material) where the structure collapses as a result of the cross-linking of carbon atoms, which reduces the space between them [
46]. Nevertheless,
Table 1 shows that for CAGs, the temperature has a lower effect on the carbon surface area when lower heating rates and shorter dwell times are used, probably because the energy-to-time ratio is not enough to remove high amounts of solid mass to create more space between atoms and, consequently, higher porosity.
Several authors have reported the use of CAGs as catalyst supports for the conversion of organic compounds that can be classified as tar. Maldonado-Hódar used CAGs-supported Pt catalysts to study the adsorption and decomposition of toluene, xylene and acetone, finding that even at high temperatures, the support retains part of these compounds [
50]. Ábrahám et al. published a study about the acetic acid hydroconversion reaction over Mo/CAGs catalysts; their results show that the reaction pathways and product distributions are controlled by the accessibility of carbon surface, as well as by the amount and shape of Mo particles [
51]. Hai Woong Park et al. published an extensive work on lignin model decomposition over Pd/CAGs catalysts, showing the need of modifying the catalyst surface in order to reach the required performances [
52,
53]. These researches show that CAGs, used as catalysts support, have direct influence on activity and selectivity in tar decomposition reactions.
The use of carbon-supported catalysts for ammonia decomposition is not common. Research has mostly focused on ammonia adsorption over carbon and carbon-supported metals as a solution to emission control. Rodrigues et al. studied the influence of initial ammonia concentration and carbon bed temperature during the ammonia adsorption on activated carbon [
54]. They found a direct dependence of adsorption capacity of ammonia over carbon with the amount of ammonia in fluid phase, contrary to rise temperature dependence. Bandosz and Petit used carbon from different sources and different metal impregnation to evaluate strength and amount of ammonia adsorbed over carbons surface and it interaction with metals particles [
55]; they concluded that in unmodified carbon, porosity controls the adsorption process. More detailed studies on metal-ammonia interaction have been published using theoretical ways, in which the effect of support is not considered. Results suggest that the adsorption of ammonia on transition metal atoms, like Fe and Ni, occurs in bridge, hollow, and top positions, through the unpaired electrons in nitrogen [
56,
57,
58]. Their results suggest a chemical interaction between ammonia and metals supported by calculated thermodynamics parameters.
CAG-based catalysts can be prepared using organics and inorganics precursors. Smirnova et al. impregnated CAG with Co-mesotetramethoxy-phenylporphine in tetrahydrofuran [
59] and Hu et al. impregnated mesoporous CAG with Cu(NO
3)
2 3H
2O [
38]. Most synthesis methods entail the addition of the active metal to the support via incipient wetness impregnation [
33,
60], followed by drying for 4 to 12 h, and calcination [
61] and/or reduction for up to 7 h, at temperatures of maximum 400 to 600 °C, with slow heating rates.
In general, experimental studies show enhancement of adsorption capacity with carbon surface modification and metal impregnation independent of catalysis preparation methods. Thermodynamic parameters are defined by theoretical studies. Experimental data of thermodynamics parameters of ammonia adsorption over metallic phases over carbon are not determined.
In the present work, the adsorption of ammonia on over nickel and iron catalysts supported on cellulose-based CAG is studied, as a first step to propose a kinetic mechanism of gasification gas cleaning reactions. An experimental design was made for the synthesis of supports and catalysts used in the study of ammonia and tar decomposition reactions, the present report shows the first part related to support preparation and ammonia adsorption. The support (CAG) was produced from freeze-dried MFCs, which were carbonized at different carbonization temperatures (900, 1000, and 1100 °C), heating rates (10 and 20 °C/min), and dwell times (0, 1, and 2 h). As the support will be used for gasification gases catalytic upgrading, the lowest carbonization temperature was restricted at 900 °C, to guarantee the stability of support during operation, typically between 700 °C and 900 °C [
62]. An impregnation of MFCs with ammonium sulphate was carried out to enhance the thermal resistance and mass yield of char in carbonization processes. Catalysts were characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM) and inductively coupled plasma optical emission spectrometry (ICP-OES). Finally, the effect of temperature and initial ammonia concentration on the adsorption of ammonia over synthesized catalysts was studied.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}