Valorization of Biomass Derived Terpene Compounds by Catalytic Amination



1
Boreskov Institute of Catalysis, pr. Lavrentieva, 5, Novosibirsk 630090, Russia
2
Department of Natural Sciences, Novosibirsk State University, Pirogova 2, Novosibirsk 630090, Russia
3
Centro de Nanociencias y Nanotecnología, Universidad Nacional Autónoma de México, km. 107 Carretera Tijuana a Ensenada, Ensenada C.P. 22860, Baja California, Mexico
4
Johan Gadolin Process Chemistry Centre, Åbo Akademi University, FI-20500 Turku/Åbo, Finland
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(9), 365; https://doi.org/10.3390/catal8090365
Submission received: 30 July 2018
/
Revised: 22 August 2018
/
Accepted: 26 August 2018
/
Published: 29 August 2018
(This article belongs to the Special Issue Solid Catalysts for the Upgrading of Renewable Sources)
Abstract
:This review fills an apparent gap existing in the literature by providing an overview of the readily available terpenes and existing catalytic protocols for preparation of terpene-derived amines. To address the role of solid catalysts in amination of terpenes the same reactions with homogeneous counterparts are also discussed. Such catalysts can be considered as a benchmark, which solid catalysts should match. Although catalytic systems based on transition metal complexes have been developed for synthesis of amines to a larger extent, there is an apparent need to reduce the production costs. Subsequently, homogenous systems based on cheaper metals operating by nucleophilic substitution (e.g., Ni, Co, Cu, Fe) with a possibility of easy recycling, as well as metal nanoparticles (e.g., Pd, Au) supported on amphoteric oxides should be developed. These catalysts will allow synthesis of amine derivatives of terpenes which have a broad range of applications as specialty chemicals (e.g., pesticides, surfactants, etc.) and pharmaceuticals. The review will be useful in selection and design of appropriate solid materials with tailored properties as efficient catalysts for amination of terpenes.
1. Introduction
A vast expansion in research activities on biomass derived compounds is clearly related to a growing interest in sustainable feedstock. The current review is focused on synthesis of various amines from biomass, namely terpenes. In general amine derivatives have found important applications as corrosion inhibitors, in cosmetics and toiletries, and color reprography to name but a few. Well known is also their utilization for production of different pesticides and dyes, such as azine, azo dyes, as well as indigo dyes [1]. Besides being important platform chemicals [2,3,4], they can be also applied in synthesis of pharmaceuticals in particular anticancer agents and DNA alkylators. Unfortunately, most of the industrially relevant aliphatic and aromatic amines, as well as aminoalcohols are currently manufactured from fossil resources [5,6]. For synthesis of shorter chain amines, (e.g., ethylene diamine [5], ethanolamines [6]) ammonia and respectively 1,2-dichloroethane and ethylene oxide are used. This is rather energy-intensive also resulting in significant CO2 emissions and problems with corrosion when HCl is produced as a by-product. For such shorter chain amines apparently more sustainable reaction routes should be developed.
For longer chain amines there is a clear alternative relying on utilization of biomass feedstock, namely production of bio-based amines can be done from bio-derived alcohols obtained from carbohydrates, fats, oils, and lignins [4,7,8,9,10] (Figure 1). In particular the development of efficient heterogeneous catalysts for such syntheses starting from carbohydrates [11,12,13,14], lignin derived phenolics [4,15,16,17,18,19,20,21], fatty acid (esters) and glycerol from oleochemical sources [8,22], monomers from chitin [23], and amino acids from proteins, was comprehensively reviewed by Froidevaux et al. [10] and Pelckmans et al. [4].
Another available biomass feedstock is the family of terpenes, being present in leaves, flowers, and fruits of many plants [24]. Distillation of turpentine, a byproduct in the pulp mills making cellulose, gives different terpenes. Apart from recent reviews [10,25] terpenes, have, however, not been considered in detail as a promising feedstock for biobased amines.
This review fills the apparent existing gap in the literature giving an overview of the readily available terpenes and describing the developed catalytic protocols for preparation of terpene-derived amines using homogeneous and heterogeneous catalysts. Bio-catalysis is beyond the scope of this review, while it should be mentioned that some interesting results have been reported [26,27,28,29] for intramolecular C–H amination of carbonazidate derivatives of menthol and borneol to corresponding five-membered cyclic compounds [30].
For some biomass derived compounds amination in the presence of heterogeneous catalysts has been extensively studied as described in detail in [4]. At the same time the same concept has been scarcely applied for so called extractives, constituting ca. 5% of lignocellulosic biomass. In particular, terpenes can be considered as very valuable components of biomass because of the potential industrial application of their derivatives ranging from basic and specialty chemicals to pharmaceuticals.
In order to address the role of solid catalysts in the amination of terpenes it was important to have an overview first of the same reactions occurring with the homogeneous counterparts. Such reactions can be considered as benchmarks, which solid catalysts should match. It should be mentioned in this connection, that it was recognized many years ago that at molecular level, there is little to distinguish between homogeneous and heterogeneous catalysis, while there are clear distinctions at the industrial level [31].
The subsequent sections consider respectively the significance of terpenes and their amine derivatives and main catalytic reactions for introduction of amine functionalities.
2. Terpenes Valorization into Valuable Amines
Terpenes are hydrocarbons consisting of isoprene (C5) basic units even if they are structurally very diverse. Terpenes or more precisely monoterpenes (C10), sesquiterpenes (C15), diterpenes (C20), sesterterpenes (C25), triterpenes (C30), and rubber (C5)n can be either branched or cyclic unsaturated molecules. Being typically extracted from the resins of coniferous trees they can be also present as acyclic or mono- to pentacyclic derivatives containing alcoxy, ether, carbonyl, keto, or ester ketone groups (i.e., “terpenoids”). These substrates present in various living species [32], particular in higher plants, are characteristic of a specific plant type.
The well-known application of natural (or even synthetic) resins of terpenes in perfumes and fragrances is related to their odor. In addition, synthesis of vitamins, insecticides, and pharmaceuticals also starts from terpenes [33,34,35,36,37]. Acyclic terpene amines are of special interest for production of insecticides, fungicides, and herbicides as well as in development of new pharmaceuticals [38,39,40,41,42,43]. Amino terpenes on the basis of (−)-menthol and (+)-3-carene were used for the preparation of potential inhibitors of γ-aminobutyric acid neuro-receptors for neurological applications [44,45]. Efficiency of limonene amino derivatives against in vitro cultures of the Leishmania (Viannia) braziliensis [46], egg hatchability, and mortality [47], as well as tobacco growth inhibitors was demonstrated [48,49]. Another interesting synthetic option is to use the amine group as a suitable protecting group, when there is a need to selectively hydrate some bonds in terpenes (e.g., myrcene) containing several double bonds. This strategy was applied in the synthesis of myrcenol, hydroxycitronellal [33] as well as terpenol [50]. It is also possible to use amino derivatives of terpenes as ligands in enantioselective reactions, such as catalytic asymmetric transfer hydrogenation of aromatic alkyl ketones [51] or enantioselective alkynyl zinc additions to aromatic and aliphatic aldehydes [52]. Amino terpenes on the basis of dihydromyrcenol were applied in synthesis of surfactants [53].
The history of plant terpenoids application in traditional herbal remedies is very extensive, therefore it is not surprising that they are currently under investigation due to their different therapeutic properties [54]. Even simple terpenes such as D-limonene, farnesol, and geraniol were reported to possess some chemotherapeutic activity against human cancer [55]. Carboranes with cinamyl, prenyl, and geranyl terpenoid fragments [55] were used to enhance boron delivery in boron neutron capture therapy [56,57,58,59]. Treatment with an alkyl halide of citronellal after amination with dimethyl or diethyl amines gives the corresponding chiral ionic liquids [60].
In general, a larger scale production of chemicals from plant extracts is limited, even if there are some examples when aminoterpenes play an important role in asymmetric and chemoselective catalysis. The Takasago Perfumery Company produces optically pure (−)-citronellal and pure (−)-menthol (1500 t/a) using N,N-diethylnerylamine [33]. SCM Corporation utilizes amination of myrcene for the synthesis of an insect repellent possessing insecticidal activity against the American flour beetle and the German cockroach [61].
This short overview illustrates a diverse scope of potential applications of terpenes-based amines in synthesis of valuable products including pharmaceuticals.
3. Possible Catalytic Tools for Synthesis of Terpene-Based Amines
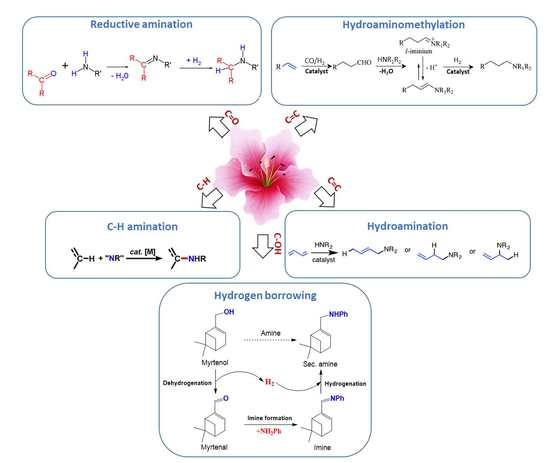
This section is devoted to several reaction routes available to form C–N bonds in the terpenes of interest. Classical approaches for amine synthesis by a direct reaction of ammonia with alkyl halides or alternatively reduction of nitro or nitrile compounds are not considered here, instead the main focus is on hydroamination, hydroaminomethylation, reductive amination, and alcohols coupling with amines [1]. As mentioned above terpenes are highly functionalized molecules that contain double bonds, while their derivatives bear carbonyl and alkoxy or hydroxyl groups (Figure 2) that can be readily involved in various amination strategies.
Thus, five possible strategies in the formation of C–N bonds in terpenes and their derivatives were distinguished (Figure 3):
- (1)
- reductive amination of aldehydes and ketones
- (2)
- hydroaminomethylation
- (3)
- hydroamination of double C=C bonds
- (4)
- hydrogen borrowing methodology for amination of alcohols
- (5)
- C–H amination of terpenes, which is a very specific case.
3.1. Reductive Amination of Terpenes with Carbonyl Moiety
3.1.1. Reductive Amination of Aldehydes
As an example a particular terpenoid containing an aldehyde function is considered.
Citronellal (3,7-dimethyloct-6-en-1-al, 1) (Figure 4) is well known as a flavoring agent and an insect repellent. The (R)-isomer of citronellal is typically found in citronella while the essential oil of kaffir lime contains the (S)-isomer. Citronellyl amine can be synthesized from the amide [62], oxime [62], and from geranylnitrile [63]. An issue related to reductive amination of aldehydes with ammonia using transition metals as catalysts is the need to suppress side reactions (Figure 4). Reductive amination of citronellal with aqueous ammonia giving primary amines was described by Behr et al. [62]. In this atom efficient method [Rh(cod)Cl]2/TPPTS (TPPTS = 3,3′,3′′-phosphanetriyl benzenesulfonic acid) as a homogenous catalyst was used in a biphasic solvent system. The organic compounds (substrate and product) are located in the apolar solvent phase of the biphasic solvent system, following an established concept applied for hydroformylation. This approach allowed a high yield of primary amines (4, 5) up to 87% effectively suppressing side reactions. These yields were obtained at 60 bar of hydrogen and 130 °C. Such high pressure was required as selectivity was seen to be dependent on pressure (Figure 5).
All details of the experimental conditions are reported in [62]. The biphasic solvent inevitably requires efficient mass transfer, therefore surfactants, including ionic liquids or native cyclodextrins and their derivatives such as 1-decyl-3-methylimidazolium bromide ([DecMIM]Br) and methylcyclodextrin were applied [64]. Another option to increase selectivity towards the desired primary alcohols is to cleave the secondary imine formed as an undesired by-product [65].
3.1.2. Reductive Amination of Ketones
Reductive amination of d-fenchone to prepare fenchylamine, which are intermediates for some biologically active compounds [66] has been studied already long time ago [67] applying heterogeneous catalysts. In particular in the gas-phase amination of D-fenchone (1) with aliphatic nitriles (acetonitrile, acrylonitrile. or butyronitrile) performed at 220–260 °С under pressure of hydrogen ranging from 10 to 15 bar over copper on alumina modified with LiOH а mixture of isomeric endo-N-alkyl-l,3,3-trimethylbicyclo[2.2.l]hept-2-ylamines (2) and the corresponding exo compounds (3) with a ratio of 3:1 and yield of 50–60% was formed along with the intermediate N-fenchylidenalkylamine (4) (Figure 6). The main side products were α-fenchol (5) and β-fenchol (6).
An interesting feature of this reaction is the generation of а primary amine from the initial nitrile on the metal sites. This primary amine then reacts with the substrate giving N-fenchylidenalkylamine (4) which is followed by hydrogenation into diastereomeric secondary amines (2, 3). This reaction competes with the intermolecular dehydration of the alcohols (5, 6). Formation of secondary amines is, however, predominant.
A systematic study on reductive amination of carbonyl terpenoids (camphor, carvone, hexahydropseudoionone, isocamphone) with different nitriles (acetonitrile, propionitrile, benzonitrile) over 15% Cu/Al2O3 modified with 2–6% LiOH resulting in both unsaturated and completely hydrogenated amines of diverse structure was conducted by Kozlov and co-workers [68].
Another example reported in the literature for reductive amination was related to camphor as a substrate. Influence of the heterogeneous catalysts type on the amination product yields in the reductive amination of camphor (1) with methylamine (Figure 7) was investigated. When Raney nickel was used as a catalyst, N-methylbornan-2-imine (2) (yield 82.8%) was predominantly formed, whereas the reaction over 5% Pd/C yielded a mixture of the imine (2) and N-methylbornan-2-ylamine (3) (30.4% and 65.7%, respectively). When platinum oxide was used as a catalyst, the yield of the amine 3 reached 92.7%.
After reduction the promoted fused iron catalyst was applied for camphor (1) conversion (Figure 7) exhibiting high stereoselectivity to endobornan-2-ylamines (3), which is somewhat unusual for metal heterogeneous catalysts. In particular conversion of d,l-camphor (1) into endo- and exobornan-2-ylamines (3) during hydroamination reached 92%, with the endo to exo ratio being (1.4–1.8):1. Apparently, this stereoselectivity is due to the “imine-enamine” tautomerization occurring on the acid–base sites of the catalyst [68].
3.2. Hydroaminomethylation of Olefin Bonds in Terpenes
Somewhat related to reductive amination described above is hydroaminomethylation (HAM), which in fact is a tandem reaction consisting of hydroformylation followed by reductive amination [69]. This one-pot process proceeds on the same catalyst responsible for hydroformylation of C=C double bond making first an aldehyde followed by amination and hydrogenation of the imine/enamine intermediate [70,71,72,73] finally giving a secondary or a tertiary amine. The only by-product in this very efficient process with good atom economy is water. Apparently a careful choice of reaction conditions is needed to satisfy requirements for all reactions comprising a complex reaction network [70,74].
A few examples of hydroaminomethylation reaction with terpenes were reported including α-pinene [75], β-pinene [74], camphene [74], limonene [74,76,77], β-myrcene and β-farnesene [78] and naturally occurring allyl benzenes such as eugenol [79] and estragole [80]. Hydroformylation of the internal double bonds is much more difficult than the terminal bonds, thus it is not surprising that the examples mentioned above are related to isolated terminal double bonds. In the only reported hydroaminomethylation of a conjugated terpene [81] regioselectivity in hydroformylation was low along with low catalytic activity per se explained by formation of relatively stable η3-allyl-Rh complexes [78].
Hydroaminomethylation of limonene (1) with secondary amines (n- and i-propylamine, benzylamine), cyclic amines (piperidine, morpholine, piperazine,) aromatic amine (aniline) and diamines (ethylenediamine, propilenediamine, tetramethylenediamine) was reported by Graebin et al. [76]. The yields of products varied from 50% to 89% and are presented in Figure 8. In the case of diamines only isomerization products were obtained for tetramethylenediamine, while no products were formed when ethylenediamine was used. A plausible explanation could be inactivation of rhodium because of formation of stable chelated compounds of diamine with the catalyst.
Olefin hydroformylation was selective towards the linear aldehydes compared to the branched ones [76] which can be ascribed to the catalyst itself along with the steric hindrance of the terpene isopropenyl group [82,83,84,85].
Amination of aldehydes is more difficult with ammonia than with primary amines. Nevertheless Behr et al. [77] applied ammonia in HAM of limonene (1) (Figure 9). The reaction proceeds through hydroformylation of limonene (1) to the corresponding aldehyde (2) in the first step followed by condensation with ammonia giving an aldehyde observed experimentally and subsequent hydrogenation of the latter to a primary amine (3).
The desired amine (3) reacted also with the aldehyde 2 resulting in formation of secondary and tertiary amines. Moreover, limonene (1) underwent isomerization to its isomer isoterpinolene (4) [25,77].
[Rh(cod)(μ-OMe)]2 as a pre-catalyst in the presence or absence of triphenylphosphine or tribenzylphosphine as ligands was applied in HAM of R-(+)-limonene (1) (Figure 10), camphene (5) (Figure 11), and (−)-β-pinene (9) (Figure 12) with di-n-butylamine, n-butylamine, morpholine, triphenylphosphine, and tribenzylphosphine using toluene as a solvent [74]. The reaction giving moderate to good yields (75–94%) was performed at 100 °C and 60 bar with an equimolar mixture of CO and H2.
Hydroaminomethylation of estragole (1) (Figure 13), a bio-renewable starting material, with di-n-butylamine was studied in [80]. Estragole being a primary constituent of essential oil of tarragon (60–75%) is also present in other sources, such as pine oil, turpentine, fennel or anise (2%) [86]. HAM consists of alkene hydroformylation followed by reductive amination of aldehydes. Different ligands were used with rhodium(I) catalysts including phosphine, phosphites, and phospholes. The latter were the most efficient not only in hydroformylation, but also in reductive amination. Three isomeric amines (9–11) were generated as final products (Figure 13). Along with these imines aldehydes (3–5) and enamines (6–8) were observed depending on conditions. Side reactions included for example aldol condensation. Some other hydrogenation products as well as unidentified products were also formed.
Hydroaminomethylation of eugenol (1) with di-n-butylamine (Figure 14) involved bis[(1,5-ciclooctadiene)(µ-methoxy)rhodium(I)] as a pre-catalyst [79]. The presence of phosphines was needed to improve chemoselectivity in hydroformylation, being detrimental for hydrogenation of enamine intermediates. Similar to the cases described above mainly linear aldehyde was obtained in hydroformylation. Efficiency of HAM could be also improved by addition of triflic acid as a promoter [79].
As can be seen from Figure 14 hydroaminomethylation of eugenol with di-n-butylamine gives three isomeric amines (9–11) of which compound 9 is predominant. Similar to estragole the intermediate aldehydes (3–5) and enamines (6–8) were also observed. Table 1 and Table 2 contain the results for HAM of eugenol for different catalysts and reaction conditions.
Triflic acid, being more stable than HBF4, was reported to be an efficient promoter for eugenol hydroaminomethylation in the presence of phosphines as ancillaries [79].
Rh/1,2-bis(diphenylphosphino)ethane was used [78] for hydroaminomethylation of industrially available β-myrcene and β-farnesene. The reaction network is basically the same as for other terpenes presented above (Figure 15) with also high regioselectivity towards the linear aldehyde. Such efficient synthetic protocol allows preparation of environmentally friendly biobased surfactants.
The final example of this section is related to a two-step synthesis protocol when hydroformylation of a-pinene was done either using rhodium or cobalt based catalysts. Chiral aminomethyl pinane was prepared in 100 g scale [75] with rhodium as a catalyst giving (+)-3-formylpinane and subsequent reductive amination with ammonia. On the contrary Co2(CO)8 led to (−)-2-formylborane.
3.3. Hydroamination on Olefin Bonds of Terpenes
Regioselective hydroamination of alkenes is more challenging than hydroamination of aldehydes. In particular a product of anti-Markovnikov addition is desired, while known synthetic protocols are mainly selective towards the product of Markovnikov addition [87,88,89].
Pd(cod)Cl2 in combination with bis(2-diphenylphosphinophenyl) ether (DPEphos) [89] afforded selective hydroamination of acyclic and cyclic dienes with several aromatic and aliphatic amines not requiring presence of any additive. Significant efforts in [89] were concentrated on transformations of isoprene. More relevant in the context of this review is hydroamination of myrcene catalyzed either by alkali metals or transition metals [33]. Following the same pattern as for other 1,3-dienes, predominantly 1,4-amines were formed (Figure 16).
The reactive double bond is the terminal one (denoted as tail in Figure 16) resulting in linear amines, which in fact are the desired products. Such a method is rather efficient compared to alternatives, which rely on the corresponding acetates as the starting compounds and allylic amination catalyzed by palladium complexes [90,91,92,93].
Superior atom efficiency of hydroamination (100%) is a reason for a plentitude of studies on addition of amines, in particular diethylamine, to myrcene (Table 3).
N,N-Diethylgeranyl- and nerylamine are valuable starting compounds for synthesis of a variety of terpenoids (Figure 17) including (−)-menthol, myrcenol, hydroxycitronellol, nerol, geraniol, linalool, (+)-citronellal, and (+)-citronellol.
The hydroamination reaction is mostly catalyzed by alkali metal-based systems such as sodium or lithium (Table 3, entries 1–9). More expensive transition metals, nevertheless provide higher TON values being effectively recycled. Nowadays two routes are known using transition metal catalysts (Rh and Pd complexes) developed by Rhone–Poulenc [94] and Berh [33] with TOF values of 27 and 124 h−1, respectively (Table 3, entries 10–11).
Preparation of optically pure (−)-citronellal and (−)-menthol (route A) developed by Takasago Perfumery Company is the largest application of asymmetric catalysis. It includes enantioselective isomerization to an optically active enamine with high enantioselectivity (ee = 95–99%) [95]. Subsequent hydrolysis results in (+)-citronellal, which then undergoes cyclization in the presence of ZnBr2 and forms (−)-isopulegol. This reaction can also be performed over heterogeneous catalysts [96]. Hydrogenation of isopulegol gives finally (−)-menthol.
In route B (Figure 17) the amine group serves as a suitable protecting group allowing selective hydration of the isolated double bond of myrcene. Further transformations using Pd complexes [97,98,99] or sodium hydride [100] give respectively myrcenol and hydroxycitronellal.
Oxidation of N,N-diethylnerylamine with hydrogen peroxide (route C in Figure 17) is the first step in the synthesis of geraniol, nerol, or linalool [101].
Amination of myrcene with 2-amino-2-methyl-1-propanol developed by SCM Corporation leads to products acting as repellents against the American flour beetle and the German cockroach [61].
Hydroamination of myrcene into diethylgeranylamine using palladium complexes with bidentate ligands such as bis(diphenylphosphino)butane (DPPB) or bis(2-diphenylphosphinophenyl) ether (DPEphos) [102] raises an issue of palladium recovery combining the advantages of homogeneous catalysis (high selectivity and activity) with those of heterogeneous catalysis (catalyst reuse and simple separation). Application of thermomorphic solvent systems, such as for example dimethylformamide/heptane and acetonitrile (ACN)/heptane, allowed simple catalyst separation. ACN/heptane turned out to be a more suitable solvent mixture, permitting efficient product extraction with negligible catalyst leaching. This method is based on the temperature dependent miscibility gap of the solvent system components. Thus, when a mixture of two liquid components, immiscible at room temperature, is heated to a higher reaction temperature, a single liquid phase is formed. In this state no mass transfer limitations occur. Cooling down under the critical solution temperature leads to a biphasic system, from which the catalyst phase can be simply separated from the extract phase providing easy catalyst recycling. Overall 90% yield of diethylgeranylamine was reported [102].
3.4. Amination of Terpene Alcohols
Hydrogen borrowing amination of terpene alcohols has attracted a lot of attention generating only water as the byproduct [7,103,104,105,106,107]. While this method has been adapted industrially [108] for production of low alkyl chain amines such as N-methyl-, N,N-dimethyl-, and N,N,N-trimethyl-amines in the presence of Bronsted and Lewis acid catalysts, rather high temperatures are needed exceeding 300 °C and moreover mixtures of N-substituted amines are often produced.
In this section, we discuss recent progress in the development of efficient homogeneous and heterogeneous catalysts, capable of carrying out selective synthesis of desirable amines, especially taking into account that selective amination of rather labile terpene alcohols requires milder conditions and thus more efficient catalytic systems. Along with the hydrogen borrowing reactions, less atomic efficient homogeneous transition metal-catalyzed allylic substitution reactions with functionalized allylic terpene alcohols are considered.
3.4.1. Homogeneous Catalysts
A broad range of homogeneous catalysts based on transition metal complexes was applied for N-alkylation, including Rh, Pd, Au, Ag, Pt, Os, and Re. Moreover, even some systems with non-noble metals (Ni, Cu, Fe, and Co) have been proven to be efficient catalysts in N-alkylation [109].
In particular contribution of Milstein and co-workers in development of direct homogeneous catalytic amination of primary alcohols with ammonia should be acknowledged [104]. Transformations of secondary alcohols to primary amines with ammonia following the so-called hydrogen borrowing methodology were described extensively in the literature [7,64,105,110,111,112,113,114,115,116,117,118,119,120,121,122].
Specific applications of this approach to transformations of terpene derivatives were also reported. For instance, Pingen et al. [113] utilized [Ru3(CO)12] and different phosphor containing ligands to selectively convert various primary and secondary terpene alcohols to primary and secondary amines. This particular catalyst was considered to be an exception, as in fact the hydrogen borrowing approach rarely results in synthesis of amines and diamines with optimal yields using homogeneous catalysts based on Ru and Ir complexes combined with P-ligands. Some examples of reasonable yields of primary and secondary terpene amines exceeding 80% were reported for such primary and secondary terpene alcohols as myrtenol [123,124], citronellol, nerol, geraniol, farnesol, and fenchol [9,125].
Some examples of N-alkylation of terpene alcohols with ammonia are presented in Table 4 and Table 5. Screening of different P-ligands showed [9] that the acridine-based diphosphine was the only bidentate ligand affording excellent results. This ligand was used in couple with [RuHCl(CO)(PPh3)3] for the amination of primary alcohols with ammonia. This combination, however, appeared to be inactive for secondary amines under the same conditions [104,125].
While palladium-catalyzed amination of allylic functionalized simple alkenes has been studied extensively, application of natural terpenic alkenes derivatives is much less common [91,92]. In this context an interesting example is the synthesis of N,N-diethylgeranylamine (2) and N,N-diethylneranylamine (4) from diethylamine and linalyl acetate (1) or nerolidyl acetate (3) (Figure 18) using Pd(PPh3)4 [91].
Besides diethylamine other amines can be applied for such transformations. For example, N-geranylaniline was obtained by reaction of aniline with linalyl acetate (1) displaying 100% stereoselectivity.
Analogously nerolidyl acetate (3) was transformed into amine derivatives [91]. Dependence of the yield and selectivity (E to Z ratio) for nerolidyl acetate (3) is presented in Table 6.
Similarly to the example above for linalyl acetate utilization of linalyl methylcarbonate with the same Pd(0) catalyst and diethylamine results also in N,N-diethylgeranylamine (Table 7).
Reaction of linalyl methylcarbonate (5) with aniline or morpholine gives selectively (E)-isomers (E:Z = 100:0) (Table 7).
Pd(0)-catalyzed amination of allylic natural functionalized terpenes—myrtenyl acetate (1), perillyl acetate (3), geranyl acetate (5), mertynyl alkyl carbonate (9), perillyl alkyl carbonate (10), geranyl alkyl carbonate (11)—was studied in [124]. The reaction scheme is shown in Figure 19, while Table 8 illustrates the catalytic results for myrtenyl acetate with and without Et3N.
This substrate was used in combination with different secondary amines (pyrrolidine, morpholine, and Me2NH) and the Pd(dba)2/PPh3 catalytic system (Figure 19). As seen from Table 8 high yields could be reached under mild conditions. Application of an acceptor base, e.g., Et3N was beneficial for pyrrolidine as the amine source. The nature of the ligand in case of morpholine as nucleophile and Et3N as the acceptor base influenced significantly the catalytic behavior (Table 9). Presence of ligands was essential as in their absence there was practically no reaction.
Some other examples of Pd(0)-catalyzed amination of terpenic allylic esters, namely perillyl acetate (3), geranyl acetate (5), mertynyl alkyl carbonate (9), perillyl alkyl carbonate (10), and geranyl alkyl carbonate (11), are summarized in Table 10 [124].
Lyubimov et al. [55] conducted Pd-catalyzed amination of allylic carbonates of terpenoids: (E)-cinnamyl ethyl carbonate (1), ethyl prenyl carbonate (2), ethyl geranyl carbonate (3), in supercritical CO2 with N-(ortho-carboran-3-yl)-N-methylamine (4) (Figure 20). Corborane amines, which are promising agents for cancer therapy, were obtained at complete conversion with excellent regioselectivity using sodium bicarbonate as the acceptor base [55].
3.4.2. Heterogeneous Catalysts
Although various transition-metal complexes can provide reasonable selectivity to the desired products, application of expensive noble metal catalysts results in complicated reaction systems. Homogeneous transition metal catalysts based on noble metals are typically expensive and toxic, not stable and prone to contaminate the products and finally difficult to recover. Moreover, application of sophisticated and unpredictable organic ligands has obvious disadvantages, such as difficulties in their synthesis and recovery. Not surprisingly to increase the probability of industrial implementation significant efforts were devoted to heterogenization of homogeneous catalysts [126,127,128,129].
One example is related to introduction of a chiral ferrocenyl-based ligand to the surface of a mesoporous material MCM-41 [130]. The active catalytic species (Figure 21) were used in allylic amination of cinnamyl acetate with benzylamine with the aim of achieving high yields of the branched product (reaching 50%) with the highest possible ee. In fact, the enantiomeric excess was close to 100%, while the homogeneous catalyst gave exclusively a straight chain product with no ee [131].
Even though immobilization of organic ligands or metal complexes by covalent binding [131,132] has been a subject of extensive research there are a number of technological challenges including complexity of the synthesis procedure and eventual leaching of the immobilized species. Advantages of heterogeneous catalysts in product and catalyst isolation, and catalyst reuse prompted their application in amination [133,134]. Specificity of heterogeneous catalysts sometimes displaying activity for only a particular substrate restricts their more widespread application. As an example sulfonamide alkylation with alcohols over Ru on iron oxide can be mentioned of such specificity, as the catalysts are not active in alkylation of carboxamides or amines [135]. Therefore, it is required to develop more active and general heterogeneous catalysts for the terpene alcohols amination without addition of organic ligands.
N-alkylation follows the hydrogen borrowing mechanism with first dehydrogenation of an alcohol to the corresponding aldehyde followed by condensation with an amine giving an imine. The latter is then hydrogenated by hydrogen “stored” on the catalyst resulting in the final alkylated amine products (Figure 22) [109]. The method is thus mainly suitable for primary amines and alcohols. According to this general mechanism, hydrogen transfer from alcohols to catalysts and then from adsorbed hydrogen to intermediate imines are the typical key processes of the methods. It should also be pointed out that inorganic bases were usually required in hydrogen autotransfer reactions for deprotonation of the alcohols to facilitate their coordination with transition metal catalysts. Since bases were usually used in large excess amounts in the early transition metal-free methods, they were used only as additives but not as catalysts in transition metal-catalyzed reactions, even though in many cases they were also used in minor (well below stoichiometry) amounts [109].
Hydrogen transfer steps (dehydrogenation of alcohols and hydrogenation of imines) are considered as the rate controlling steps. Primary amines and alcohols are thus preferred displaying better results in these key steps. Inorganic bases as mentioned above are typically used as additives to facilitate deprotonation of alcohols and coordination to transition metals [109].
First application of typical heterogeneous catalysts in direct terpene alcohol amination was demonstrated in a series of recent studies by Simakova and co-workers [123,136,137,138,139,140]. Liquid-phase amination of myrtenol [123,136,137,138,139], nopol, and perillyl alcohol [140] was carried out over supported Au catalysts (Au 1.4 mol% to substrate) in toluene at 180 °C under 9 bar nitrogen pressure using equimolar amounts of substrates without any bases as additives. The reaction network for myrtenol amination is presented in Figure 23. In addition to the expected products, myrtenal and the corresponding imines (1 and 2), also myrtanol as well as myrtanal with the saturated C–C bond were formed subsequently resulting also in imines (3) and (4). It should be noted that prior to the work using heterogeneous catalysts only amine (2) was obtained by interacting myrtenol with PBr3 giving a bromide which was then put in contact with aniline leading to amine (2) [141].
The authors showed that the support played a crucial role in this reaction. The product distribution during the reaction was found to depend strongly on the type of the support (Figure 24). A nearly complete conversion of myrtenol was achieved only in the presence of Au/ZrO2 and Au/Al2O3 catalysts.
Gold supported on ceria, magnesia, and lanthana showed a relatively high alcohol conversion, even if the reaction rate was lower than for gold on alumina and zirconia. The non-basic supports Au/Al2O3 and Au/ZrO2 promoted much faster interactions of the aldehyde with aniline as well as hydrogen transfer [123].
The authors underlined the necessity of a certain balance between the different acid–base sites of the metal oxide for efficient alcohol amination. The initial myrtenol activation was shown to require the presence of the basic sites on metal oxide surfaces whereas availability of the protons on the support surface was suggested to be important for the target amine formation. The highest activity in one-pot myrtenol amination among the tested catalysts was obtained over Au/ZrO2 with both acidic and basic sites.
Moreover, the complex influence of redox treatment on catalytic behavior of Au/ZrO2, Au/CeO2, and Au/La2O3 was studied (Figure 25) elucidating formation of the active species. Au/ZrO2 treated under an oxidizing atmosphere was shown to be more effective in terms of the target secondary amine yield [136].
Demidova et al. [123] found that myrtenal condensation with aniline per se was non-catalytic being, however, noticeably accelerated in the presence of a catalyst. The first step of alcohol deprotonation was concluded to be promoted at the basic sites of the support giving an alkoxide intermediate on the support surface. This is followed by β-hydride elimination catalyzed by Au to form myrtenal. The adsorbed myrtenal and aniline interact to form hemiaminal, which then undergoes an attack by the hydride ion from Au nanoparticles and a proton from the support surface resulting in production of the final amine. Formally in the last step the H+ and H− transfer to the hemiaminal is accompanied by dehydration. According to the literature data in the case of homogeneous catalysts a cooperative mechanism of a coordinately unsaturated metal center and adjacent acid/base center is widely accepted [123]. H− in the metal hydrides and H+ of OH or NH groups of the ligand are transferred to carbon and nitrogen of the C–N bond, respectively. Taking into account this model as well as the regularities obtained for other heterogeneous catalysts [106], the authors proposed [123] that the hemiaminal undergoes dehydration and subsequent H+ addition resulting in formation of an immonium cation, which is then attacked by hydride ion from Au nanoparticles to form the final product [106,142]. The mechanism is illustrated in Figure 26.
It was found that Au on metal oxides was slightly deactivated during amination predominantly due to imine adsorption. Therefore, kinetic modelling of myrtenol amination was done for the mechanism which also incorporated the catalyst deactivation step [123,136].
Based on the kinetic data of myrtenol amination with aniline published in [123] it was proposed that introducing additional hydrogen is beneficial for the overall process by improving in particular hydrogenation of the imine. Such approach is of general interest for the so-called hydrogen borrowing reactions, when hydrogen generated in the dehydrogenation step is transferred to an intermediate imine.
In [138] the effect of hydrogen addition was thus explored to increase selectivity. Hydrogen addition timing depending on myrtenol conversion and hydrogenation temperature affected selectivity for the reaction products. Hydrogen addition (1 bar) after an almost complete myrtenol conversion at 100 °C increased the yield to amine up to 68% preserving the C=C bond in the initial myrtenol structure. Hydrogen addition at 180 °C independent on the level of myrtenol conversion promoted reduction of both C=C and C=N bonds with formation of two diastereomers (yield up to 93%). Formation of the trans-isomer was preferred when hydrogen was added at almost complete myrtenol conversion. As a result it was shown in [138], that in the presence of a gold catalyst controlled hydrogenation of competitive C=C and C=N groups can be performed during one-pot alcohol amination by regulating the reaction conditions (Table 11).
The same authors studied the application of the more safe hydrogen sources and often more readily available in the fine chemical industry than molecular hydrogen, namely alcohols (methanol, 2-propanol) or formic acid [139]. It was found that small amounts of 2-propanol or formic acid (additive/myrtenol molar ratio equal to 0.5 and 0.25, respectively) helped to improve the yield of the target amine elevating it to 68% and 65%, respectively, compared to 52% amine yield in the absence of additives. However, a further increase of the additive amount decreased amine formation because 2-propanol itself reacted with aniline competing with myrtenol. Introduction of formic acid into the reaction mixture suppressed activity of the Au/ZrO2 catalyst due to a strong adsorption of formic acid and decomposition products on the support basic sites required for activation of the initial alcohol. In comparison with other hydrogen additives methanol was found not to be as effective for one-pot alcohol amination. The catalytic activity and selectivity to the reaction products obtained using different external hydrogen sources are compared in Figure 27.
Finally, the structure effect of terpene alcohols selected based on their pharmaceutical relevance was studied over Au/ZrO2 [140] (Table 12 and Table 13). Primary bicyclic (myrtenol and nopol, bearing an unconjugated –OH group) and monocyclic (perillyl) alcohols were chosen for aniline amination under comparable conditions. The rate of alcohol dehydrogenation decreased 10-fold using nopol which after dehydrogenation gave an unconjugated aldehyde group. Selectivity to the desired amine in the latter case increased via selective hydrogen transfer to C=N bonds. Monocyclic perillyl alcohol was more reactive than myrtenol, giving a complex product mixture at 180 °C with the amine present only in small amounts. A decrease of the reaction temperature resulted in a more controlled transformation of perillyl alcohol to imines and amines, with predominant hydrogenation of the C=C bond (Table 12).
Myrtenol amination was also investigated for a range of aliphatic and aromatic amines, showing that the primary amine structure affected both the initial dehydrogenation rate and the selectivity to the desired amine (Table 13). Important issues to be considered were the substrate accumulation on the catalyst surface as well as the reactivity of the substrates and intermediates. Stronger adsorption of more basic amines on the cationic gold species can retard dehydrogenation of an alcohol, which is the first step of the overall process. The nature of the substituent and the reactivity of intermediates is even more important. This was confirmed, as a good correlation was found between the substrate structure and reactivity using the Hammett equation [140].
These data indicated that while an increase of hydrogen availability is an evident method of improving the yields, fine tuning of the amines reactivity can be an even more efficient tool.
3.5. C–H Amination of Terpenes
The final section of this review is devoted to C–H amination. Sulfonimidoylnitrene species (1) (Figure 28) allow the intermolecular chemoselective C–H amination of various complex molecules [143] giving a possibility to synthesize enantiopure aminated derivatives not easily accessible by classical organic synthesis. Allylic methylene units of terpenes and enol ethers have been efficiently aminated with the yields close to 98% and very high diastereomeric ratios (up to ca. 200 to 1) in the presence of a rhodium catalyst Rh2(S-nta)4 (nta = N-(1,8-naphthoyl)-alanine) (Figure 29) [143]. More importantly, the combination of steric, inductive, and conformational factors leads to chemoselective functionalization in the allylic positions allowing site-selective amination with yields of up to 88%. Steric and stereoelectronic effects, in this case, favor amination of tertiary equatorial C-H bonds.
Catalytic C–H amination of terpene substrates was highly efficient allowing isolation of C–H aminated products (3a–3e) in high yields (Table 14). In Table 14 compounds (3a), (3b) stand for derivatives of α-pinene, while (3c–3d), and (3e) correspond to derivatives of respectively limonene, nopol trichloroacetate, and carene. An important feature of amination is chemoselectivity in activation of the C–H bond, while the double bond is inert.
4. Learning from Homogeneous Catalysis and Future Outlook for Heterogeneous Catalysis
The current review focuses on terpene amine synthesis in the presence of solid catalysts rather than with homogeneous catalysts even if the latter are also discussed including immobilized ones.
Terpene alcohol amination represents an interesting example of where catalytic synthesis might reflect different mechanistic views: the hydrogen borrowing pathway in general and the allylic substitution in a particular case of functionalized allylic alcohols substrates. The hydrogen borrowing pathway is a highly atom efficient approach matching green chemistry requirements and providing selective C–N bond formation while keeping the initial terpene moiety. In this connection close attention in the current review was paid to this very promising approach realized over homogeneous and heterogeneous catalysts. Amination of myrtenol over supported gold catalysts was reliably documented to proceed through hydrogen borrowing methodology [123]. Amination of various other allylic and non-allylic terpene alcohols with homogeneous Ru complexes was shown to occur via a hydrogen borrowing pathway as well [125]. Thus [Ru3(CO)12]/L9 catalyzed amination of secondary and primary terpene alcohols (Table 4 and Table 5 in the manuscript) was reported to proceed through intermediate carbonyl compounds indicating a hydrogen borrowing pathway rather than an allylic substitution. The corresponding amine compounds were formed both in the case of substrates with an allylic –OH group (carveol, verbenol, geraniol, nerol, farnesol) and a non-allylic OH group (menthol, borneol, fenchol, citronellol). Reactivity of terpene alcohols depends rather on the presence of steric hindered substituents than on the conjugation with the double bond. Thus in the case of substrates with bulky substituents such as menthol, verbenol, and fenchol the intermediate ketones were the major products regardless of OH group conjugation with the C=C bond.
Along with hydrogen borrowing reactions less atomic efficient catalytic methodologies were also demonstrated in the review. In particular, homogeneous transition metal-catalyzed allylic substitution reactions with functionalized allylic terpene alcohols, discussed in the review (Figure 18, Figure 19 and Figure 20, Table 6, Table 7, Table 8, Table 9 and Table 10) [55,92,124], resulted in stoichiometric by-products formation. Allylic substitution reactions typically utilize an activated allylic substrate (i.e., an allylic alcohol protected as an acetate or ester acting as a leaving group), a transition metal catalyst (usually palladium), and a nucleophile. In a very good recent review [145], which unfortunately did not present examples of terpene allylic alcohols amination, it was demonstrated that allylic substitution in general is possible for unactivated allylic alcohols. In the current review, allylic substitution type of transformations are related to amination of several functionalized allylic alcohols catalyzed by Pd complexes, namely linalyl acetate (Figure 18), nerolidyl acetate (Figure 18, Table 6), linalyl methylcarbonate (Figure 18, Table 7), myrtenyl acetate (Figure 19, Table 8 and Table 9), perillyl acetate, geranyl acetate, mertynyl alkyl carbonate, perillyl alkyl carbonate, and geranyl alkyl carbonate (Table 10) [92,124], as well as cinnamyl ethyl carbonate, ethyl prenyl carbonate, and ethyl geranyl carbonate (Figure 20) [55].
Mechanistic aspects of myrtenol amination in the presence of supported gold catalysts were discussed in [123] suggesting an important role of the hydride ion. In this context it is interesting to find a common denominator for heterogeneous and homogeneous catalytic amination.
Palladium catalyzed allylic amination typically involves formation of neutral or cationic palladium π-allyl complexes via SN2 reaction. A soft nucleophile attacks from the back side of the metal allowing retention of configuration in the product [144]. According to DFT calculations for palladium-catalyzed allylation of primary amines by allylic alcohols [146] one potential pathway involves formation of cationic Pd hydride species while in the second option decomplexation of the coordinated allylammonium can occur. In [124] it was assumed that both amination of allyl acetates and carbonates involves generation of a (π-allyl)-palladium complex (2) (Figure 29). Experimentally observed formation of the racemic product (3) was thus rationalized considering that the nucleophile can attack both allylic positions of the (π-allyl)-palladium complex (2).
Formation of cationic π-allyl-Pd-complex intermediate B was supposed [89] to proceed through the initial formation of transient Pd–H species A, followed by their reaction with the diene. The nucleophilic attack of aniline on the less-substituted carbon of the intermediate species B (Figure 30) [89] results in a regioselective 1,4-hydroamination product.
As mentioned above analysis of available literature shows that there are just a few examples when terpene amines were synthesized using heterogeneous systems, comprising reductive amination over Ni Raney, Pt/C, Pd/C, copper on alumina modified with LiOH, hydroamination on alkali metals, and hydrogen borrowing reactions over Au and AuPd. In fact, there is a clear trend in the more widespread application of heterogeneous catalysts. It is interesting that complexes of precious metals are mainly applied as homogeneous catalysts, while despite utilization of noble heterogeneous catalysts (e.g., carbon supported Pt and Pd in amination of camphor), other metals such as supported Cu and Au were considered to be efficient. Moreover, while addition of Pd to heterogeneous catalysts deteriorates the overall performance by decreasing selectivity towards complex amines at the expense of hydrogenation, similar behavior was not observed for homogeneous catalysts. This is even more striking as according to the available data the mechanisms of amination in the presence of transition metal complexes discussed above and heterogeneous catalysts are similar. In particular, catalytically active species are formed by generation of the metal hydrides in the case of Pd–H from homogeneous Pd chloride complexes. Similar suggestions follow from the work of the authors of this review on amination of myrtenol with aniline over gold catalysts. Obviously, there is a need for more detailed studies of the nature of active sites in homogeneous catalysts to fully explore this knowledge in the design of heterogeneous systems. Alternatively if the mechanisms of homogeneous and heterogeneous catalysis are different, a significant amount of work should be devoted to heterogeneous catalysts in a quest for understanding the reaction mechanism. This and many other questions do not have clear answers at the moment urging on one hand more in depth and on the other more broader studies on amination of terpenes over heterogeneous catalysts.
5. Conclusions
Although amination of terpenoids has been extensively studied since the early decades of the last century, industrial implementation of biomass-based terpenes as starting materials is still in its infancy. Catalytic systems based on transition metals complexes have been developed for performing such reactions. However, to reduce the production costs, either easily recoverable homogeneous systems based on cheaper metals operating by nucleophilic substitution, as well as supported metal nanoparticles (Ni, Co, Cu, Pd, Au) on low alkaline supports should be developed. These catalysts will provide synthesis of amine derivatives of terpenes having a broad range of applications as specialty chemicals, surfactants, pharmaceuticals, etc.
Author Contributions
I.L.S. performed the literature search and drafted the manuscript; A.V.S. and D.Y.M. contributed to the writing and editing of the manuscript. All the authors revised and approved the manuscript.
Acknowledgments
This research was supported by RFBR Grant # 18-53-45013 IND_a.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lawrence, S.A. Amines: Synthesis, Properties and Applications; Cambridge University Press: Cambridge, UK, 2004; p. 372. [Google Scholar]
- Shi, F.; Cui, X. Catalytic Amination for N-Alkylamine Synthesis; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Maxwell, G.R. Synthetic Nitrogen Products: A Practical Guide to the Products and Processes; Kluwer Academic Publishers: New York, NY, USA, 2004. [Google Scholar]
- Pelckmans, M.; Renders, T.; Van de Vyver, S.; Sels, B.F. Bio-based amines through sustainable heterogeneous catalysis. Green Chem. 2017, 19, 5303–5331. [Google Scholar] [CrossRef]
- Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. Amines, aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2012; pp. 647–698. [Google Scholar]
- Frauenkron, M.; Melder, J.-P.; Ruider, G.; Rossbacher, R.; Höke, H. Ethanolamines and Propanolamines. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2012; pp. 405–431. [Google Scholar]
- Bähn, S.; Imm, S.; Neubert, L.; Zhang, M.; Neumann, H.; Beller, M. The catalytic amination of alcohols. ChemCatChem 2011, 3, 1853–1864. [Google Scholar] [CrossRef]
- Kimura, H. Progress in one-step amination of long-chain fatty alcohols with dimethylamine: Development of key technologies for industrial applications, innovations, and future outlook. Catal. Rev. Sci. Eng. 2011, 53, 1–90. [Google Scholar] [CrossRef]
- Pera-Titus, M.; Shi, F. Catalytic amination of biomass-based alcohols. ChemSusChem 2014, 7, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Froidevaux, V.; Negrell, C.; Caillol, S.; Pascault, J.-P.; Boutevin, B. Biobased amines: From synthesis to polymers; present and future. Chem. Rev. 2016, 116, 14181–14224. [Google Scholar] [CrossRef] [PubMed]
- Geboers, J.; Van de Vyver, S.; Carpentier, K.; de Blochouse, K.; Jacobs, P.; Sels, B. Efficient catalytic conversion of concentrated cellulose feeds to hexitols with heteropoly acids and Ru on carbon. Chem. Commun. 2010, 46, 3577–3579. [Google Scholar] [CrossRef] [PubMed]
- Geboers, J.; Van de Vyver, S.; Carpentier, K.; Jacobs, P.; Sels, B. Efficient hydrolytic hydrogenation of cellulose in the presence of Ru-loaded zeolites and trace amounts of mineral acid. Chem. Commun. 2011, 47, 5590–5592. [Google Scholar] [CrossRef] [PubMed]
- Geboers, J.A.; Van de Vyver, S.; Ooms, R.; Op de Beeck, B.; Jacobs, P.A.; Sels, B.F. Chemocatalytic conversion of cellulose: Opportunities, advances and pitfalls. Catal. Sci. Technol. 2011, 1, 714–726. [Google Scholar] [CrossRef]
- Van de Vyver, S.; Geboers, J.; Jacobs, P.A.; Sels, B.F. Recent Advances in the Catalytic Conversion of Cellulose. ChemCatChem 2011, 3, 82–94. [Google Scholar] [CrossRef]
- Tinikul, R.; Chenprakhon, P.; Maenpuen, S.; Chaiyen, P. Biotransformation of plant-derived phenolic acids. Biotechnol. J. 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, J.; Lindström, M.E. Modification of industrial softwood kraft lignin using Mannich reaction with and without phenolation pretreatment. Ind. Crop. Prod. 2014, 52, 729–735. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, S.; Schutyser, W.; Vanholme, R.; Driessen, T.; Koelewijn, S.F.; Renders, T.; De Meester, B.; Huijgen, W.J.J.; Dehaen, W.; Courtin, C.M.; et al. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ. Sci. 2015, 8, 1748–1763. [Google Scholar] [CrossRef] [Green Version]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chena, T.-Y.; Wanga, H.-M.; Li, H.-Y.; Liub, C.-F.; Wena, J.-L. Amination of biorefinery technical lignins using Mannich reaction synergy with subcritical ethanol depolymerization. Int. J. Biol. Macromol. 2018, 107, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Sun, G.; Zhao, T. Synthesis and characterization of aminated lignin. Int. J. Biol. Macromol. 2013, 59, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Kondo, A.; Noda, H. Biodiesel fuel production by transesterification of oils. J. Biosci. Bioeng. 2001, 92, 405–416. [Google Scholar] [CrossRef]
- Zargar, V.; Asghari, M.; Dashti, A. A review on chitin and chitosan polymers: Structure, chemistry, solubility, derivatives. ChemBioEng. Rev. 2015, 2, 204–226. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A Sustainable platform for production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Behr, A.; Wintzer, A. From terpenoids to amines: A critical review. In New Developments in Terpenes Research; Hu, J., Ed.; Nova Science Publishers: New York, NY, USA, 2014; Chapter 6; pp. 113–134. [Google Scholar]
- Kroutil, W.; Fischereder, E.-M.; Fuchs, C.S.; Lechner, H.; Mutti, F.G.; Pressnitz, D.; Rajagopalan, A.; Sattler, J.H.; Simon, R.C.; Siirola, E. Asymmetric preparation of prim-, sec-, and tert-amines employing selected biocatalysts. Org. Process Res. Dev. 2013, 17, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.J.; Truppo, M.D. Biocatalytic routes to nonracemic chiral amines. In Chiral Amine Synthesis: Methods, Developments and Applications, 2nd ed.; Nugent, T.C., Ed.; Wiley: Hoboken, NJ, USA, 2010; p. 523. [Google Scholar]
- Schrewe, M.; Ladkau, N.; Buehler, B.; Schmid, A. Direct terminal alkylamino-functionalization via multistep biocatalysis in one recombinant whole-cell catalyst. Adv. Synth. Catal. 2013, 355, 1693–1697. [Google Scholar] [CrossRef]
- Song, J.-W.; Lee, J.-H.; Bornscheuer, U.T.; Park, J.-B. Microbial synthesis of medium-chain α,ω-dicarboxylic acids and ω-aminocarboxylic acids from renewable long-chain fatty acids. Adv. Synth. Catal. 2014, 356, 178–1788. [Google Scholar] [CrossRef]
- Singh, R.; Kolev, J.N.; Sutera, P.A.; Fasan, R. Enzymatic C(sp3)-H amination: P450-catalyzed conversion of carbonazidates into oxazolidinones. ACS Catal. 2015, 5, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Deutschmann, O.; Knozinger, H.; Kochloefl, K.; Turek, T. Heterogeneous catalysis and solid catalysts. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2009; pp. 1–110. [Google Scholar]
- Harrewijn, P.; van Oosten, A.M.; Piron, P.G.M. Natural Terpenoids as Messengers. A Multidisciplinary Study of their Production, Biological Functions and Practical Applications; Kluwer Academic Publishers: South Holland, The Netherlands, 2001. [Google Scholar]
- Behr, A.; Johnen, L. Myrcene as a natural base chemical in sustainable chemistry: A critical review. ChemSusChem 2009, 2, 1072–1095. [Google Scholar] [CrossRef] [PubMed]
- Mäki-Arvela, P.; Simakova, I.L.; Salmi, T.; Murzin, D.Y. Catalytic transformations of extractives. In Catalytic Process Development for Renewable Materials; Hardcover Handbook; Wiley: Weinheim, Germany, 2013; Chapter 13; 450p. [Google Scholar]
- Murzin, D.Y.; Simakova, I.L. Catalysis in biomass conversion. In Comprehensive Inorganic Chemistry II; Schlogl, R., Niemantsverdriet, J.W., Eds.; Elsevier: New York, NY, USA, 2013; Chapter 7.27; pp. 2–32. [Google Scholar]
- Murzin, D.Y.; Simakova, I.L. Catalysis in biomass processing. Catal. Ind. 2011, 3, 218–249. [Google Scholar] [CrossRef]
- Murzin, D.Y.; Demidova, Y.; Hasse, B.; Etzold, B.; Simakova, I.L. Synthesis of fine chemicals using catalytic nanomaterials: Structure sensitivity. In Producing Fuels and Fine Chemicals from Biomass Using Nanomaterials; Luque, R., Balu, A.M., Eds.; CRC Press: Boca Raton, FL, USA, 2013; pp. 267–281. [Google Scholar]
- Salakhutdinov, N.F.; Volcho, K.P.; Yarovaya, O.I. Monoterpenes as a renewable source of biologically active compounds. Pure Appl. Chem. 2017, 89, 1105–1118. [Google Scholar] [CrossRef]
- Kapitsa, I.G.; Suslov, E.V.; Teplov, G.V.; Korchagina, D.V.; Komarova, N.I.; Volcho, K.P.; Voronina, T.A.; Shevela, A.I.; Salakhutdinov, N.F. Synthesis and anxiolytic activity of 2-aminoadamantane derivatives containing monoterpene fragments. Pharm. Chem. J. 2012, 46, 263–265. [Google Scholar] [CrossRef]
- Teplov, G.V.; Suslov, E.V.; Zarubaev, V.V.; Shtro, A.A.; Karpinskaya, L.A.; Rogachev, A.D.; Korchagina, D.V.; Volcho, K.P.; Salakhutdinov, N.F.; Kisilev, O.I. Synthesis of new compounds combining adamantanamine and monoterpene fragments and their antiviral activity against influenza virus A(H1N1)pdm09. Lett. Drug Des. Discov. 2013, 10, 477–485. [Google Scholar] [CrossRef]
- Volcho, K.P.; Laev, S.S.; Ashraf, G.M.; Aliev, G.; Salakhutdinov, N.F. Application of monoterpenoids and their derivatives for treatment of neurodegenerative disorders. Curr. Med. Chem. 2017, 24, 3283–3309. [Google Scholar]
- Silva, R.O.; Salvadori, M.S.; Sousa, F.B.M.; Santos, M.S.; Carvalho, N.S.; Sousa, D.P.; Gomes, B.S.; Oliveira, F.A.; Barbosa, A.L.R.; Frietas, R.M.; et al. Evaluation of the anti-inflammatory and antinociceptive effects of myrtenol, a plant-derived monoterpene alcohol, in mice. Flavour Fragr. J. 2014, 29, 184–192. [Google Scholar] [CrossRef]
- Sarmento-Neto, J.F.; do Nascimento, L.G.; Felipe, C.F.B.; de Sousa, D.P. Analgesic potential of essential oils. Molecules 2016, 21, 20. [Google Scholar] [CrossRef] [PubMed]
- Lochynski, S.; Kuldo, J.; Frackowiak, B.; Holband, J.; Wojcik, G. Stereochemistry of terpene derivatives. Part 2: Synthesis of new chiral amino acids with potential neuroactivity. Tetrahedron Asymmetry 2000, 11, 1295–1302. [Google Scholar] [CrossRef]
- Gajcy, K.; Pekala, J.; Frackowiak-Wojtasek, B.; Librowski, T.; Lochynski, S. Stereochemistry of terpene derivatives. Part 7: Novel rigidified amino acids from (+)-3-carene designed as chiral GABA analogues. Tetrahedron Asymmetry 2010, 21, 2015–2020. [Google Scholar] [CrossRef]
- Ferrarini, S.R.; Graebin, C.S.; Limberger, J.; Canto, R.F.S.; Dias, D.O.; da Rosa, R.G.; Madeira, M.D.F.; Eifler-Lima, V.L. Synthesis of limonene β-amino alcohol derivatives in support of new antileishmanial therapies. Mem. Inst. Oswaldo Cruz 2008, 103, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrarini, S.R.; Duarte, M.O.; da Rosa, R.G.; Rolim, V.; Eifler-Lima, V.L.; von Poser, G.; Ribeiro, V.L.S. Acaricidal activity of limonene, limonene oxide and β-amino alcohol derivatives on Rhipicephalus (Boophilus) microplus. Vet. Parasitol. 2008, 157, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Strong, J. N-[3-(4-methyl-3-cyclohexenyl)butyl]amines. U.S. Patent 3,890,384, 27 January 1975. [Google Scholar]
- Strong, J. N-[3-(4-methyl-3-cyclohexenyl)butyl]amines and Their Use as Plant Growth Regulators. U.S. Patent 4,030,908, 27 January 1972. [Google Scholar]
- Keim, W.; Kurtz, K.R.; Roeper, M. Palladium catalyzed telomerization of isoprene with secondary amines and conversion of the resulting terpene amines to terpenols. J. Mol. Catal. 1983, 20, 129–138. [Google Scholar] [CrossRef]
- Watts, C.C.; Thoniyot, P.; Cappuccio, F.; Verhagen, J.; Gallagher, B.; Singaram, B. Catalytic asymmetric transfer hydrogenation of ketones using terpene-based chiral β-amino alcohols. Tetrahedron Asymmetry 2006, 17, 1301–1307. [Google Scholar] [CrossRef]
- Watts, C.C.; Thoniyot, P.; Hirayama, L.C.; Romano, T.; Singaram, B. Enantioselective alkynylations of aromatic and aliphatic aldehydes catalyzed by terpene derived chiral amino alcohols. Tetrahedron Asymmetry 2005, 16, 1829–1835. [Google Scholar] [CrossRef]
- Alves, M.-H.; Sfeir, H.; Tranchant, J.-F.; Gombart, E.; Sagorin, G.; Caillol, S.; Billon, L.; Save, M. Terpene and dextran renewable resources for the synthesis of amphiphilic biopolymers. Biomacromolecules 2014, 15, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Ajikumar, P.K.; Tyo, K.; Carlsen, S.; Mucha, O.; Phon, T.H.; Stephanopoulos, G. Terpenoids: Opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol. Pharm. 2008, 5, 167–190. [Google Scholar] [CrossRef] [PubMed]
- Lyubimov, S.E.; Kuchurov, I.V.; Verbitskaya, T.A.; Rastorguev, E.A.; Kalinin, V.N.; Zlotin, S.G.; Davankov, V.A. Pd-catalyzed allylic amination in supercritical carbon dioxide: Synthesis of carborane-containing terpenoids. J. Supercrit. Fluids 2010, 54, 218–221. [Google Scholar] [CrossRef]
- Armstrong, A.F.; Valliant, J.F. The bioinorganic and medicinal chemistry of carboranes: From new drug discovery to molecular imaging and therapy. Dalton Trans. 2007, 4240–4251. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Griesbach, U.; Bothe, U.; Nakamura, H.; Yamamoto, Y. Novel carboranes with a DNA binding unit for the treatment of cancer by boron neutron capture therapy. ChemBioChem 2002, 3, 219–225. [Google Scholar] [CrossRef]
- Di Meo, C.; Panza, L.; Capitani, D.; Mannina, L.; Banzato, A.; Rondina, M.; Renier, D.; Rosato, A.; Crescenzi, V. Hyaluronan as carrier of carboranes for tumor targeting in boron neutron capture therapy. Biomacromolecules 2007, 8, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Barth, R.F.; Coderre, J.A.; Vicente, M.G.H.; Blue, T.E. Boron neutron capture therapy of cancer: Current status and future prospects. Clin. Cancer Res. 2005, 11, 3987–4002. [Google Scholar] [CrossRef] [PubMed]
- Nageshwar, D.; Rao, D.M.; Acharyulu, P.V.R. Terpenes to ionic liquids: Synthesis and characterization of citronellal-based chiral ionic liquids. Synth. Commun. 2009, 39, 3357–3368. [Google Scholar] [CrossRef]
- Bordenca, C.; Dorschner, K.P.; Johnson, R.P. Insect Repellent Compositions and Process Having an N-substituted Hydroxyalkyl Amine as an Active Ingredient. U.S. Patent 3,933,915, 23 June 1972. [Google Scholar]
- Behr, A.; Wintzer, A.; Lübke, C.; Müller, M. Synthesis of primary amines from the renewable compound citronellal via biphasic reductive amination. J. Mol. Catal. A Chem. 2015, 404–405, 74–82. [Google Scholar] [CrossRef]
- Kukula, P.; Koprivova, K. Structure-selectivity relationship in the chemoselective hydrogenation of unsaturated nitriles. J. Catal. 2005, 234, 161–171. [Google Scholar] [CrossRef]
- Bahn, S.; Imm, S.; Neubert, L.; Zhang, M.; Neumann, H.; Beller, M. Selective ruthenium-catalyzed alkylation of indoles by using amines. Chem. Eur. J. 2011, 17, 4705–4708. [Google Scholar] [PubMed]
- Fuchs, S.; Rösler, T.; Grabe, B.; Kampwerth, A.; Meier, G.; Strutz, H.; Behr, A.; Vorholt, A.J. Synthesis of primary amines via linkage of hydroaminomethylation of olefins and splitting of secondary amines. Appl. Cat. A Gen. 2018, 550, 198–205. [Google Scholar] [CrossRef]
- Donetti, A.; Casadio, S.; Bonardi, G.; Omodei-Sale, A. Terpene compounds as drugs. 13. o-Terpenylaminomethylphenols and their N-methyl derivatives. J. Med. Chem. 1972, 15, 1089–1091. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, N.G.; Kalechits, G.V.; Vyalimyae, T.K. Terpene amines. IV. Synthesis and study of the structure of amines from d-fenchone. Khimiya Prirodnych Soedinenii (Chem. Nat. Comp.) 1983, 4, 483–488. [Google Scholar] [CrossRef]
- Tarasevich, V.A.; Kozlov, N.G. Reductive amination of oxygen-containing organic compounds. Russ. Chem. Rev. 1999, 68, 55–72. [Google Scholar] [CrossRef]
- Kalck, P.; Urrutigoïty, M. Tandem hydroaminomethylation reaction to synthesize amines from alkenes. Chem. Rev. 2018, 118, 3833–3861. [Google Scholar] [CrossRef] [PubMed]
- Eilbracht, P.; Barfacker, L.; Buss, C.; Hollmann, C.; Kitsos-Rzychon, B.E.; Kranemann, C.L.; Rische, T.; Roggenbuck, R.; Schmidt, A. Tandem reaction sequences under hydroformylation conditions: new synthetic applications of transition metal catalysis. Chem. Rev. 1999, 99, 3329–3366. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Seayad, A.M.; Jackstell, R.; Beller, M. Highly selective synthesis of enamines from olefins. Angew. Chem. Int. Ed. 2003, 42, 5615–5619. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Seayad, A.M.; Jackstell, R.; Beller, M. Amines made easily: a highly selective hydroaminomethylation of olefins. J. Am. Chem. Soc. 2003, 125, 10311–10318. [Google Scholar] [CrossRef] [PubMed]
- Fogg, D.E.; dos Santos, E.N. Tandem catalysis: A taxonomy and illustrative review. Coord. Chem. Rev. 2004, 248, 2365–2379. [Google Scholar] [CrossRef]
- Melo, D.S.; Pereira-Júniora, S.S.; dos Santosa, E.N. An efficient method for the transformation of naturally occurring monoterpenes into amines through rhodium-catalyzed hydroaminomethylation. Appl. Catal. A Gen. 2012, 411–412, 70–76. [Google Scholar] [CrossRef]
- Börner, A.; Franke, R. Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2016; 736p. [Google Scholar]
- Graebin, C.S.; Eifler-Lima, V.L.; da Rosa, R.G. One-pot synthesis of secondary and tertiary amines from R(+)-limonene by tandem hydroformylation/reductive amination (hydroaminomethylation). Catal. Commun. 2008, 9, 1066–1070. [Google Scholar] [CrossRef]
- Behr, A.; Wintzer, A. Hydroaminomethylation of the renewable limonene with ammonia in an aqueous biphasic solvent system. Chem. Eng. Technol. 2015, 38, 2299–2304. [Google Scholar] [CrossRef]
- Faßbach, T.A.; Gaide, T.; Terhorst, M.; Behr, A.; Vorholt, A.J. Renewable surfactants through the hydroaminomethylation of terpenes. ChemCatChem 2017, 9, 1359–1362. [Google Scholar] [CrossRef]
- Oliveira, K.C.B.; Santos, A.G.; dos Santos, E.N. Hydroaminomethylation of eugenol with di-n-butylamine catalyzed by rhodium complexes: Bringing light on the promoting effect of Bronsted acids. Appl. Catal. A Gen. 2012, 445–446, 204–208. [Google Scholar] [CrossRef]
- Oliveira, K.C.B.; Carvalho, S.N.; Duarte, M.F.; Gusevskaya, E.V.; dos Santos, E.N.; Karroumi, J.E.; Gouygou, M.; Urrutigoïty, M. Phospholes as efficient ancillaries for the rhodium-catalyzed hydroformylation and hydroaminomethylation of estragole. Appl. Catal. A Gen. 2015, 497, 10–16. [Google Scholar] [CrossRef]
- Behr, A.; Reyer, S.; Manz, V. Hydroaminomethylation of isoprene: Recycling of the homogeneous rhodium catalyst in aqueous biphasic systems. Chem. Ing. Tech. 2012, 84, 108–113. [Google Scholar] [CrossRef]
- Sirol, S.; Kalck, P. Hydroformylation of optically pure monoterpenes catalyzed by dinuclear thiolato-bridged rhodium complexes. New J. Chem. 1997, 21, 1129–1137. [Google Scholar]
- Foca, C.M.; Barros, H.J.V.; dos Santos, E.N.; Gusevskaya, E.V.; Bayon, J.C. Hydroformylation of myrcene: Metal and ligand effects in the hydroformylation of conjugated dienes. New J. Chem. 2003, 27, 533–539. [Google Scholar] [CrossRef]
- Halligudi, S.B.; Bhatt, K.N.; Venkatasubramanian, K. Hydroformylation of olefins catalyzed by rhodium complex anchored on clay matrices. React. Kinet. Catal. Lett. 1993, 51, 459–464. [Google Scholar] [CrossRef]
- Barros, H.J.V.; Ospina, M.L.; Arguello, E.; Rocha, W.R.; Gusevskaya, E.V.; dos Santos, E.N. Rhodium catalyzed hydroformylation of β-pinene and camphene: Effect of phosphorous ligands and reaction conditions on diastereoselectivity. J. Organomet. Chem. 2003, 671, 150–157. [Google Scholar] [CrossRef]
- Estragol. Available online: https://en.wikipedia.org/wiki/Estragole#cite_note-2 (accessed on 28 June 2018).
- Haggin, J. Chemists seek greater recognition for catalysis. Chem. Eng. News 1993, 71, 23–27. [Google Scholar] [CrossRef]
- Beller, M.; Seavad, J.; Tillack, A.; Jiao, H. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: Recent developments and trends. Angew. Chem. Int. Ed. 2004, 43, 3368–3398. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Junge, K.; Beller, M. Palladium-catalysed regioselective hydroamination of 1,3-dienes: Synthesis of allylic amines. Org. Chem. Front. 2014, 1, 368–372. [Google Scholar] [CrossRef]
- Watson, I.D.G.; Yudin, A.K. New insights into the mechanism of palladium-catalyzed allylic amination. J. Am. Chem. Soc. 2005, 127, 17516–17529. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Fujita, T.; Sakamoto, M.; Haga, T.; Kuramochi, T. Palladium-catalyzed addition of dialkylamines to linalyl acetate and related compounds. J. Essent. Oil Res. 1994, 9, 441–445. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Urrutigoïty, M.; Fihri, A.; Hierso, J.-C.; Meunier, P.; Kalck, P. Efficient palladium-ferrocenylphosphine catalytic systems for allylic amination of monoterpene derivatives. Appl. Organomet. Chem. 2006, 20, 845–850. [Google Scholar] [CrossRef]
- Fihri, A.; Meunier, P.; Hierso, J.-C. Performances of symmetrical achiral ferrocenylphosphine ligands in palladium-catalyzed cross-coupling reactions: A review of syntheses, catalytic applications and structural properties. Coord. Chem. Rev. 2007, 251, 2017–2055. [Google Scholar] [CrossRef]
- Mignani, G.; Morel, D. Processes Amination of Conjugated Dienes. Patent FR 2,569,403, 23 August 1986. [Google Scholar]
- Akutagawa, S. Asymmetric synthesis by metal BINAP catalysts. Appl. Catal. A Gen. 1995, 128, 171–207. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Kumar, N.; Nieminen, V.; Sjöholm, R.; Salmi, T.; Murzin, D.Y. Cyclization of citronellal over zeolites and mesoporous materials for production of isopulegol. J. Catal. 2004, 225, 155–169. [Google Scholar] [CrossRef]
- Kumobayashi, H.; Mitsuhashi, S.; Akutagawa, S.; Ohtsuka, S. A practical synthesis of myrcenol by palladium complex-catalyzed elimination reaction. Chem. Lett. 1986, 15, 157–160. [Google Scholar] [CrossRef]
- Chalk, A.J.; Magennis, S.A.; Wertheimer, V.S.; Naipawer, R.E. Process for the Catalytic Synthesis of Conjugated Dienes from Dialkylallylamines. U.S. Patent 4,467,118, 21 August 1984. [Google Scholar]
- Chalk, A.J.; Wertheimer, V.; Magennis, S.A. A new palladium catalyzed equivalent of hofmann elimination for allylic amines. J. Mol. Catal. 1983, 19, 189–200. [Google Scholar] [CrossRef]
- Hata, G.; Tanaka, M. Terpene Hydrocarbons. JP Patent 50,123,605, 29 September 1975. [Google Scholar]
- Murata, A.; Tsuchiya, S.; Suzuki, H.; Ikeda, H. A Method for Producing of Chain Terpene Alcohols. DE Patent 2720839, 14 May 1977. [Google Scholar]
- Behr, A.; Johnen, L.; Rentmeister, N. Novel Palladium-catalysed hydroamination of myrcene and catalyst separation by thermomorphic solvent systems. Adv. Synth. Catal. 2010, 352, 2062–2072. [Google Scholar] [CrossRef]
- Hamid, M.H.S.A.; Slatford, P.A.; Williams, J.M.J. Borrowing hydrogen in the activation of alcohols. Adv. Synth. Catal. 2007, 349, 1555–1575. [Google Scholar] [CrossRef]
- Gunanathan, C.; Milstein, D. Applications of acceptorless dehydrogenation and related transformations in chemical synthesis. Science 2013, 341, 1229712. [Google Scholar] [CrossRef] [PubMed]
- Guillena, G.; Ramon, D.J.; Yus, M. Hydrogen autotransfer in the N-alkylation of amines and related compounds using alcohols and amines as electrophiles. Chem. Rev. 2010, 110, 1611–1641. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.-I.; Kon, K.; Onodera, W.; Yamazaki, H.; Kondo, J.N. Heterogeneous Ni catalyst for direct synthesis of primary amines from alcohols and ammonia. ACS Catal. 2013, 3, 112–117. [Google Scholar] [CrossRef]
- Dang, T.T.; Ramalingam, B.; Shan, S.P.; Seayad, A.M. Reductive N-Alkylation of nitro compounds to N-alkyl and N,N-dialkyl amines with glycerol as the hydrogen source. ACS Catal. 2013, 3, 2536–2540. [Google Scholar] [CrossRef]
- Murzin, D.Y. Chemical Reaction Technology; De Gruyter: Berlin, Germany, 2015; 428p. [Google Scholar]
- Ma, X.; Su, C.; Xu, Q. N-Alkylation by hydrogen autotransfer reactions. In Hydrogen Transfer Reactions: Reductions and Beyond; Guillena, G., Ramon, D.J., Eds.; Springer International Publishing: Basel, Switzerland, 2016; pp. 291–364. [Google Scholar]
- Imm, S.; Bahn, S.; Neubert, L.; Neumann, H.; Beller, M. An efficient and general synthesis of primary amines by ruthenium catalyzed amination of secondary alcohols with ammonia. Angew. Chem. Int. Ed. 2010, 49, 8126–8129. [Google Scholar] [CrossRef] [PubMed]
- Imm, S.; Bahn, S.; Zhang, M.; Neubert, L.; Neumann, H.; Klasovsky, F.; Pfeffer, J.; Haas, T.; Beller, M. Improved ruthenium-catalyzed amination of alcohols with ammonia: Synthesis of diamines and amino esters. Angew. Chem. Int. Ed. 2011, 50, 7599–7603. [Google Scholar] [CrossRef] [PubMed]
- Lamb, G.W.; Williams, J.M.J. Borrowing hydrogen-C-N bond formation from alcohols. Chim. Oggi-Chem. Today 2008, 26, 17–19. [Google Scholar]
- Pingen, D.; Muller, C.; Vogt, D. Direct amination of secondary alcohols using ammonia. Angew. Chem. Int. Ed. 2010, 49, 8130–8133. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Navas, J.; Sabater, M.J. Advances in one-pot synthesis through borrowing hydrogen catalysis. Chem. Rev. 2018, 118, 1410–1459. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.J.A.; Maxwell, A.C.; Williams, J.M.J. Borrowing hydrogen methodology for amine synthesis under solvent-free microwave conditions. J. Org. Chem. 2011, 76, 2328–2331. [Google Scholar] [CrossRef] [PubMed]
- Saidi, O.; Blacker, A.J.; Farah, M.M.; Marsden, S.P.; Williams, J.M.J. Iridium-catalysed amine alkylation with alcohols in water. Chem. Commun. 2010, 46, 1541–1543. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Fujita, K.; Yamaguchi, R. N-Alkylation of Amines with Alcohols catalyzed by a water soluble Cp*Iridium complex: An efficient method for the synthesis of amines in aqueous media. Adv. Synth. Catal. 2011, 353, 1161–1168. [Google Scholar] [CrossRef]
- Hollmann, D.; Tillack, A.; Michalik, D.; Jackstell, R.; Beller, M. An improved ruthenium catalyst for the environmentally benign amination of primary and secondary alcohols. Chem. Asian J. 2007, 2, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Asencio, A.; Ramon, D.J.; Yus, M. N-alkylation of poor nucleophilic amines and derivatives with alcohols by a hydrogen autotransfer process catalyzed by copper (II) acetate: Scope and mechanistic considerations. Tetrahedron 2011, 67, 3140–3149. [Google Scholar] [CrossRef]
- Martinez-Asencio, A.; Yus, M.; Ramon, D.J. Palladium (II) acetate as a catalyst for the N-alkylation of aromatic amines, sulfonamides and related nitrogenated compounds with alcohols by a hydrogen autotransfer process. Synthesis 2011, 3730–3740. [Google Scholar] [CrossRef]
- Blank, B.; Kempe, R. Catalytic alkylation of methyl-N-heteroaromatics with alcohols. J. Am. Chem. Soc. 2010, 132, 924–925. [Google Scholar] [CrossRef] [PubMed]
- Michlik, S.; Hille, T.; Kempe, R. The Iridium-catalyzed synthesis of symmetrically and unsymmetrically alkylated diamines under mild reaction conditions. Adv. Synth. Catal. 2012, 354, 847–862. [Google Scholar] [CrossRef]
- Demidova, Y.S.; Simakova, I.L.; Estrada, M.; Beloshapkin, S.; Suslov, E.V.; Korchagina, D.V.; Volcho, K.P.; Salakhutdinov, N.F.; Simakov, A.V.; Murzin, D.Y. One-pot myrtenol amination over Au nanoparticles supported on different metal oxides. Appl. Catal. A Gen. 2013, 464–465, 348–356. [Google Scholar] [CrossRef]
- Houssame, S.E.; Anane, H.; Firdoussi, L.E.; Karim, A. Palladium(0)-catalyzed amination of allylic natural terpenic functionalized olefins. Cent. Eur. J. Chem. 2008, 6, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Pingen, D.; Diebolt, O.; Vogt, D. Direct amination of bio-alcohols using ammonia. ChemCatChem 2013, 5, 2905–2912. [Google Scholar] [CrossRef]
- Valkenberg, M.H.; Holderich, W.F. Preparation and use of hybrid organic–inorganic catalysts. Catal. Rev. 2002, 44, 321–374. [Google Scholar] [CrossRef]
- Huang, X.; Wu, H.; Liao, X.P.; Shi, B. Liquid phase hydrogenation of olefins using heterogenized ruthenium complexes as high active and reusable catalyst. Catal. Commun. 2010, 11, 487–492. [Google Scholar] [CrossRef]
- Cao, Y.; Hu, J.C.; Yang, P.; Dai, W.L.; Fan, K.N. CuCl catalyst heterogenized on diamide immobilized SBA-15 for efficient oxidative carbonylation of methanol to dimethylcarbonate. Chem. Commun. 2003, 908–909. [Google Scholar] [CrossRef]
- Mukhopadhyay, K.; Chaudhari, R.V. Heterogenized HRh(CO)(PPh3)3 on zeolite Y using phosphotungstic acid as tethering agent: A novel hydroformylation catalyst. J. Catal. 2003, 213, 73–77. [Google Scholar] [CrossRef]
- Johnson, B.F.G.; Raynor, S.A.; Shephard, D.S.; Mashmeyer, T.; Thomas, J.M.; Sankar, G.; Bromley, S.; Oldroyd, R.; Gladden, L.; Mantle, M.D. Superior performance of a chiral catalyst confined within mesoporous silica. Chem. Commun. 1999, 1167–1168. [Google Scholar] [CrossRef]
- Dyal, A.; Loos, K.; Noto, M.; Chang, S.W.; Spagnoli, C.; Shafi, K.V.P.M.; Ulman, A.; Cowman, M.; Gross, R.A. Activity of Candida rugose lipase immobilized on γ-Fe2O3 Magnetic Nanoparticles. J. Am. Chem. Soc. 2003, 125, 1684–1685. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, Y.; Wang, D.I.C.; Li, Z. Recyclable nanobiocatalyst for enantioselective sulfoxidation: Facile fabrication and high performance of chloroperoxidase-coated magnetic nanoparticles with iron oxide core and polymer shell. J. Am. Chem. Soc. 2009, 131, 12892–12893. [Google Scholar] [CrossRef] [PubMed]
- Shylesh, S.; Schuenemann, V.; Thiel, W.R. Magnetically separable nanocatalysts: Bridges between homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. 2010, 49, 3428–3459. [Google Scholar] [CrossRef] [PubMed]
- Campelo, M.; Luna, D.; Luque, R.; Marinas, J.M.; Romero, A.A. Sustainable preparation of supported metal nanoparticles and their applications in catalysis. ChemSusChem 2009, 2, 18–45. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Tse, M.; Zhou, S.; Pohl, M.-M.; Radnik, J.; Hubner, S.; Jahnisch, K.; Bruckner, A.; Beller, M. Green and efficient synthesis of sulfonamides catalyzed by nano-Ru/Fe3O4. J. Am. Chem. Soc. 2009, 131, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Demidova, Y.S.; Simakova, I.L.; Warne, J.; Simakov, A.; Murzin, D.Y. Kinetic modeling of one-pot myrtenol amination over Au/ZrO2 catalyst. Chem. Eng. J. 2014, 238, 164–171. [Google Scholar] [CrossRef]
- Simakova, I.L.; Demidova, Y.S.; Estrada, M.; Beloshapkin, S.; Suslov, E.V.; Volcho, K.P.; Salakhutdinov, N.F.; Murzin, D.Y.; Simakov, A. Gold catalyzed one-pot myrtenol amination: Effect of catalyst redox activation. Catal. Today 2017, 279, 63–70. [Google Scholar] [CrossRef]
- Demidova, Y.S.; Suslov, E.V.; Simakova, I.L.; Korchagina, D.V.; Mozhajcev, E.S.; Volcho, K.P.; Salakhutdinov, N.F.; Simakov, A.; Murzin, D.Y. Selectivity control in one-pot amination of Au/ZrO2 by molecular hydrogen addition. J. Mol. Catal. A Chem. 2017, 426, 60–67. [Google Scholar] [CrossRef]
- Demidova, Y.S.; Suslov, E.V.; Simakova, I.L.; Volcho, K.P.; Salakhutdinov, N.F.; Simakov, A.; Murzin, D.Y. Promoting effect of alcohols and formic acid on Au-catalyzed one-pot alcohol amination. Mol. Catal. 2017, 433, 414–419. [Google Scholar] [CrossRef]
- Demidova, Y.S.; Suslov, E.V.; Simakova, I.L.; Korchagina, D.V.; Mozhajcev, E.S.; Volcho, K.P.; Salakhutdinov, N.F.; Simakov, A.; Murzin, D.Y. One-pot monoterpene alcohol amination over Au/ZrO2 catalyst: Effect of the substrates structure. J. Catal. 2018, 360, 127–134. [Google Scholar] [CrossRef]
- Cherng, Y.-J.; Fang, J.-M.; Lu, T.-J. A new pinane-type tridentate modifier for asymmetric reduction of ketones with lithium aluminum hydride. Tetrahedron Asymmetry 1995, 6, 89–92. [Google Scholar] [CrossRef]
- Ishida, T.; Takamura, R.; Takei, T.; Akita, T.; Haruta, M. Support effects of metal oxides on gold-catalyzed one-pot N-alkylation of amine with alcohol. Appl. Catal. A Gen. 2012, 413–414, 261–266. [Google Scholar] [CrossRef]
- Lescot, C.; Darses, B.; Collet, F.; Retailleau, P.; Dauban, P. Intermolecular C–H amination of complex molecules: Insights into the factors governing the selectivity. J. Org. Chem. 2012, 77, 7232–7240. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; van Vranken, D.L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 1996, 96, 395–422. [Google Scholar] [CrossRef] [PubMed]
- Butt, N.A.; Zhang, W. Transition metal-catalyzed allylic substitution reactions with unactivated allylic substrates. Chem. Soc. Rev. 2015, 44, 7929–7967. [Google Scholar] [CrossRef] [PubMed]
- Piechaczyk, O.; Thoumazet, C.; Jean, Y.; Le Floch, P. DFT study on the palladium-catalyzed allylation of primary amines by allylic alcohol. J. Am. Chem. Soc. 2006, 128, 14306–14317. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemistry of wood.
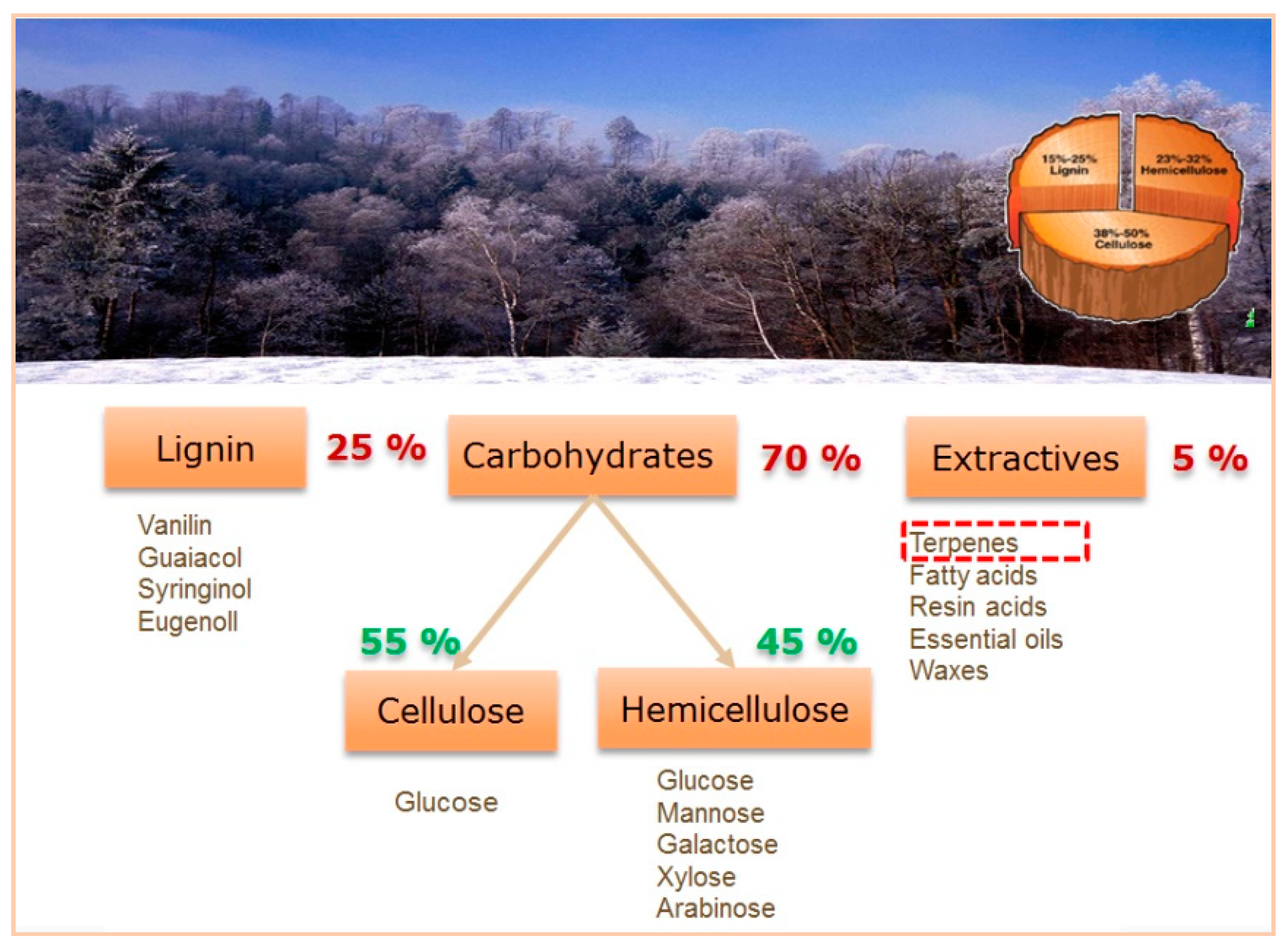
Figure 2.
The main available terpenes and terpenoids.

Figure 3.
Overview of the available tools for amination of terpenoids.
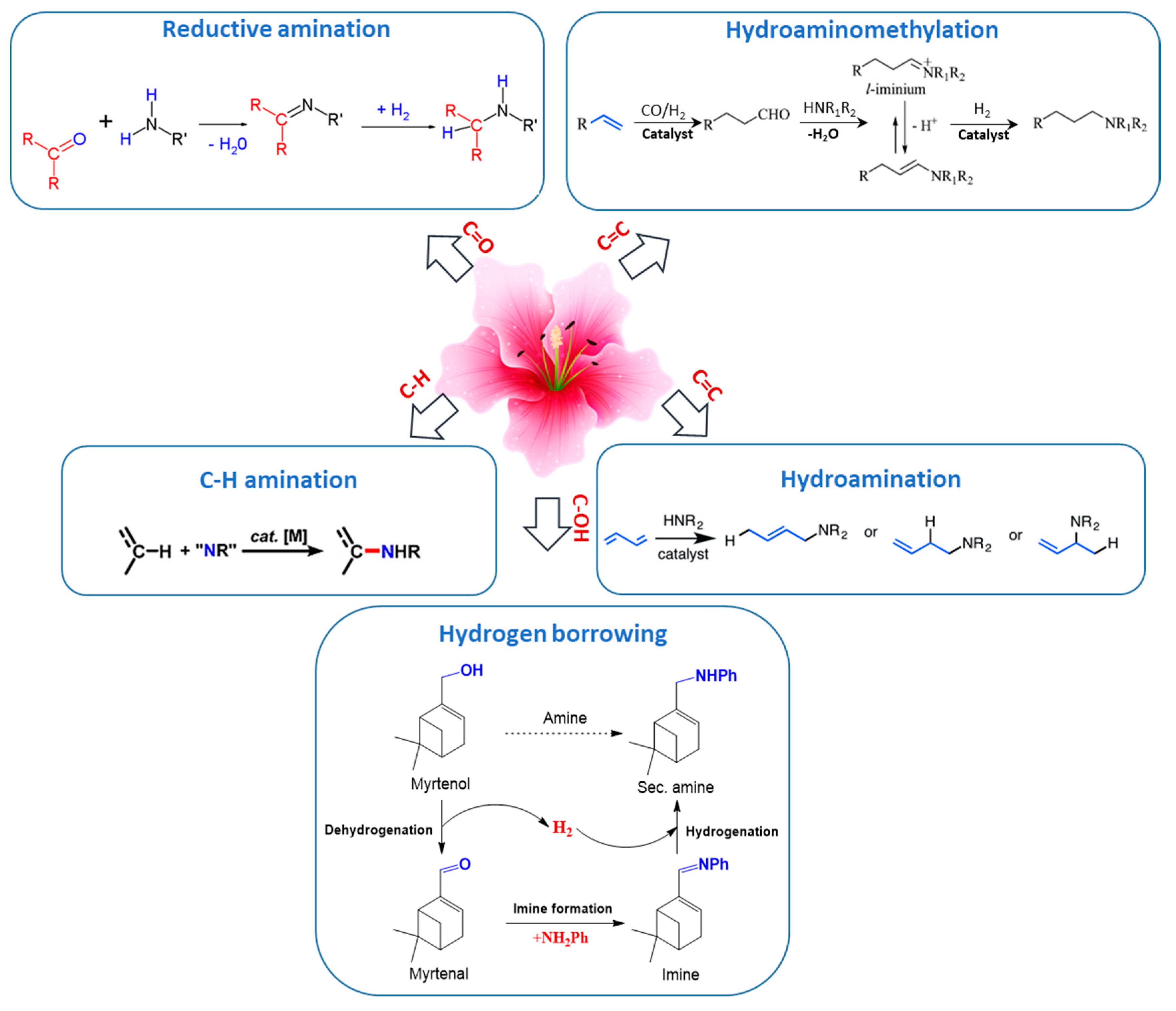
Figure 4.
Reaction network in reductive amination of citronellal (1) with ammonia. Reprinted from [62] with permission from Elsevier.
Figure 4.
Reaction network in reductive amination of citronellal (1) with ammonia. Reprinted from [62] with permission from Elsevier.

Figure 5.
Influence of hydrogen pressure on the product distribution in the reductive amination of citronellal; data points are connected to show a better comparison. Reaction conditions: 6 mmol citronellal (1), 216 mmol NH3, 0.5 mol% [Rh(cod)Cl]2, 2.0 mol% TPPTS, 0.5 mol% CTAC, 5 mL toluene, 130 °C, 800 rpm, 6 h. Reprinted from [62] with permission from Elsevier.
Figure 5.
Influence of hydrogen pressure on the product distribution in the reductive amination of citronellal; data points are connected to show a better comparison. Reaction conditions: 6 mmol citronellal (1), 216 mmol NH3, 0.5 mol% [Rh(cod)Cl]2, 2.0 mol% TPPTS, 0.5 mol% CTAC, 5 mL toluene, 130 °C, 800 rpm, 6 h. Reprinted from [62] with permission from Elsevier.

Figure 6.
Scheme of reductive amination of D-fenchone (1) by aliphatic nitriles to prepare fenchylamine. Adapted from [67].
Figure 6.
Scheme of reductive amination of D-fenchone (1) by aliphatic nitriles to prepare fenchylamine. Adapted from [67].
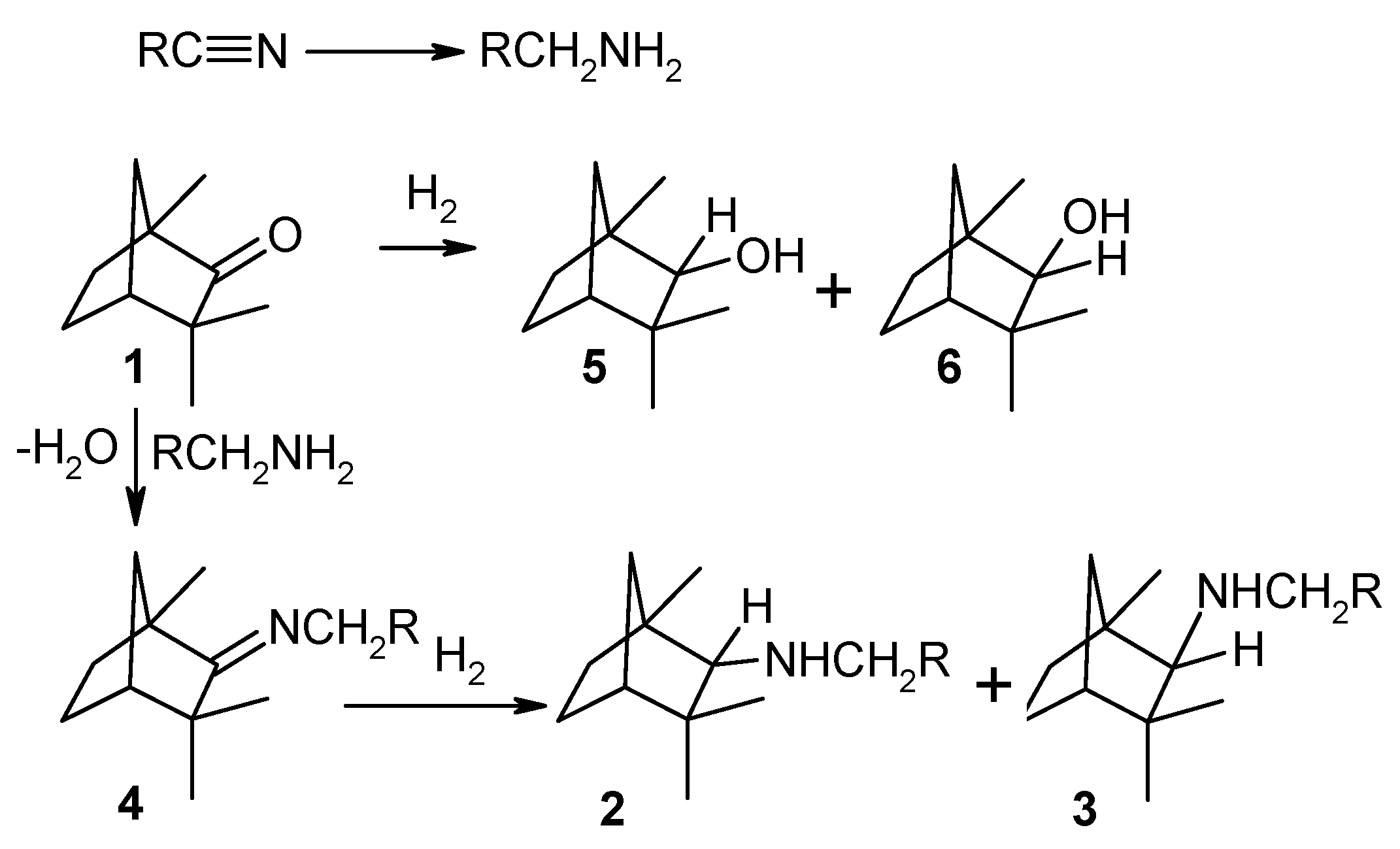
Figure 7.
Reductive amination of camphor (1) with methylamine for synthesis of N-methylbornan-2-ylamine. Adapted from [68].
Figure 7.
Reductive amination of camphor (1) with methylamine for synthesis of N-methylbornan-2-ylamine. Adapted from [68].
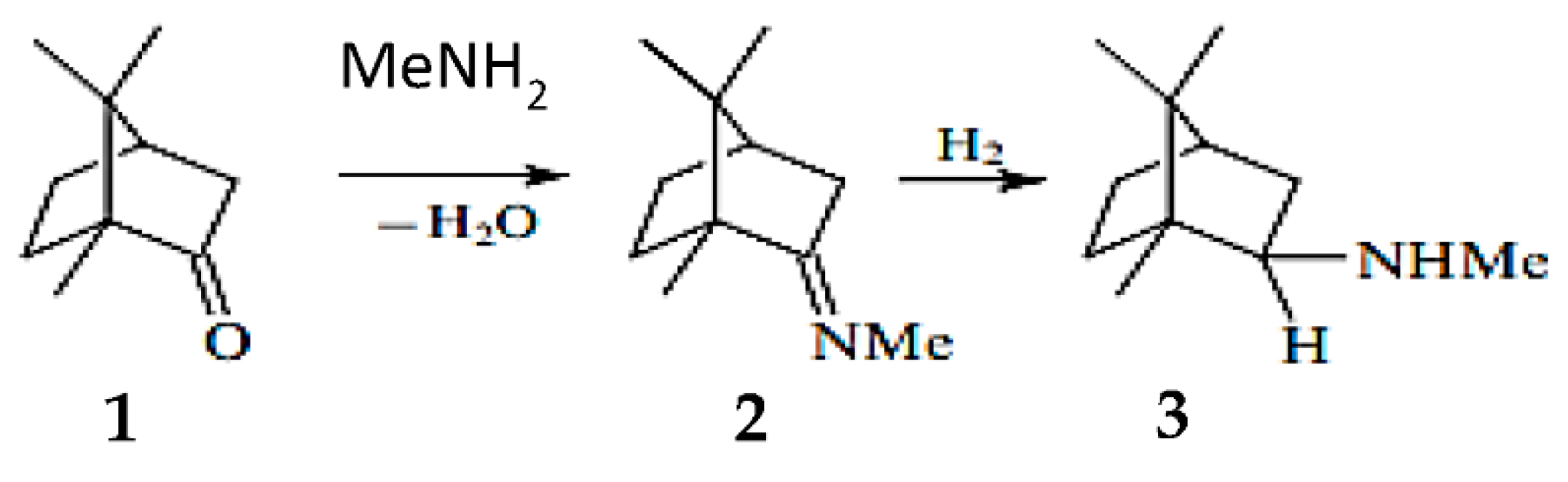
Figure 8.
Hydroaminomethylation of limonene (1) with secondary amines, Y—yield of product: 2a—R1 = n-propyl, R2 = H, Y = 85%; 2b—R1 = i-propyl, R2 = H, Y = 50%; 2c—R1 = benzyl, R2 = H, Y = 44%; 2d—R1 = R2 = piperidine, Y = 80%; 2e—R1 = R2 = morpholine, Y = 79%; 2f—R1 = R2 = piperazine, Y = 89%; 2g—R1 = phenyl, R2 = H, Y = 50%. Reprinted from [76] with permission from Elsevier.
Figure 8.
Hydroaminomethylation of limonene (1) with secondary amines, Y—yield of product: 2a—R1 = n-propyl, R2 = H, Y = 85%; 2b—R1 = i-propyl, R2 = H, Y = 50%; 2c—R1 = benzyl, R2 = H, Y = 44%; 2d—R1 = R2 = piperidine, Y = 80%; 2e—R1 = R2 = morpholine, Y = 79%; 2f—R1 = R2 = piperazine, Y = 89%; 2g—R1 = phenyl, R2 = H, Y = 50%. Reprinted from [76] with permission from Elsevier.

Figure 9.
Reaction scheme of the hydroaminomethylation of limonene (1) with ammonia using [Rh(cod)Cl]2 catalyst. Reprinted from [77] with permission from Willey.
Figure 9.
Reaction scheme of the hydroaminomethylation of limonene (1) with ammonia using [Rh(cod)Cl]2 catalyst. Reprinted from [77] with permission from Willey.

Figure 10.
Hydroaminomethylation of limonene (1). Reprinted from [74] with permission from Elsevier.
Figure 10.
Hydroaminomethylation of limonene (1). Reprinted from [74] with permission from Elsevier.

Figure 11.
Hydroaminomethylation of camphene (5). Reprinted from [74] with permission from Elsevier.
Figure 11.
Hydroaminomethylation of camphene (5). Reprinted from [74] with permission from Elsevier.
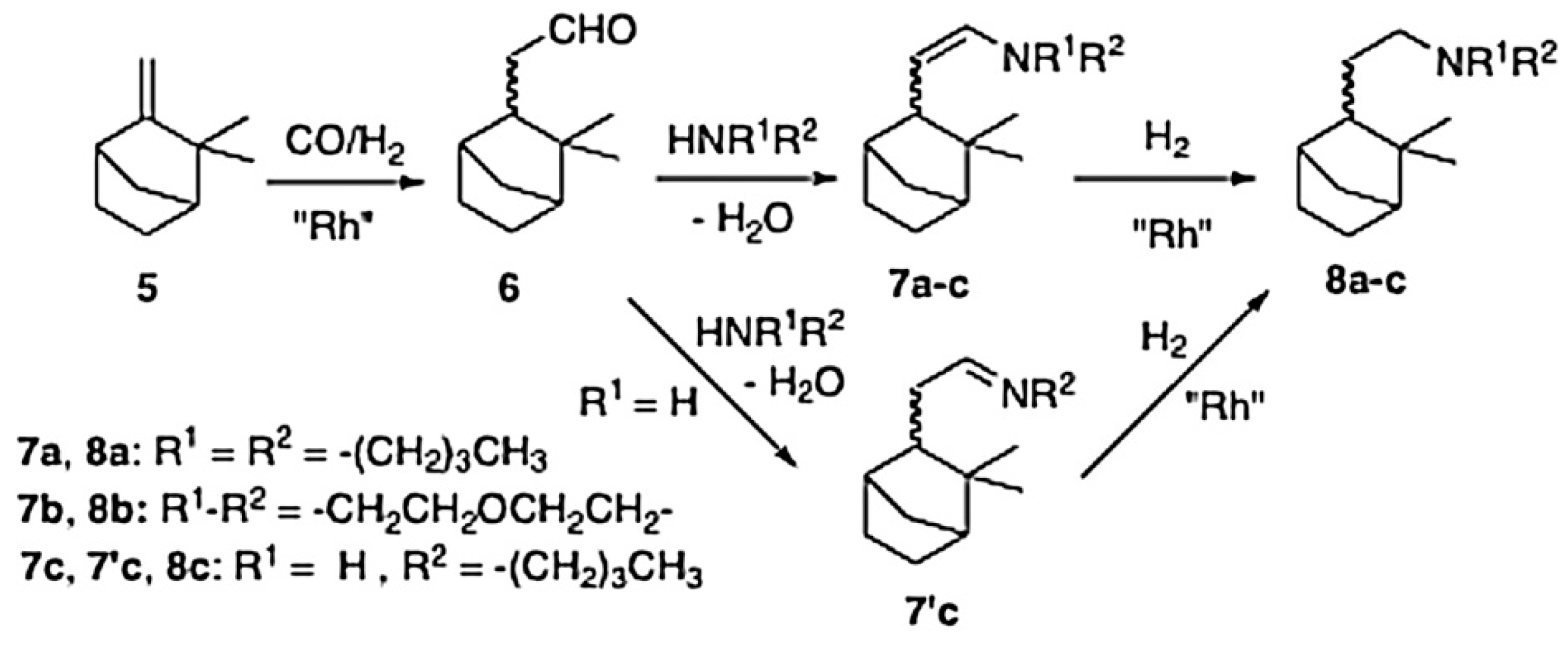
Figure 12.
Hydroaminomethylation of β-pinene (9). Reprinted from [74] with permission from Elsevier.
Figure 12.
Hydroaminomethylation of β-pinene (9). Reprinted from [74] with permission from Elsevier.
Figure 13.
Hydroaminomethylation of estragole (1). Reprinted from [80] with permission from Elsevier.
Figure 13.
Hydroaminomethylation of estragole (1). Reprinted from [80] with permission from Elsevier.

Figure 14.
Hydroaminomethylation of eugenol with di-n-butylamine. Reprinted from [79] with permission from Elsevier.
Figure 14.
Hydroaminomethylation of eugenol with di-n-butylamine. Reprinted from [79] with permission from Elsevier.

Figure 15.
Hydroaminomethylation of myrcene. GC based yields (Y) and conversions (X) are given in % of reacted myrcene. Reproduced from [78] with permission from Willey.
Figure 15.
Hydroaminomethylation of myrcene. GC based yields (Y) and conversions (X) are given in % of reacted myrcene. Reproduced from [78] with permission from Willey.

Figure 16.
Identification of tail products in hydroamination of myrcene. Reproduced from [33] with permission from Willey.
Figure 16.
Identification of tail products in hydroamination of myrcene. Reproduced from [33] with permission from Willey.
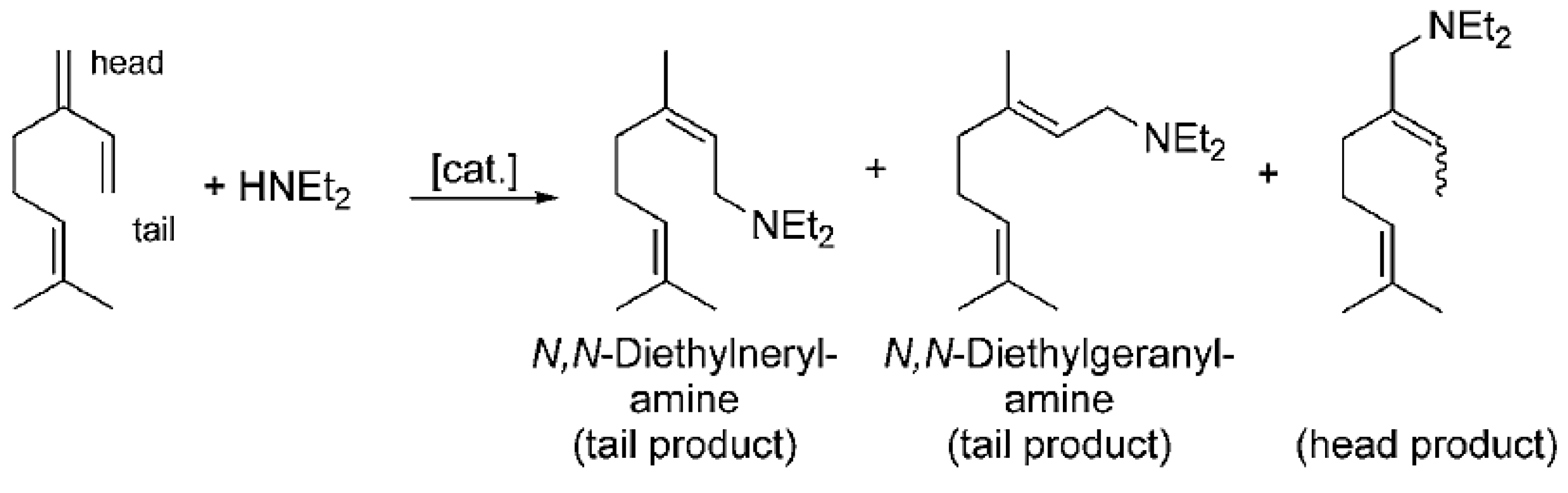
Figure 17.
Product network based on N,N-diethylnerylamine. Reproduced from [33] with permission from Willey.
Figure 17.
Product network based on N,N-diethylnerylamine. Reproduced from [33] with permission from Willey.

Figure 18.
Pd catalyzed allylic amination of linalyl acetate (1), nerolidyl acetate (3), and linalyl methylcarbonate (5). Adapted from [91].
Figure 18.
Pd catalyzed allylic amination of linalyl acetate (1), nerolidyl acetate (3), and linalyl methylcarbonate (5). Adapted from [91].

Figure 19.
A general reaction amination of myrtenyl acetate (1) with different secondary amines (morpholine, pyrrolidine, and dimethylamine). Reproduced from [124] under the terms of the Creative Commons Licenses.
Figure 19.
A general reaction amination of myrtenyl acetate (1) with different secondary amines (morpholine, pyrrolidine, and dimethylamine). Reproduced from [124] under the terms of the Creative Commons Licenses.
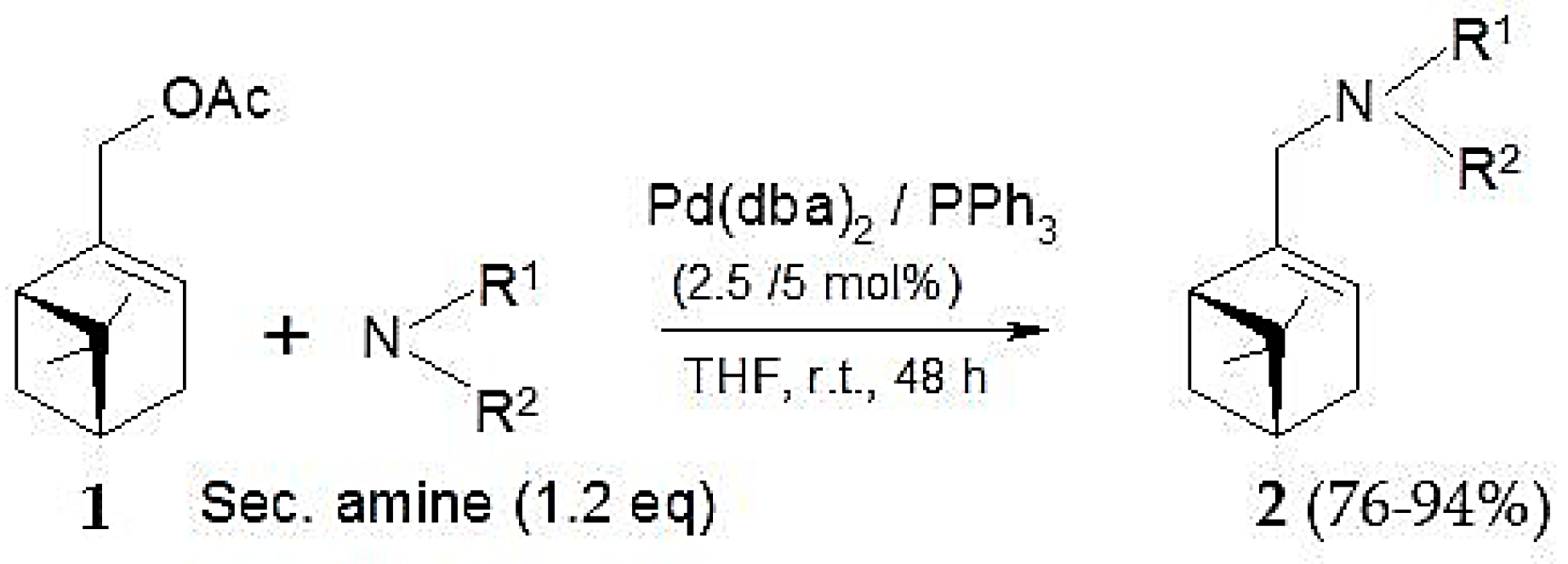
Figure 20.
Allylic amination with N-(ortho-carboran-3-yl)-N-methylamine: N-Cinnamyl-N-(ortho-carboran-3-yl)methylamine (5), N-(3-Methylbut-2-enyl)-N-(ortho-carboran-3-yl)methylamine (6), (E)-N-(3,7-Dimethylocta-2,6-dienyl)-N-(ortho-carboran-3-yl)methyl amine (7). Reprinted from [55] with permission from Elsevier.
Figure 20.
Allylic amination with N-(ortho-carboran-3-yl)-N-methylamine: N-Cinnamyl-N-(ortho-carboran-3-yl)methylamine (5), N-(3-Methylbut-2-enyl)-N-(ortho-carboran-3-yl)methylamine (6), (E)-N-(3,7-Dimethylocta-2,6-dienyl)-N-(ortho-carboran-3-yl)methyl amine (7). Reprinted from [55] with permission from Elsevier.

Figure 21.
Amination catalysts used in allylic amination of cinnamyl acetate with benzylamine. Adapted from [130].
Figure 21.
Amination catalysts used in allylic amination of cinnamyl acetate with benzylamine. Adapted from [130].

Figure 22.
Schematic view of the one-pot alcohols amination applying “hydrogen borrowing approach”, where oxidation in fact stands for dehydrogenation. Reprinted from [123] with permission from Elsevier.
Figure 22.
Schematic view of the one-pot alcohols amination applying “hydrogen borrowing approach”, where oxidation in fact stands for dehydrogenation. Reprinted from [123] with permission from Elsevier.

Figure 23.
Myrtenol amination with aniline over Au catalysts. Reprinted from [138] with permission from Elsevier.
Figure 23.
Myrtenol amination with aniline over Au catalysts. Reprinted from [138] with permission from Elsevier.

Figure 24.
Myrtenol conversion (a) and the selectivity (b) to the corresponding products 1 (blue bars), 2 (dark blue bars), 3 (green bars), and 4 (red bars) at the same myrtenol conversion (74%) for myrtenol amination in the presence of gold supported on ZrO2, Al2O3, CeO2, La2O3, and MgO. The reaction conditions: T = 180 °C, p(N2) = 9 bar, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, catalyst 1.4 mol% Au, R = aniline. Reprinted from [123] with permission from Elsevier.
Figure 24.
Myrtenol conversion (a) and the selectivity (b) to the corresponding products 1 (blue bars), 2 (dark blue bars), 3 (green bars), and 4 (red bars) at the same myrtenol conversion (74%) for myrtenol amination in the presence of gold supported on ZrO2, Al2O3, CeO2, La2O3, and MgO. The reaction conditions: T = 180 °C, p(N2) = 9 bar, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, catalyst 1.4 mol% Au, R = aniline. Reprinted from [123] with permission from Elsevier.

Figure 25.
The products distribution as a function of the reaction time during myrtenol amination over Au/ZrO2 (a), Au/CeO2 (b), Au/La2O3 (c) pre-treated in O2 (left) or in H2 (right). The reaction products: myrtenol (■), myrtenal (◄), myrtanol ( ![Catalysts 08 00365 i038]() ), myrtanal (►), 1 (●), 2(▲), 3 (
), myrtanal (►), 1 (●), 2(▲), 3 ( ![Catalysts 08 00365 i039]() ), 4 (▼). The reaction conditions: T = 180 °C, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, catalyst 1.4 mol% Au. Reprinted from [137] with permission from Elsevier.
), 4 (▼). The reaction conditions: T = 180 °C, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, catalyst 1.4 mol% Au. Reprinted from [137] with permission from Elsevier.
 ), myrtanal (►), 1 (●), 2(▲), 3 (
), myrtanal (►), 1 (●), 2(▲), 3 (  ), 4 (▼). The reaction conditions: T = 180 °C, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, catalyst 1.4 mol% Au. Reprinted from [137] with permission from Elsevier.
), 4 (▼). The reaction conditions: T = 180 °C, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, catalyst 1.4 mol% Au. Reprinted from [137] with permission from Elsevier.
Figure 25.
The products distribution as a function of the reaction time during myrtenol amination over Au/ZrO2 (a), Au/CeO2 (b), Au/La2O3 (c) pre-treated in O2 (left) or in H2 (right). The reaction products: myrtenol (■), myrtenal (◄), myrtanol ( ![Catalysts 08 00365 i038]() ), myrtanal (►), 1 (●), 2(▲), 3 (
), myrtanal (►), 1 (●), 2(▲), 3 ( ![Catalysts 08 00365 i039]() ), 4 (▼). The reaction conditions: T = 180 °C, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, catalyst 1.4 mol% Au. Reprinted from [137] with permission from Elsevier.
), 4 (▼). The reaction conditions: T = 180 °C, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, catalyst 1.4 mol% Au. Reprinted from [137] with permission from Elsevier.
), myrtanal (►), 1 (●), 2(▲), 3 ( ), 4 (▼). The reaction conditions: T = 180 °C, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, catalyst 1.4 mol% Au. Reprinted from [137] with permission from Elsevier.
Figure 26.
Proposed catalytic mechanism for myrtenol amination with aniline over Au on metal oxide MeO. Reprinted from [123] with permission from Elsevier.
Figure 26.
Proposed catalytic mechanism for myrtenol amination with aniline over Au on metal oxide MeO. Reprinted from [123] with permission from Elsevier.
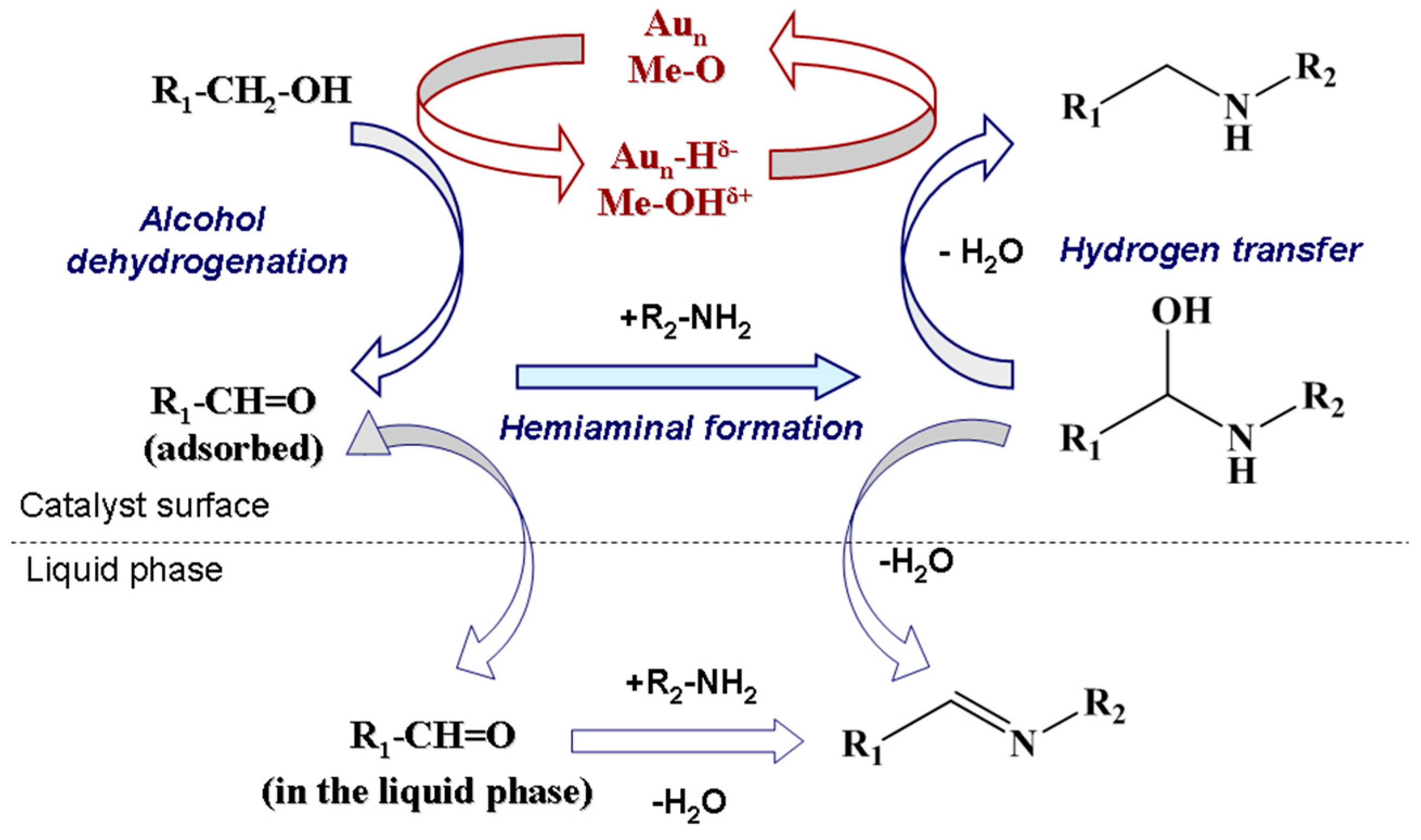
Figure 27.
The effect of H-donors addition and its amount on myrtenol conversion (a) and selectivity to amine (2) (b,c). The reaction conditions: T = 180 °C, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, Au/ZrO2 catalyst 92 mg, 9 bar N2. Reprinted from [139] with permission from Elsevier.
Figure 27.
The effect of H-donors addition and its amount on myrtenol conversion (a) and selectivity to amine (2) (b,c). The reaction conditions: T = 180 °C, myrtenol 1 mmol, aniline 1 mmol, toluene 10 mL, Au/ZrO2 catalyst 92 mg, 9 bar N2. Reprinted from [139] with permission from Elsevier.

Figure 28.
Sulfonimidamide S*NH2 (1) and the rhodium catalyst Rh2(S-nta)4 (2). Reproduced with permission from [144]. Copyright American Chemical Society.
Figure 28.
Sulfonimidamide S*NH2 (1) and the rhodium catalyst Rh2(S-nta)4 (2). Reproduced with permission from [144]. Copyright American Chemical Society.
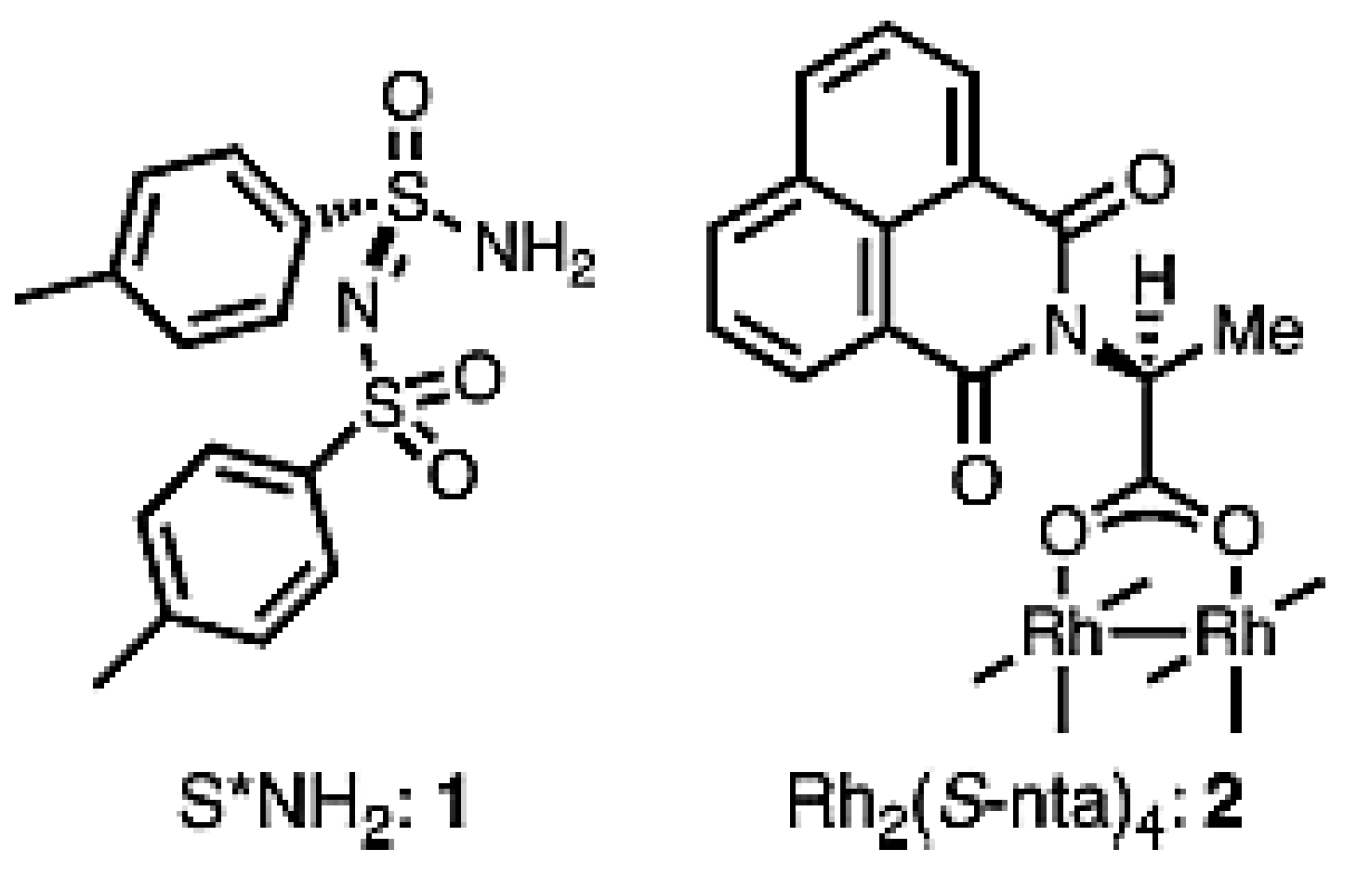
Figure 29.
Proposed mechanism of Pd(0)-allylic amination of cis-carvyl acetate (1). Reproduced from [124] under the terms of the Creative Commons Licenses.
Figure 29.
Proposed mechanism of Pd(0)-allylic amination of cis-carvyl acetate (1). Reproduced from [124] under the terms of the Creative Commons Licenses.

Figure 30.
Proposed mechanism for the intermolecular hydroamination of 1,3-dienes with aniline. Reproduced from [89] by permission of The Royal Society of Chemistry.
Figure 30.
Proposed mechanism for the intermolecular hydroamination of 1,3-dienes with aniline. Reproduced from [89] by permission of The Royal Society of Chemistry.
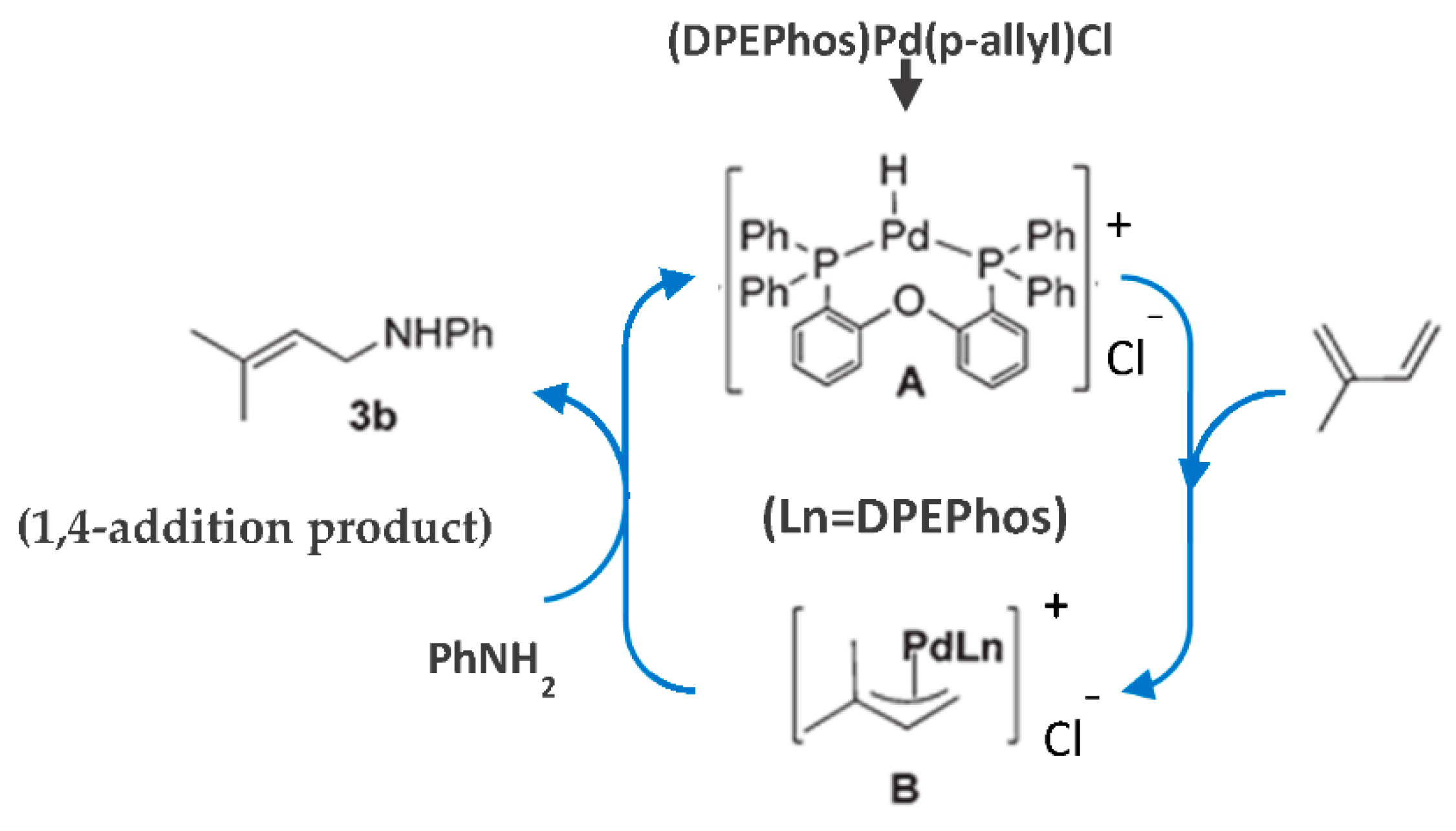

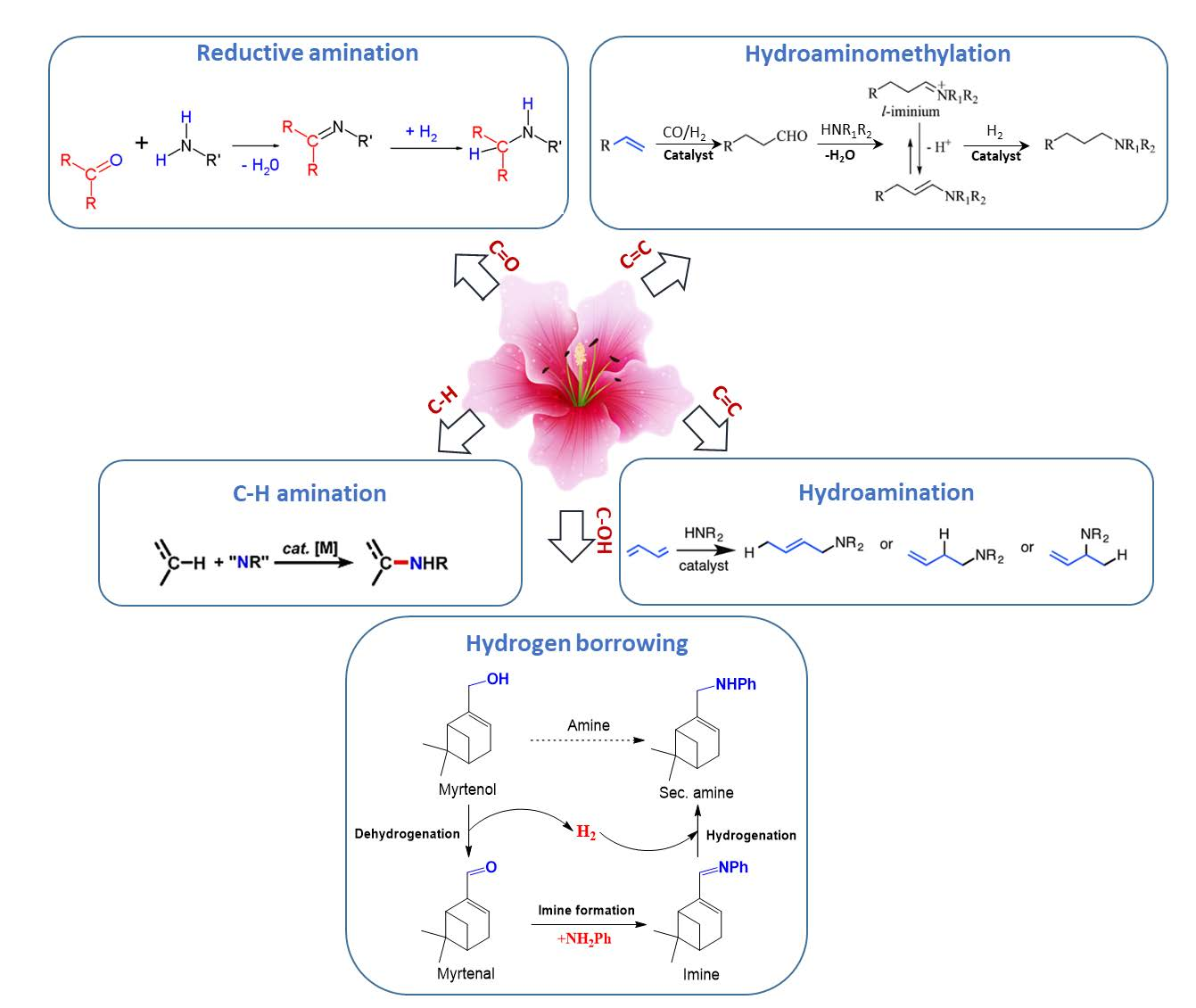{kind=link}
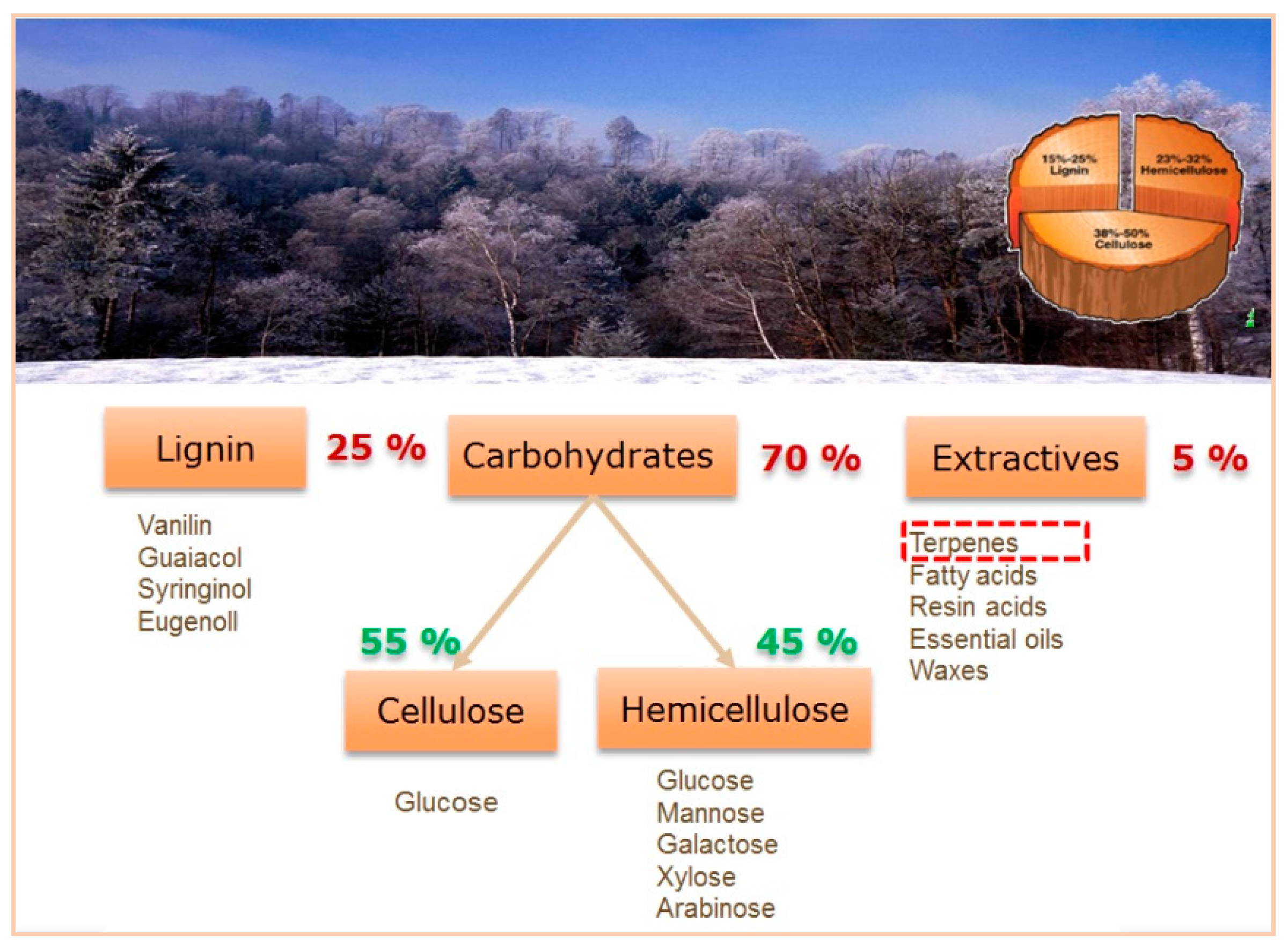{kind=link}
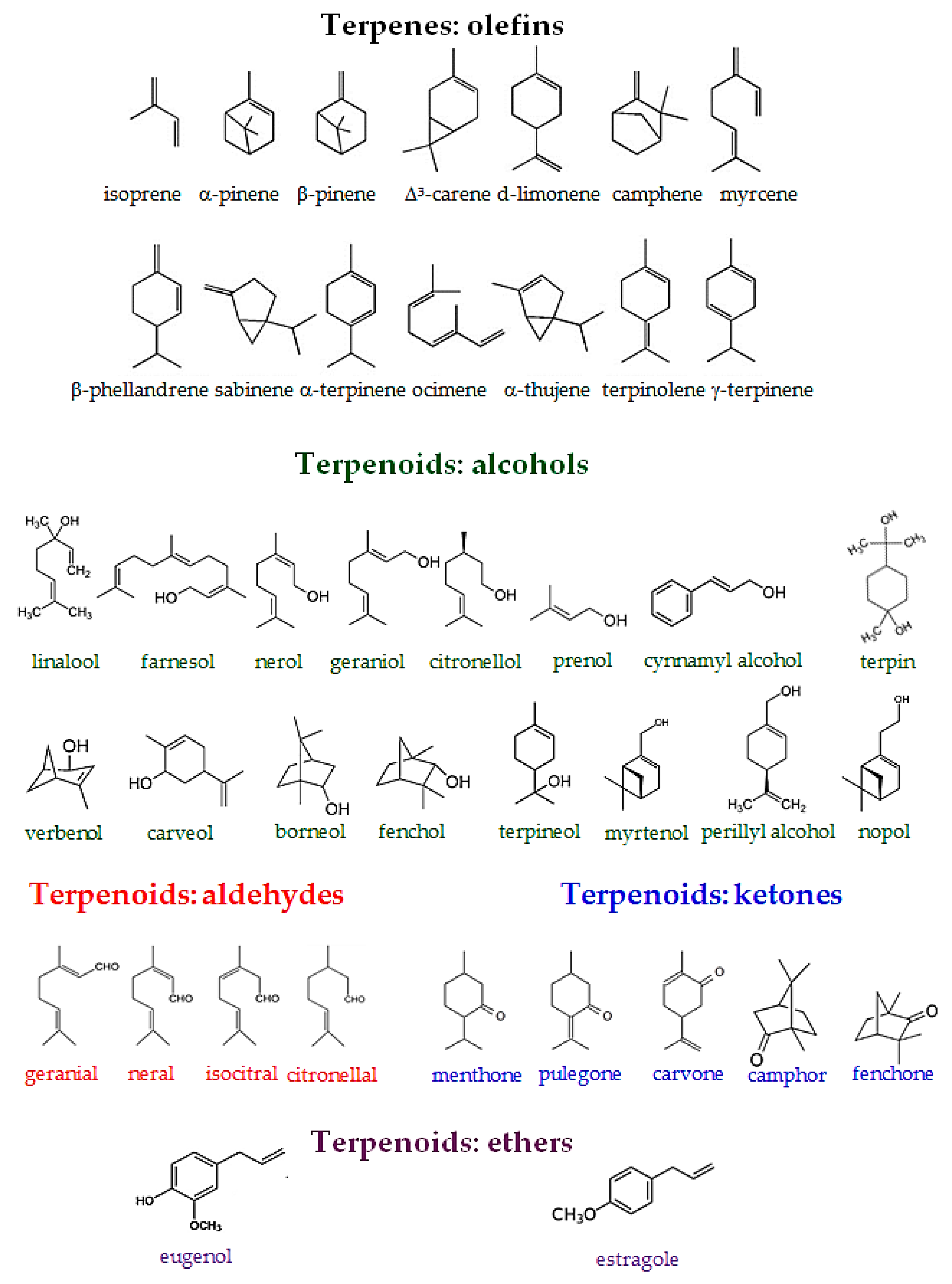{kind=link}
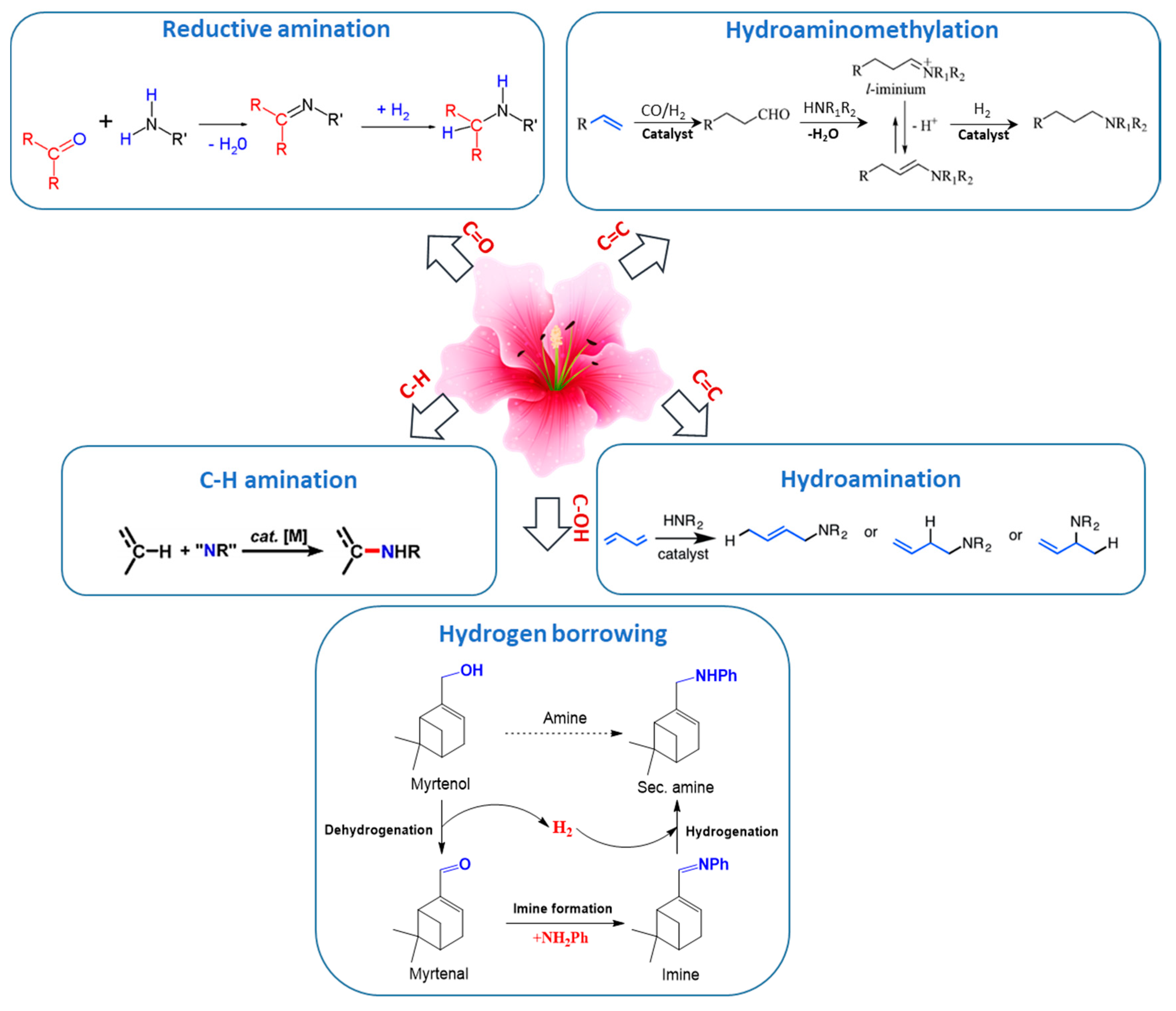{kind=link}
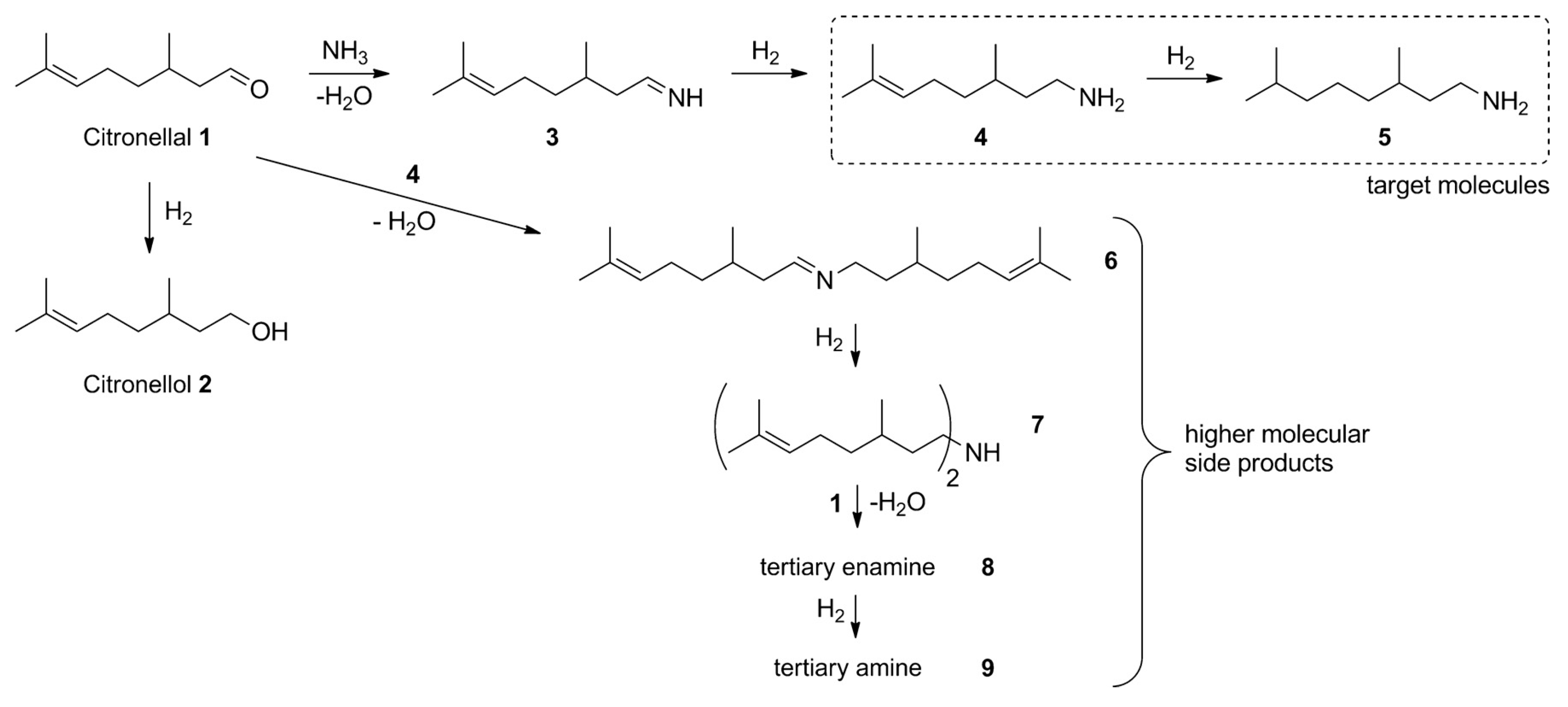{kind=link}
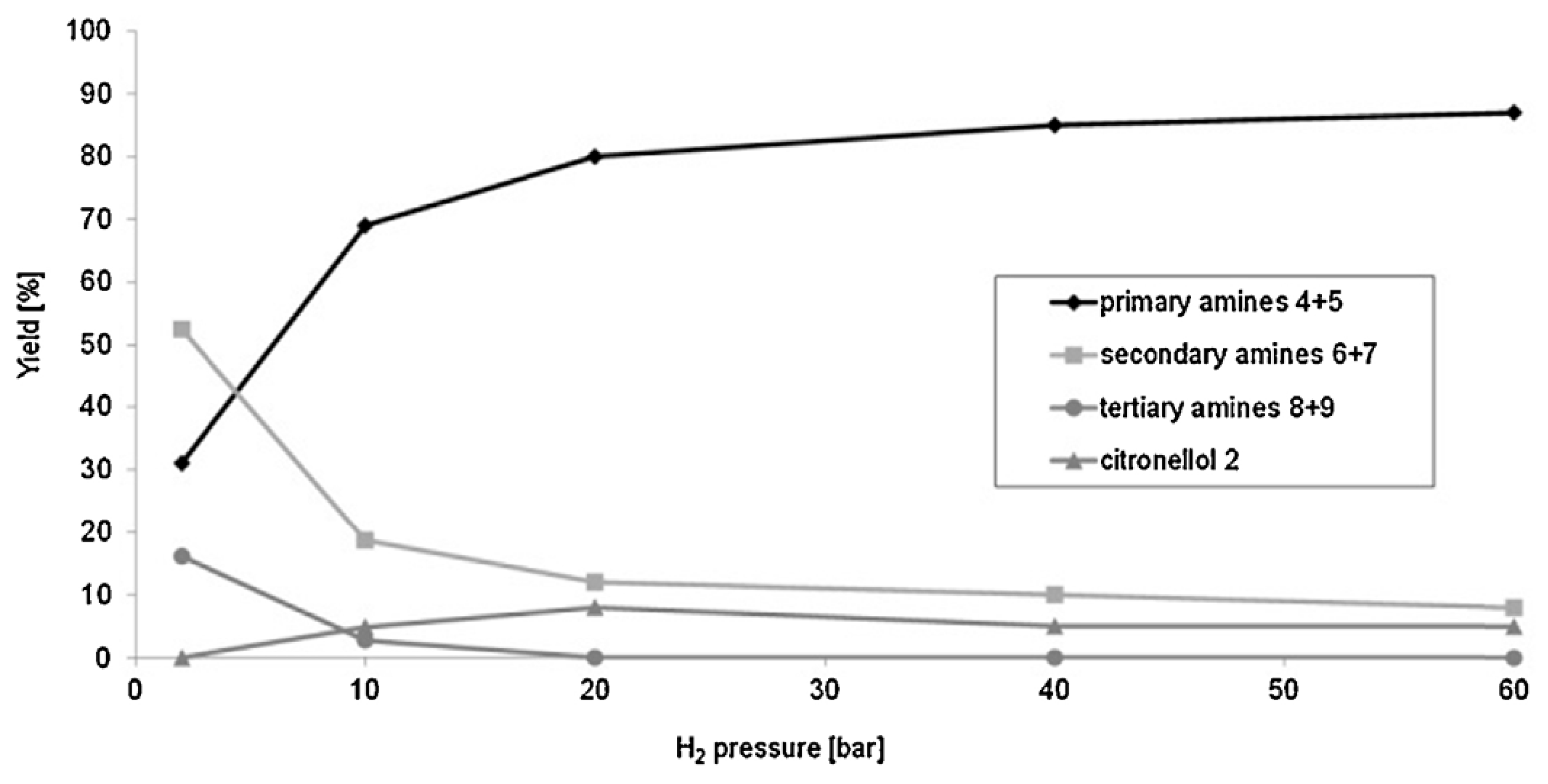{kind=link}
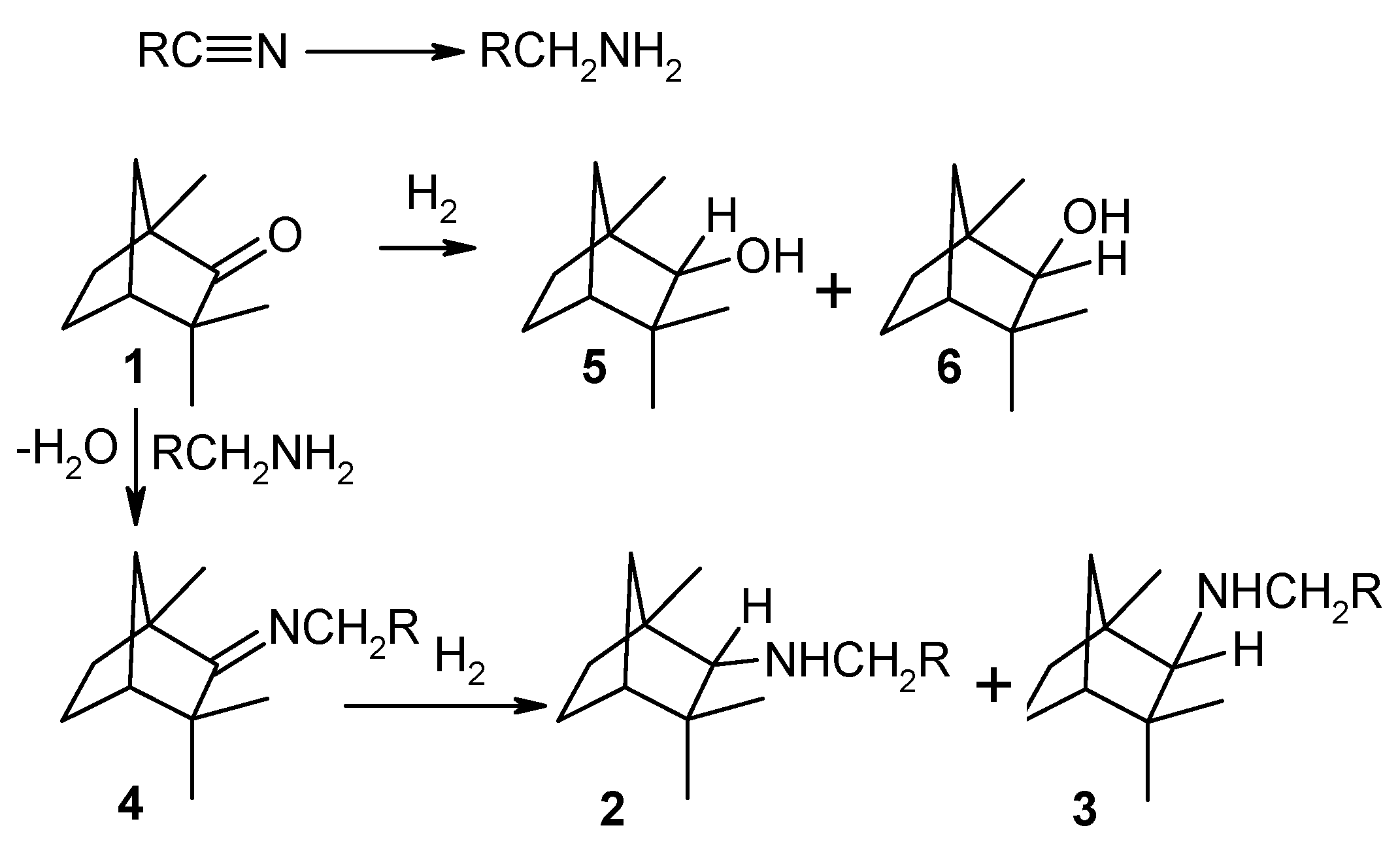{kind=link}
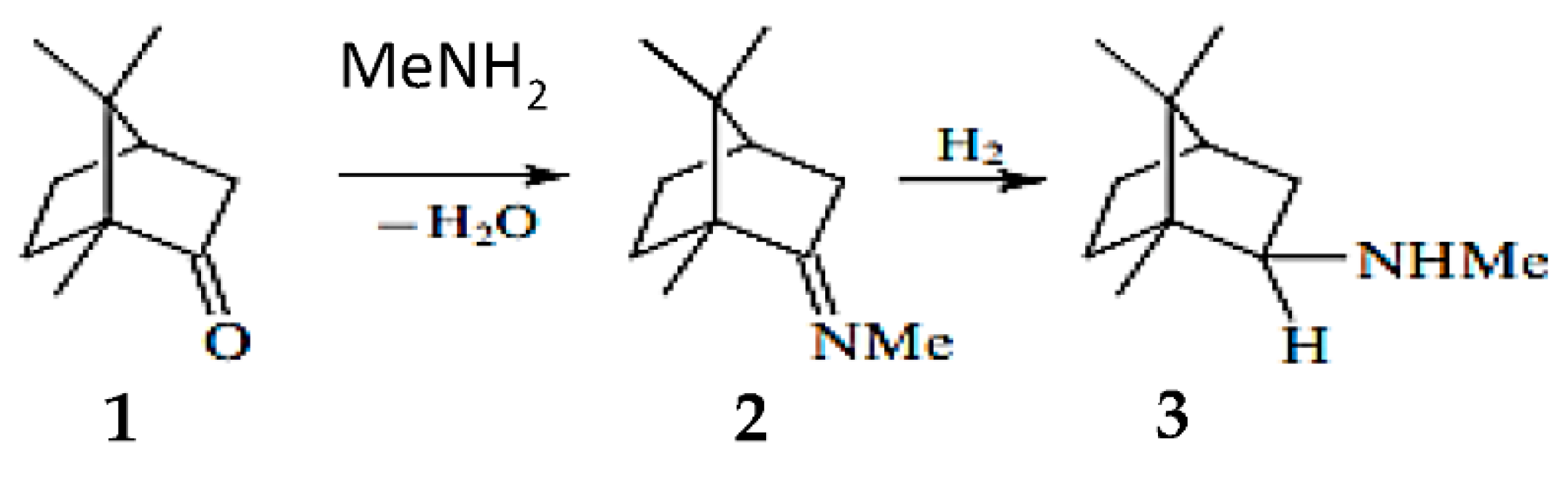{kind=link}
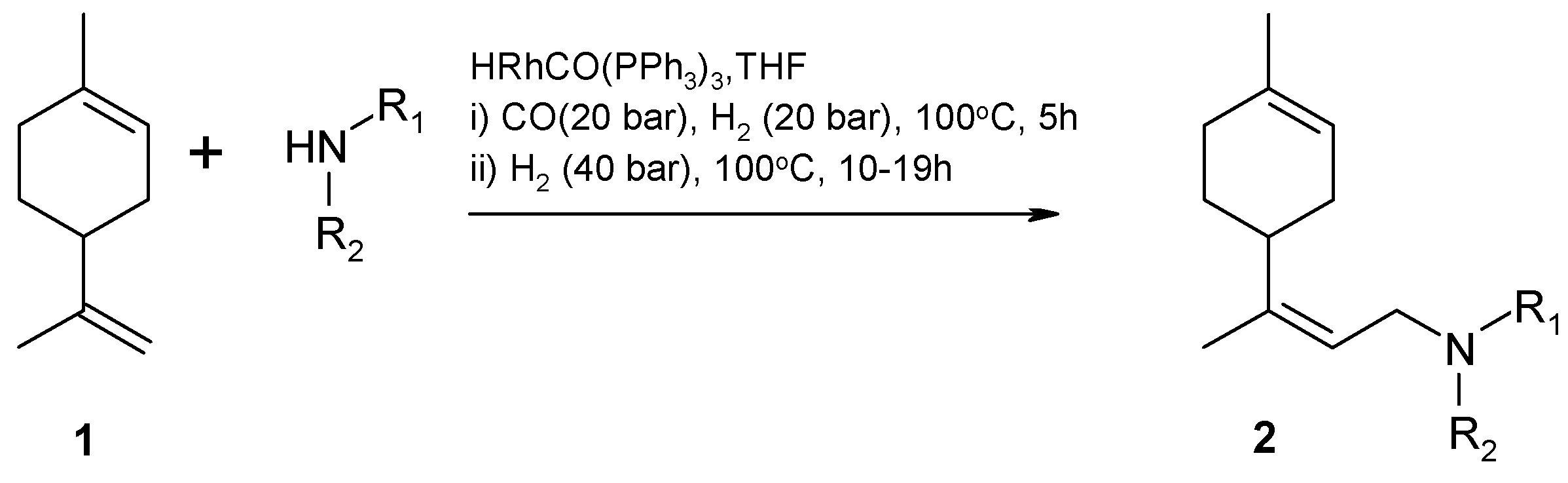{kind=link}
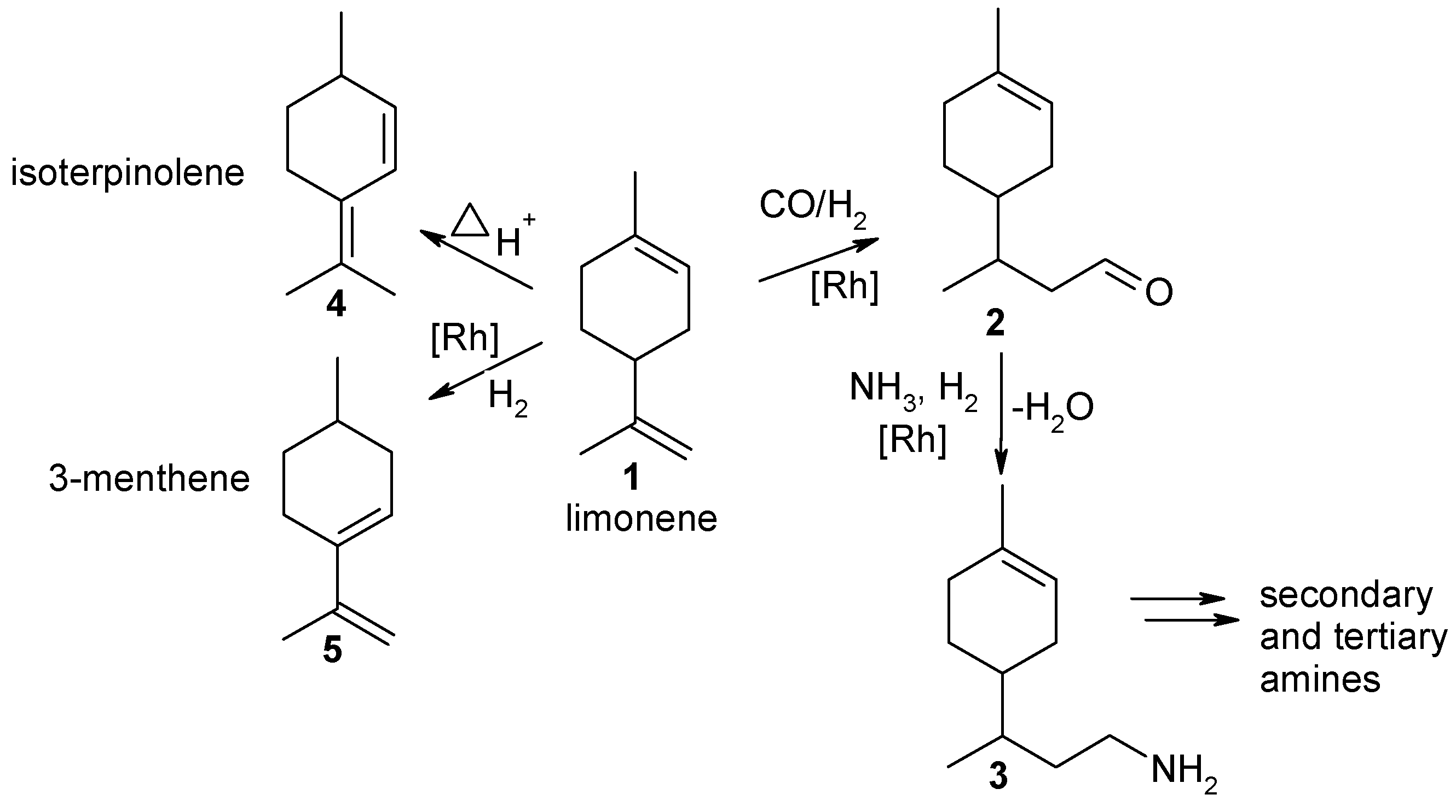{kind=link}
{kind=link}
{kind=link}
{kind=link}
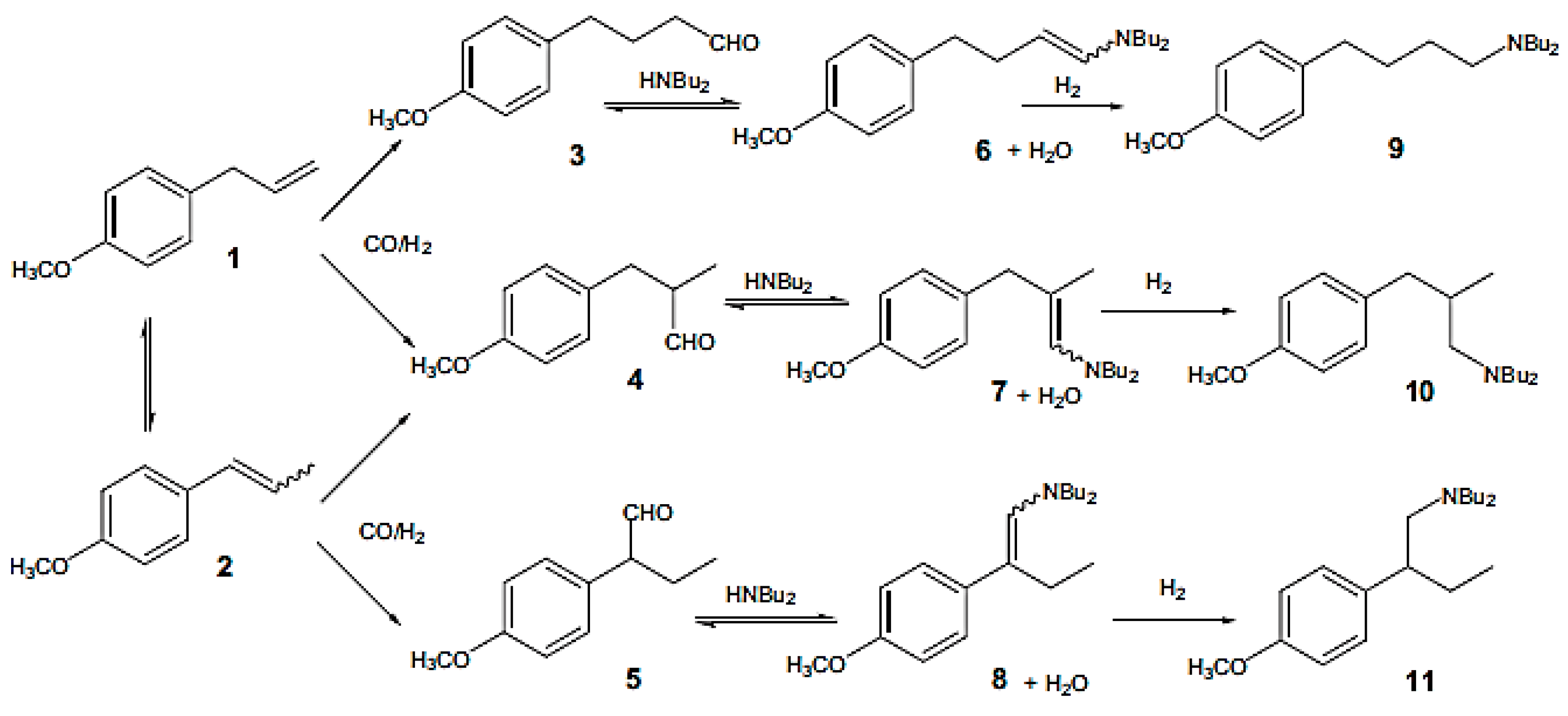{kind=link}
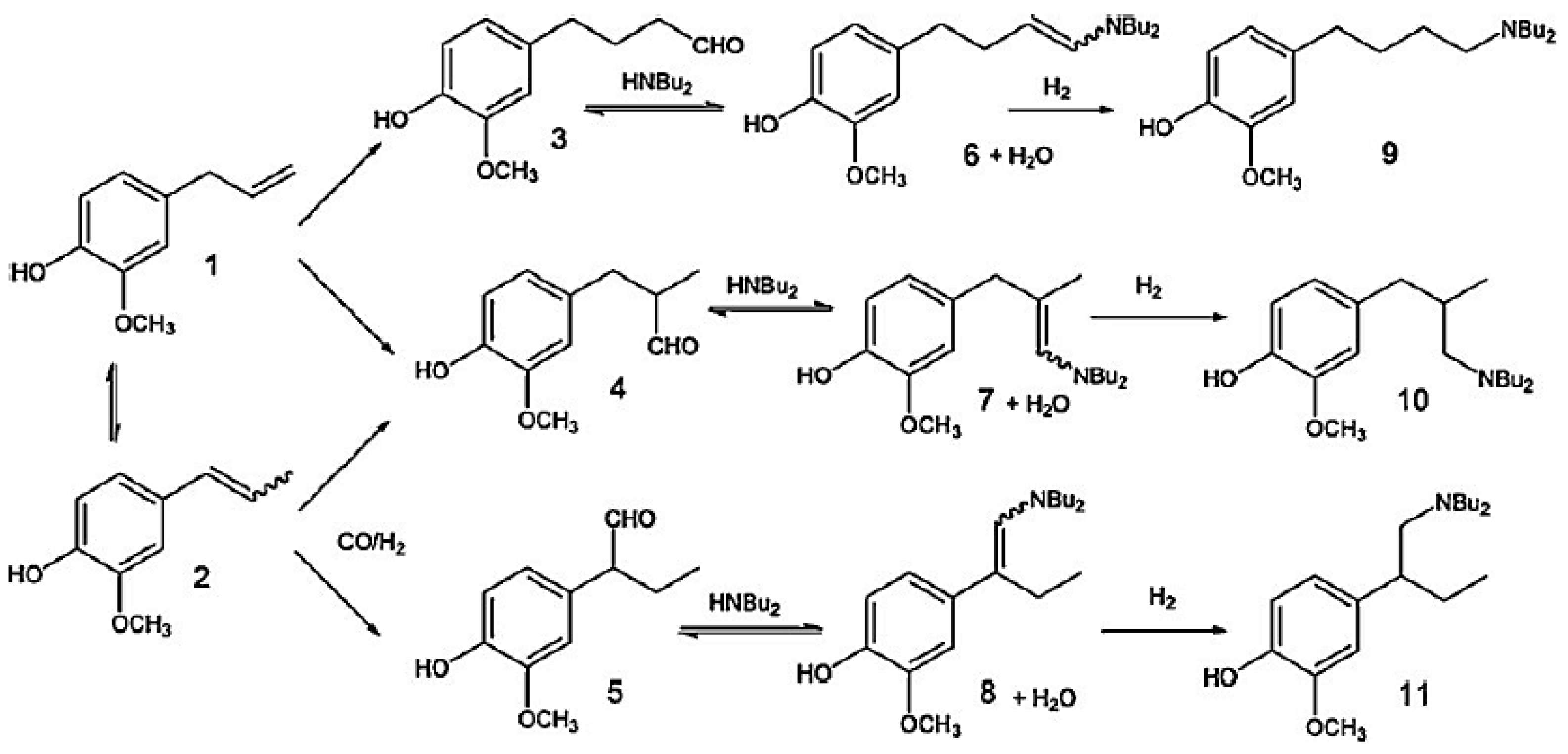{kind=link}
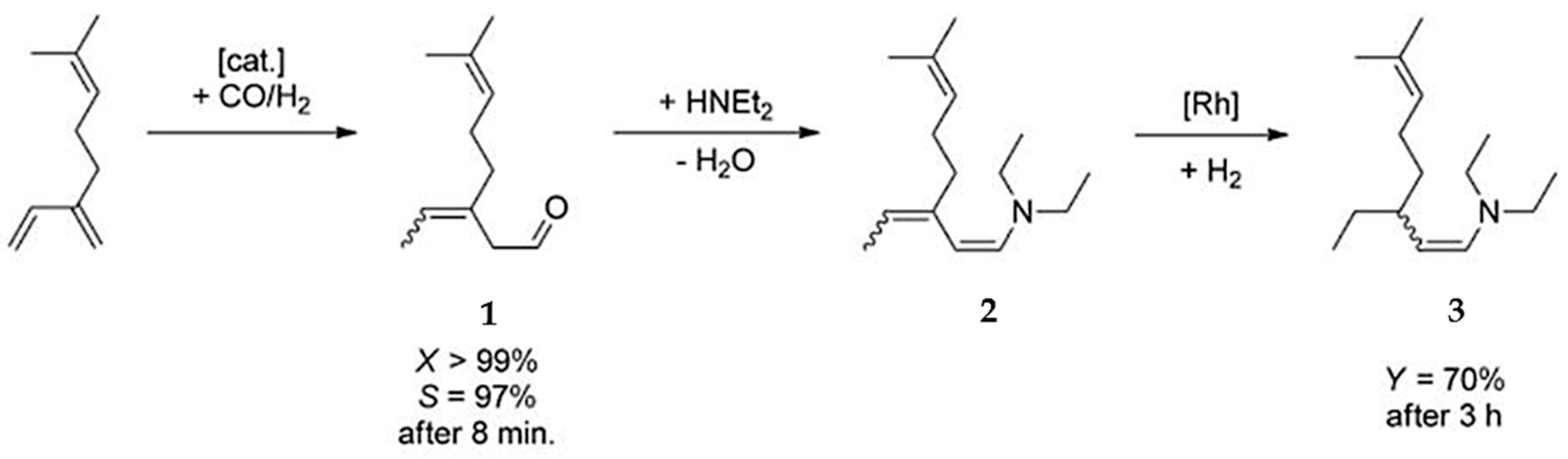{kind=link}
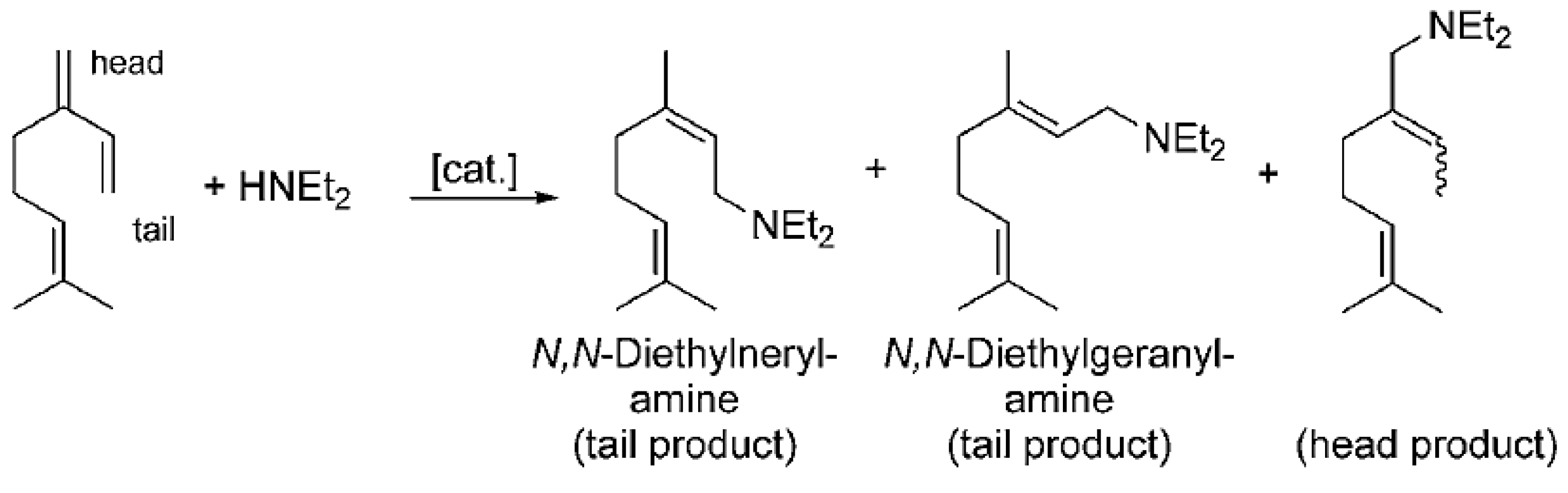{kind=link}
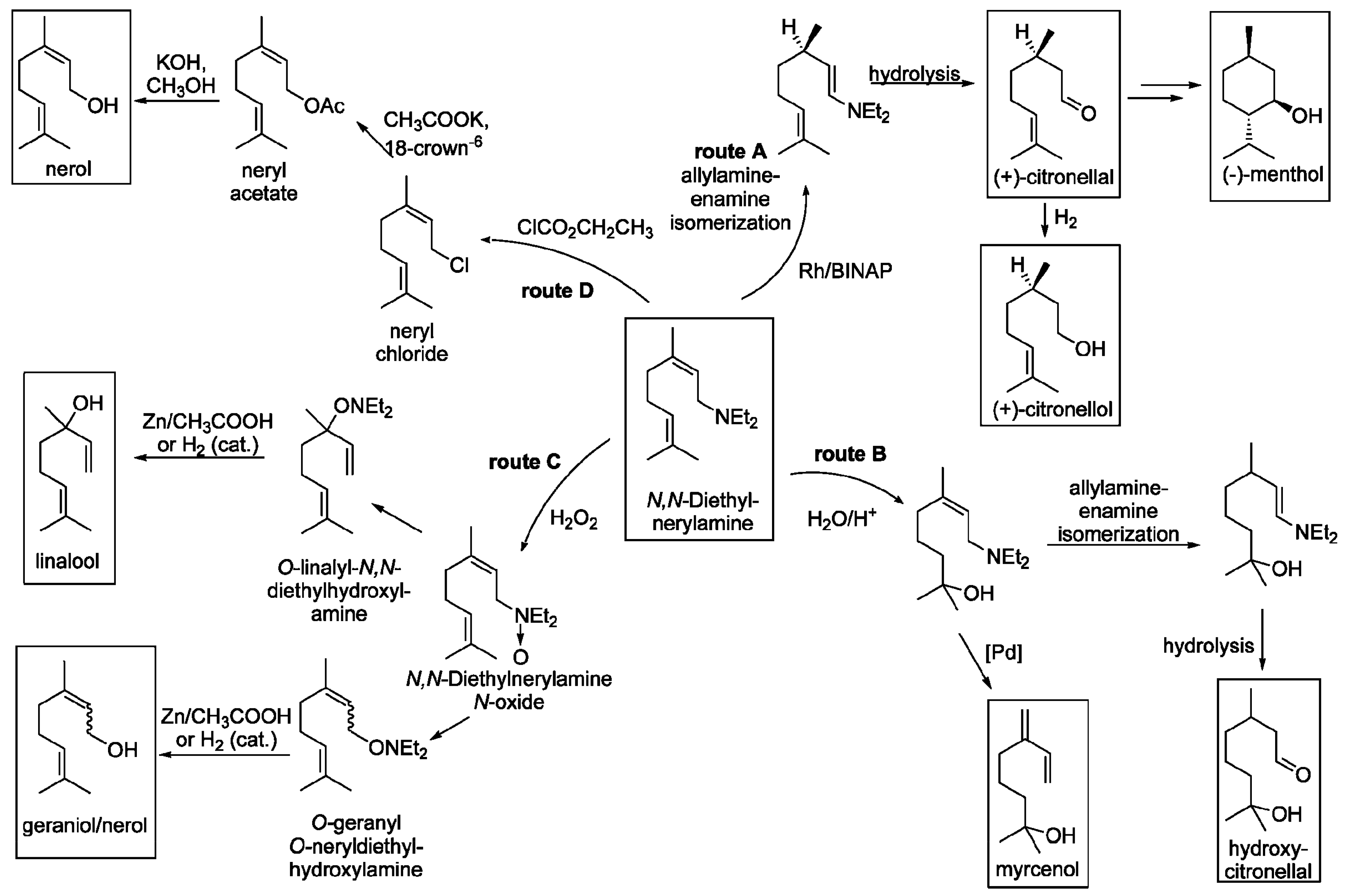{kind=link}
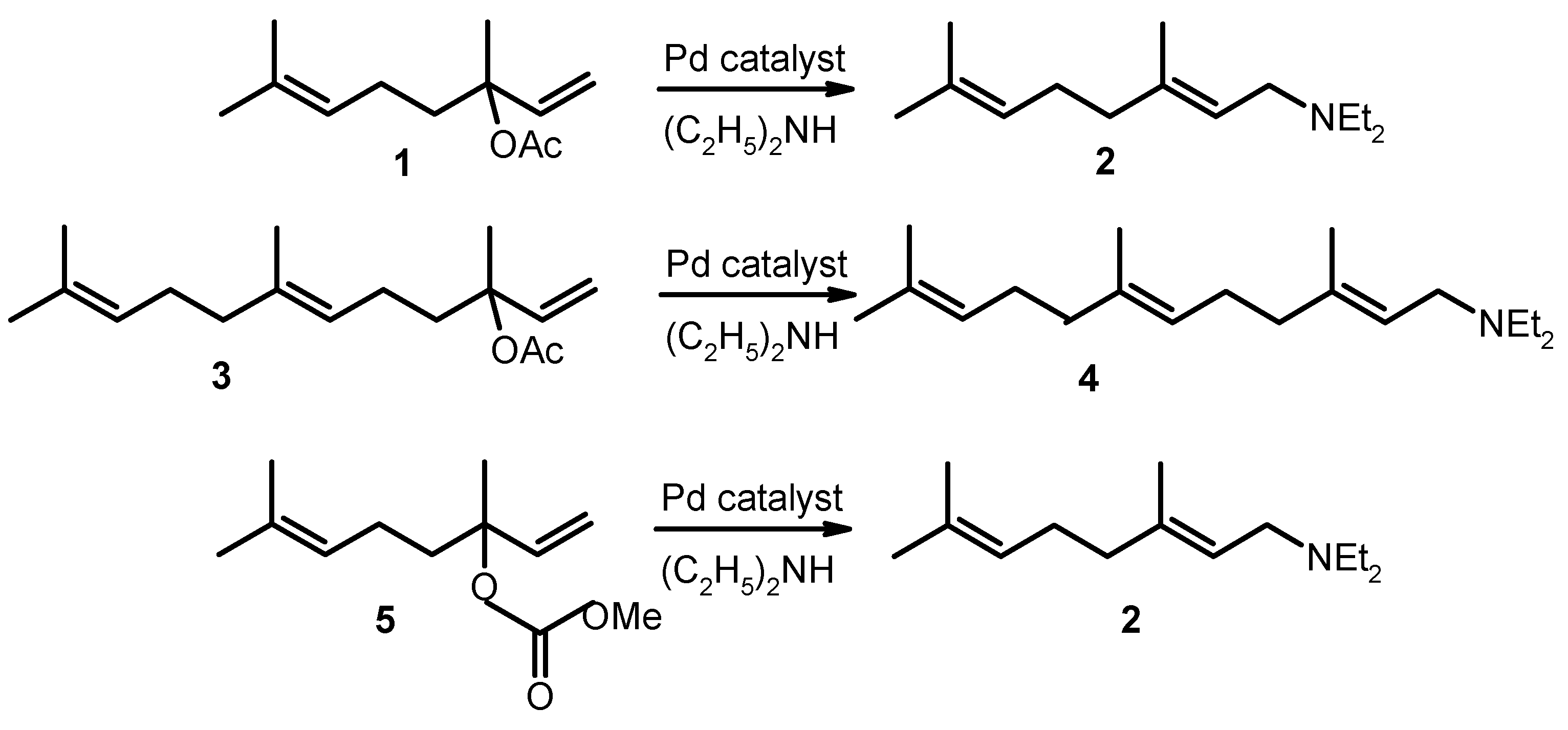{kind=link}
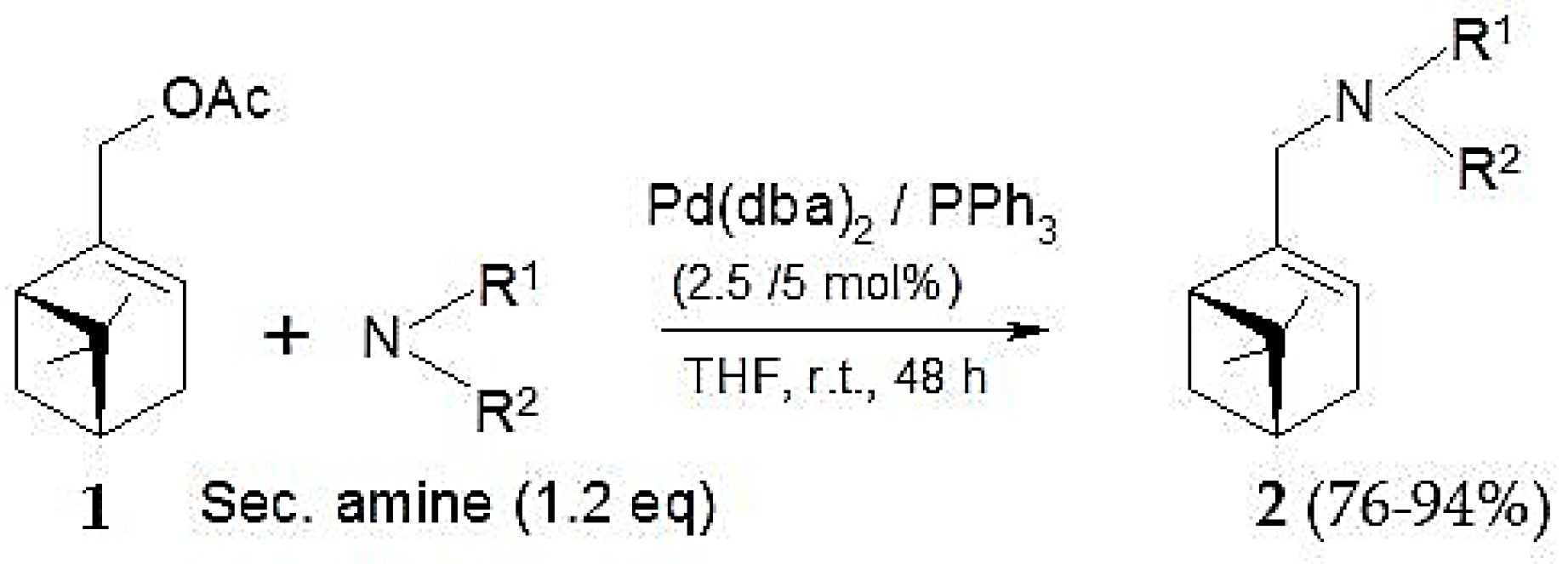{kind=link}
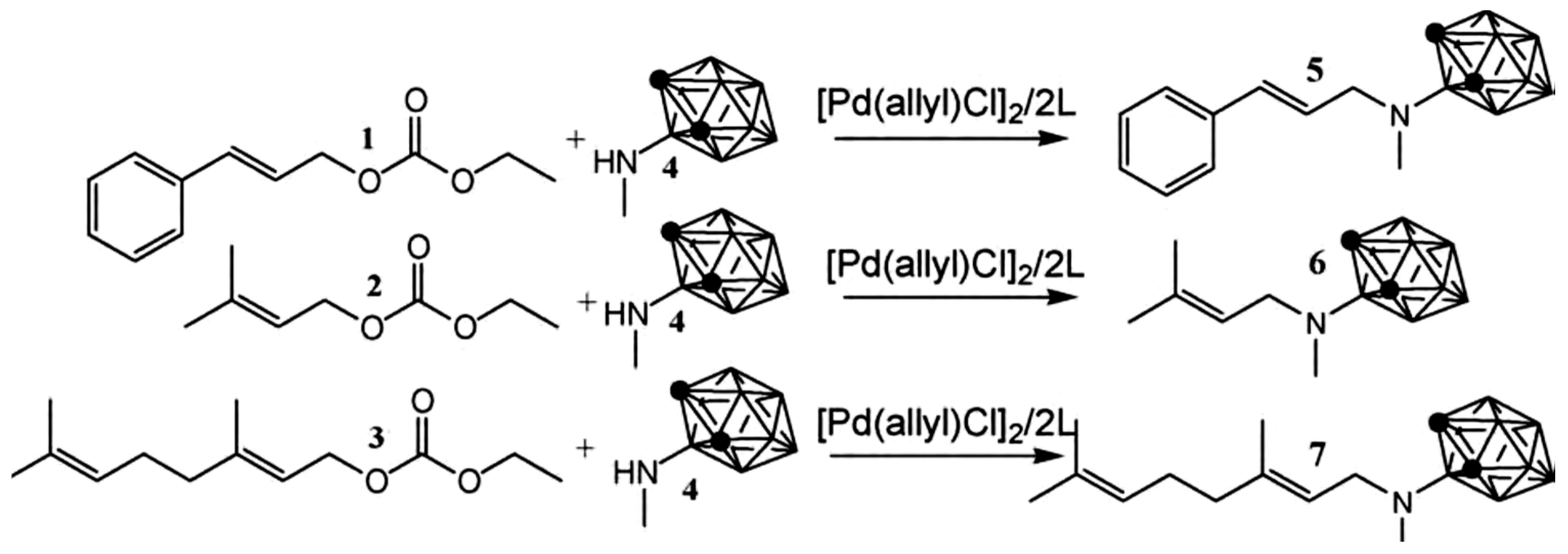{kind=link}
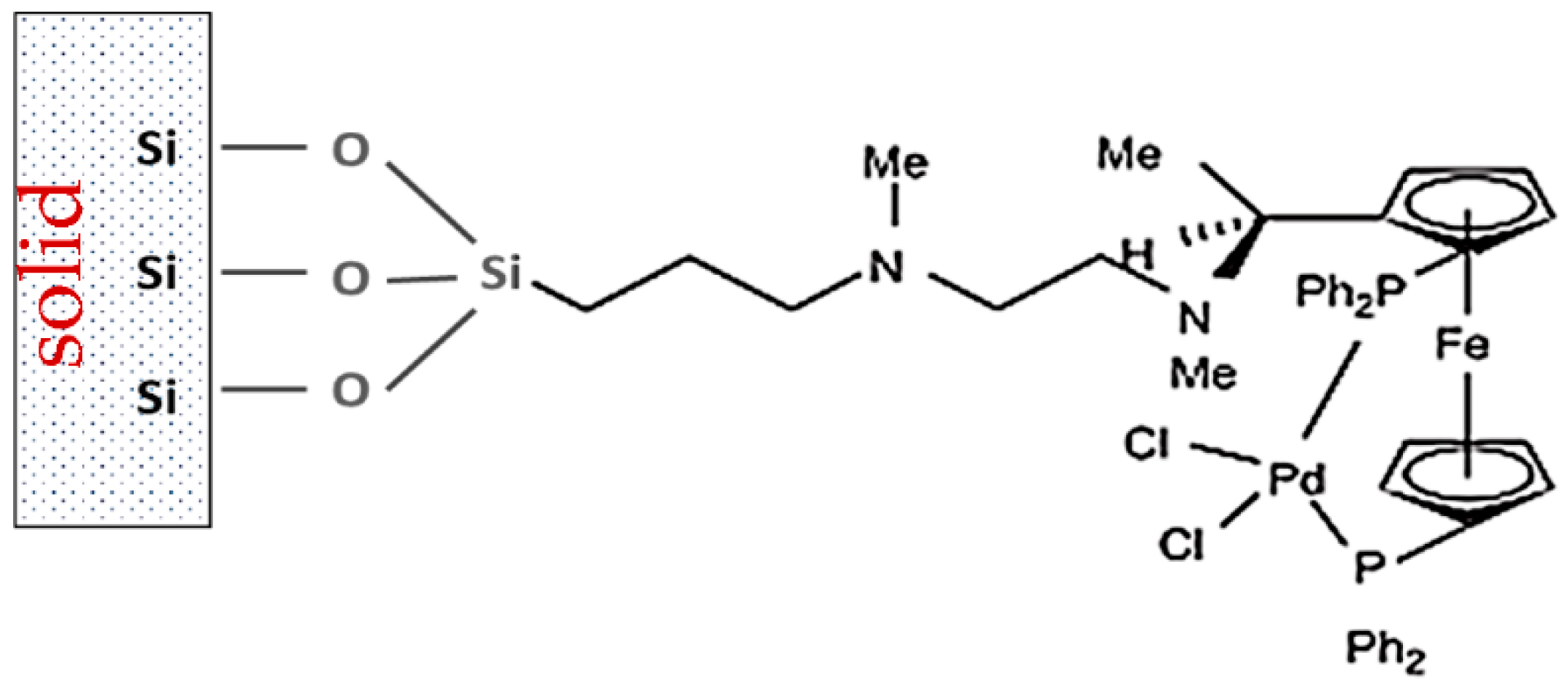{kind=link}
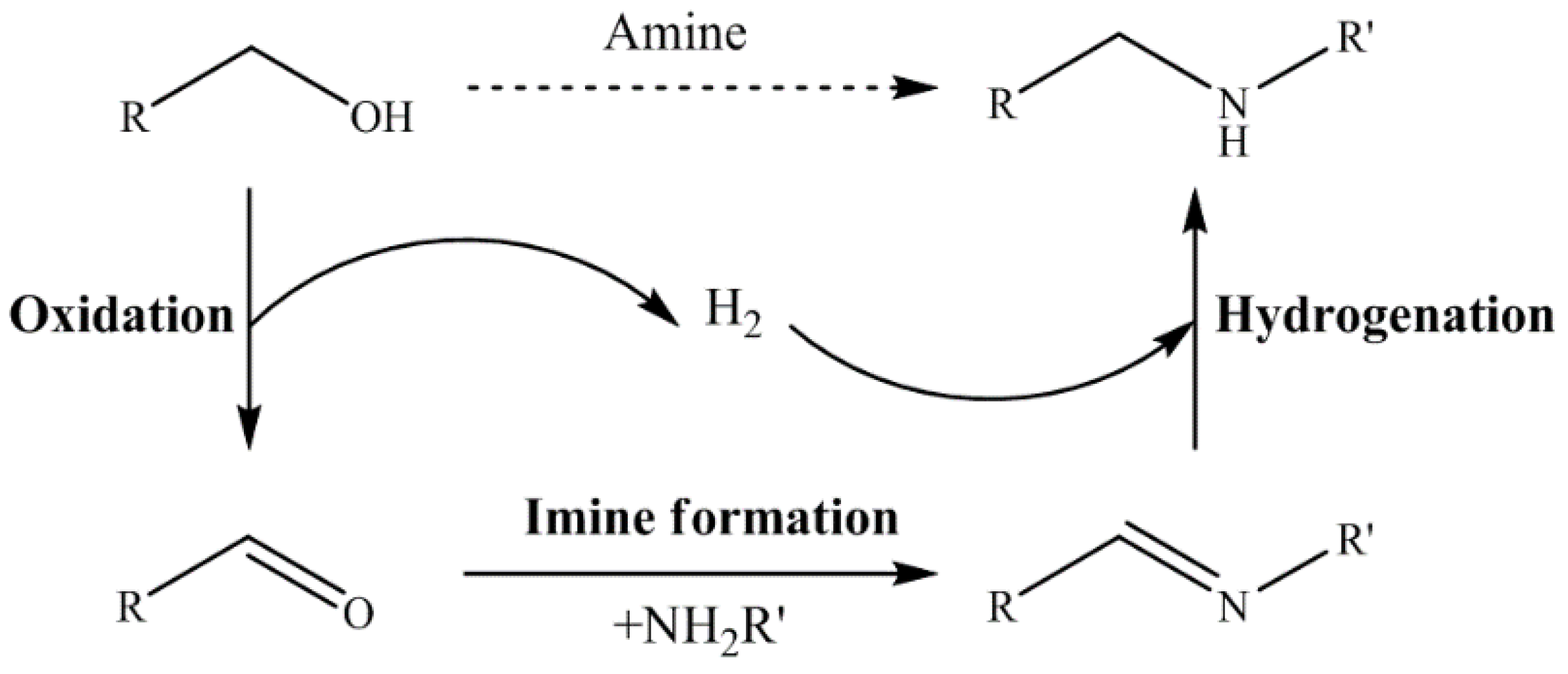{kind=link}
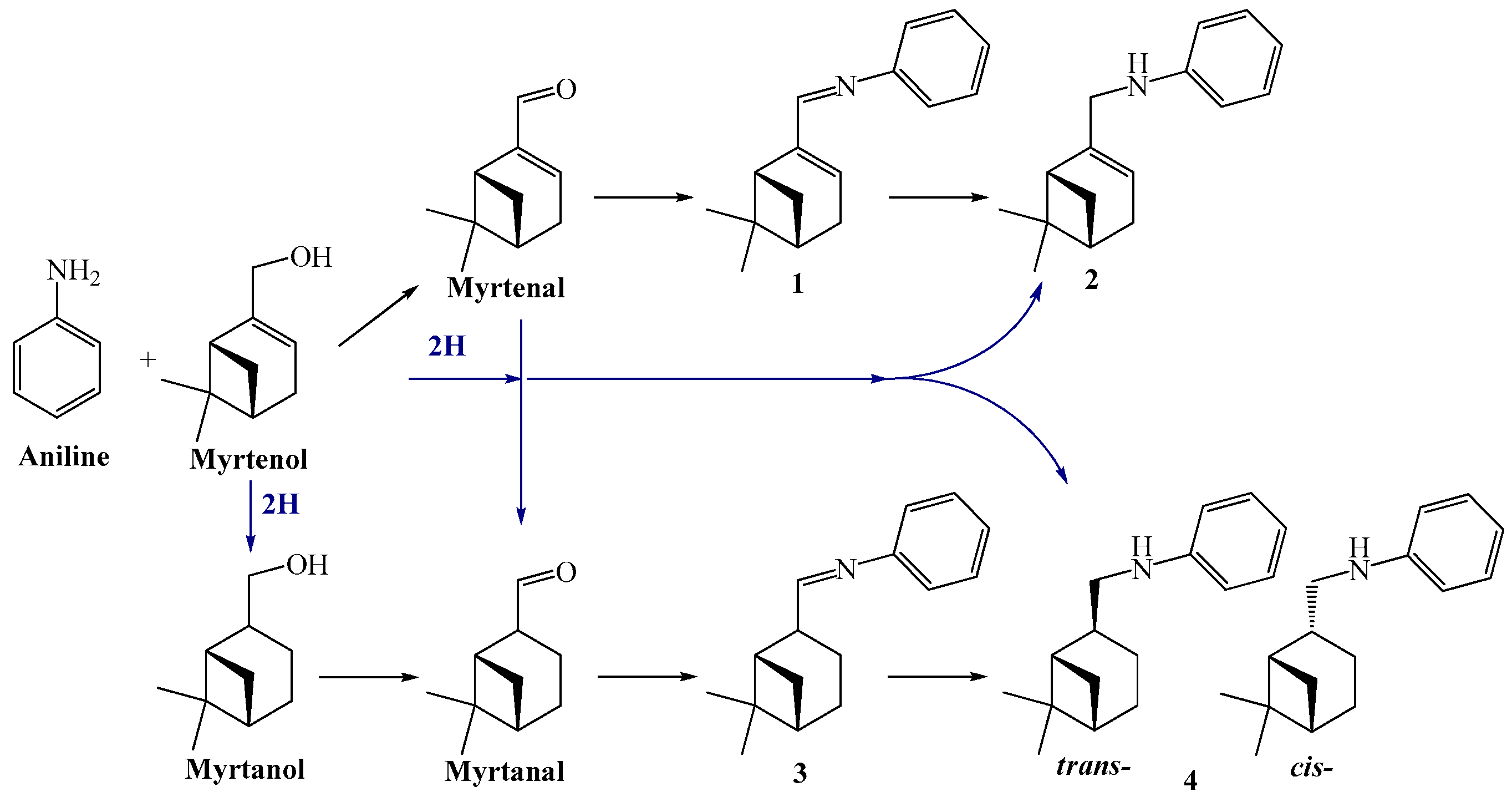{kind=link}
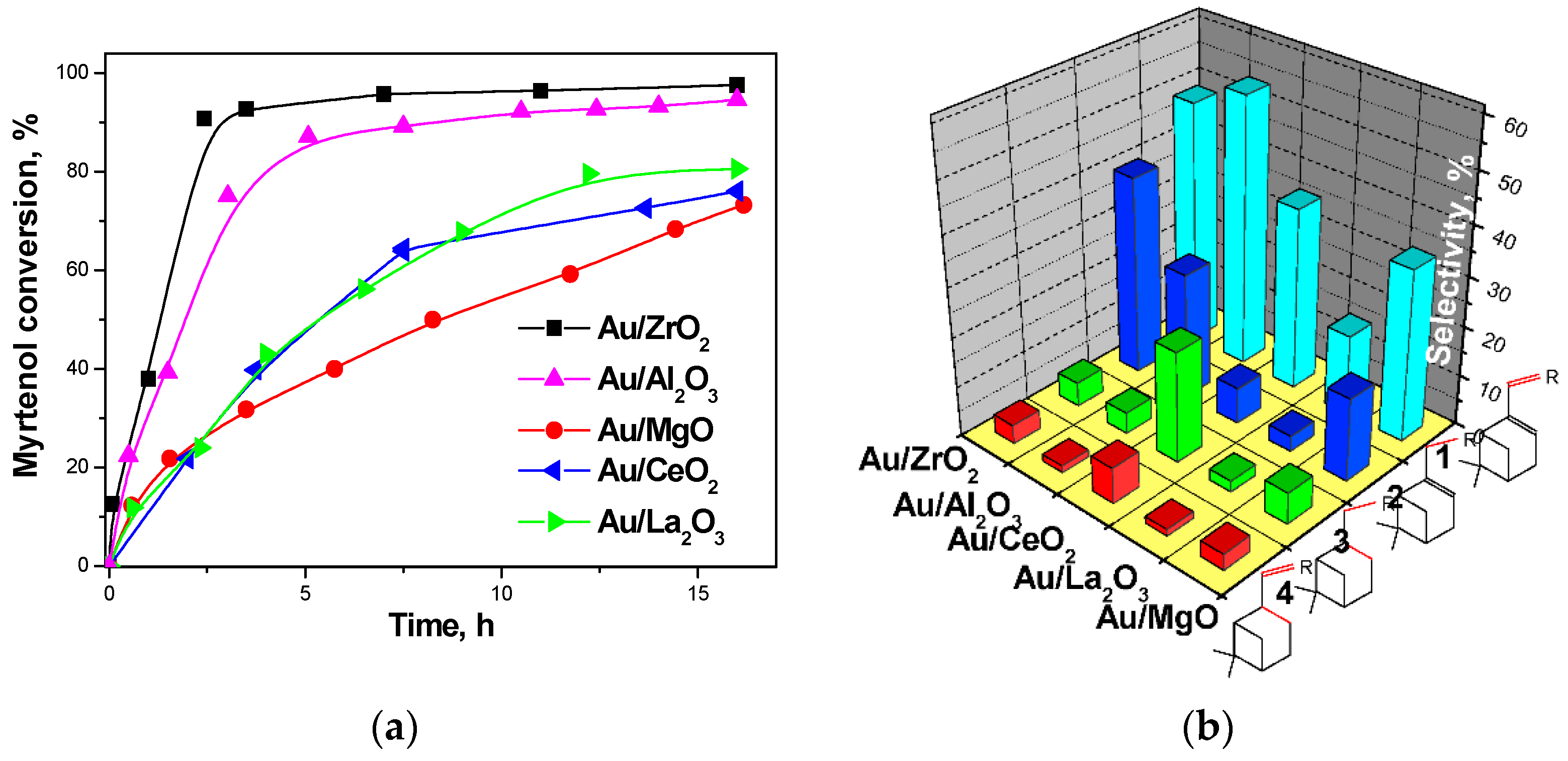{kind=link}
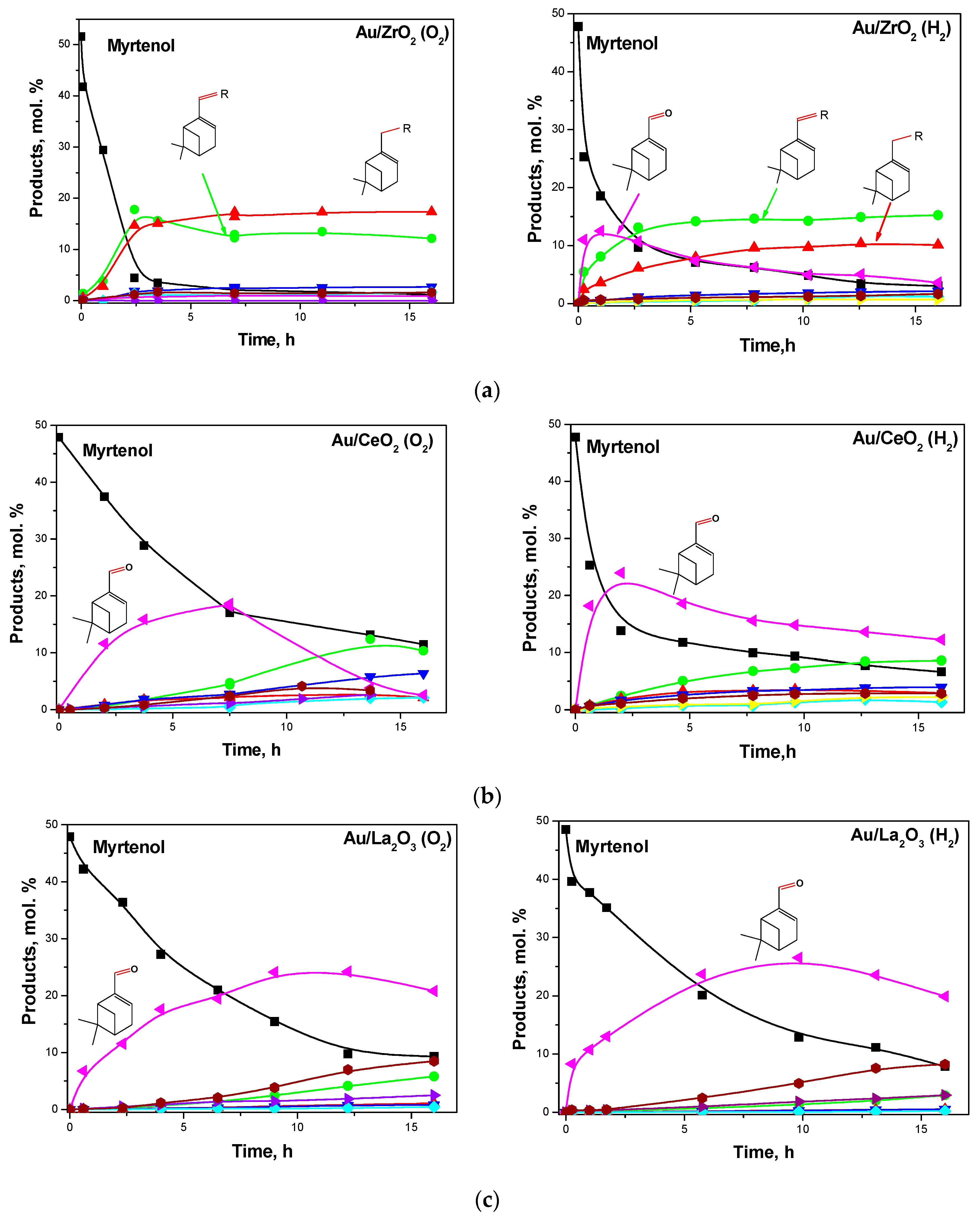{kind=link}
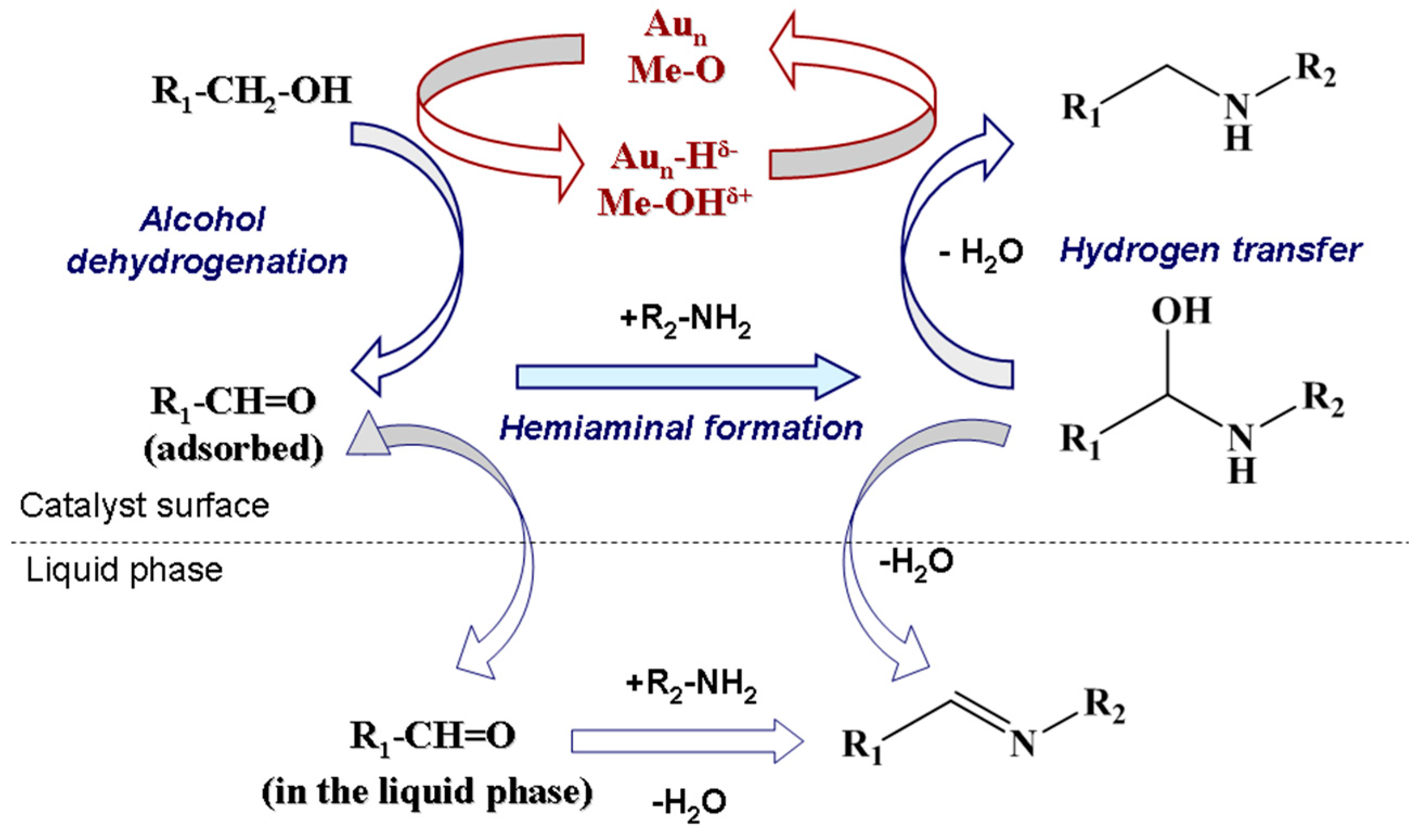{kind=link}
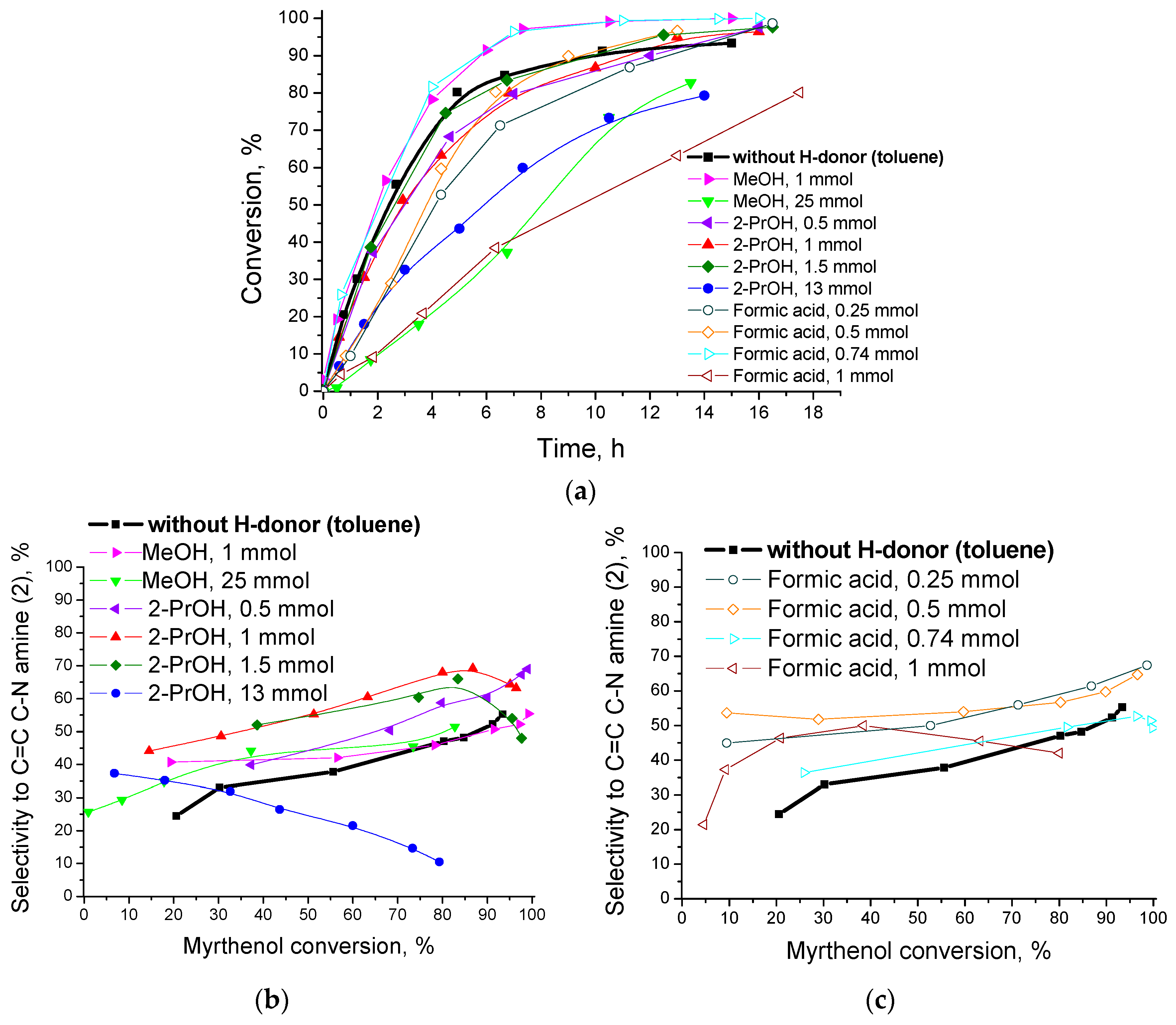{kind=link}
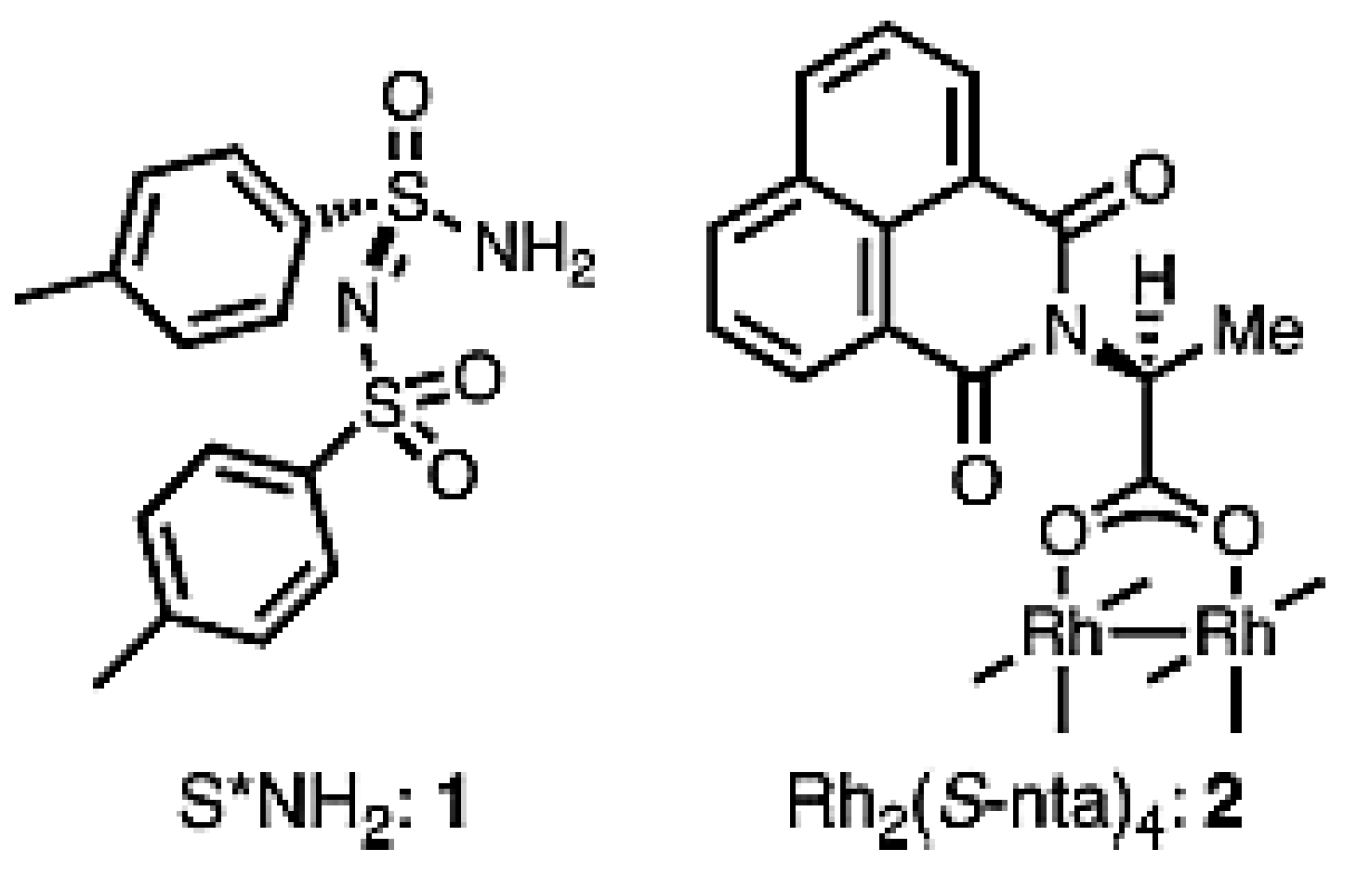{kind=link}
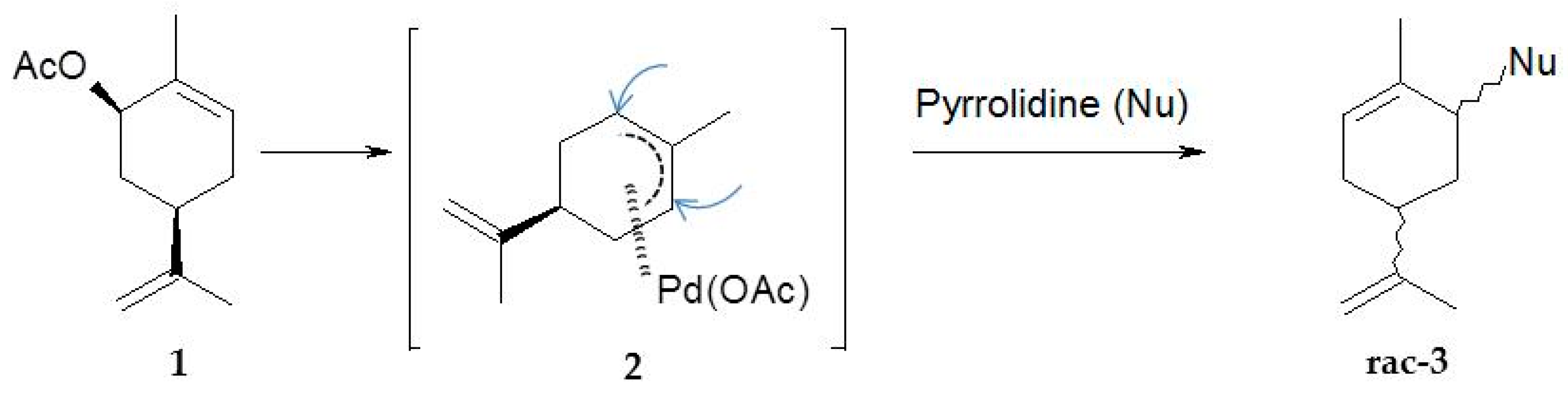{kind=link}
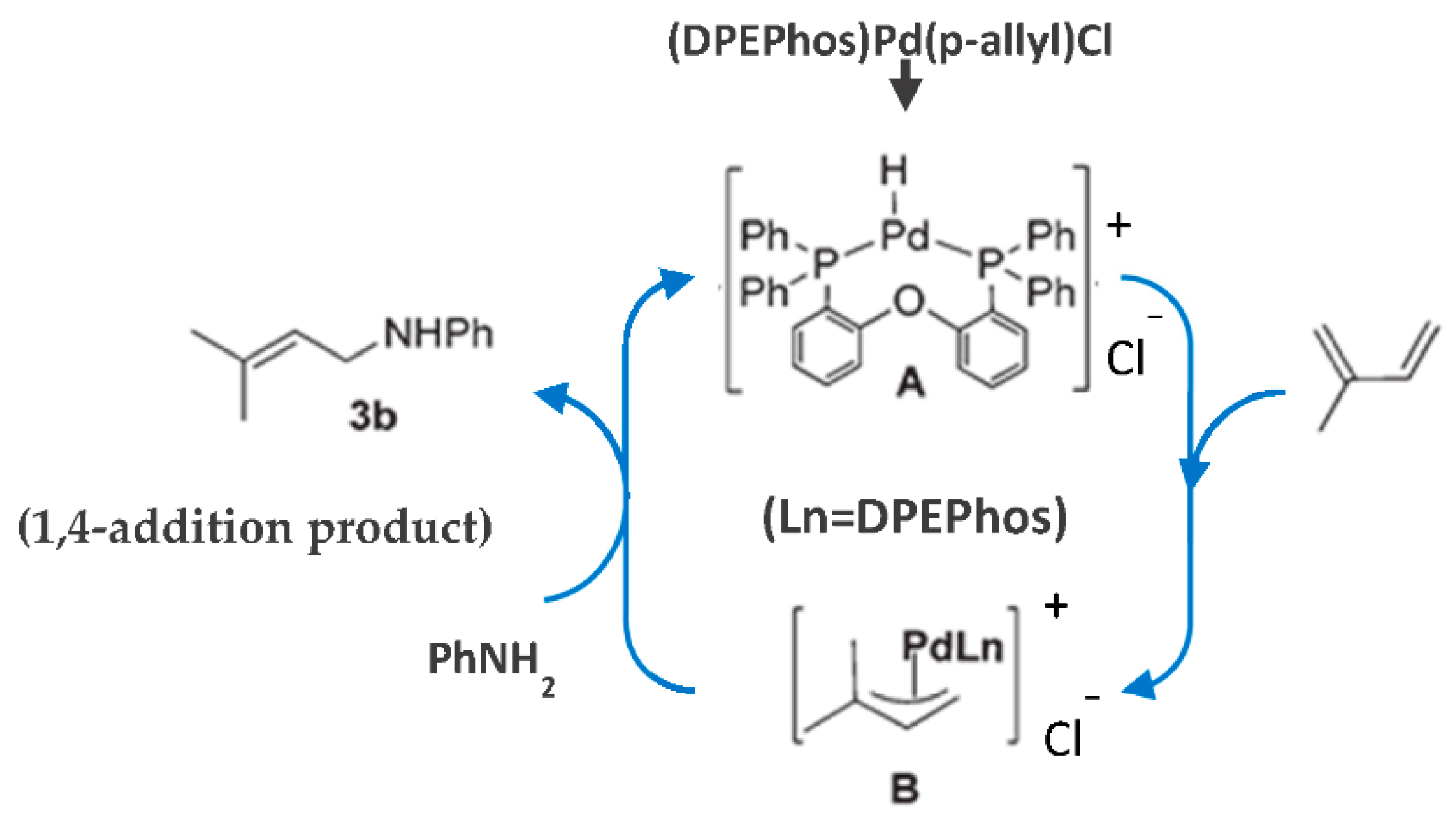{kind=link}
Table 1.
Hydroaminomethylation of eugenol (1, Figure 14) with di-n-butylamine: ligand effect a. Adapted from [79].
| Ligand | P/Rh b | Con. (%) | Product Distribution (%) | Regioselectivity (%) c | |||||
|---|---|---|---|---|---|---|---|---|---|
| 2 | Aldehydes | Enamines | Amines | 9 | 10 | 11 | |||
| None | 0 | 100 d | 4 | 1 | 0 | 90 | 61 | 33 | 6 |
| PPh3 | 2 | 100 | 0 | 24 | 3 | 73 | 96 | 4 | 0 |
| PPh3 | 10 | 100 | 0 | 32 | 10 | 58 | 94 | 5 | 1 |
| NAPHOS | 2 | 34 | 3 | 34 | 56 | 7 | >99 | 0 | 0 |
| NAPHOS e | 2 | 100 | 10 | 21 | 5 | 64 | 97 | 3 | 0 |
a Conditions: 1 (10 mmol); di-n-butylamine (10 mmol); [Rh(cod)(μ-OMe)]2 (5.0 × 10−3 mmol), toluene (30 mL), 4.0 MPa (CO:H2 = 1:3), 100 °C, 24 h. For products, the value “zero” means not observed or <0.5%. b Phosphorus/Rhodium atomic ratio. c Related to the sum of amines (9 + 10 + 11). d 5% of hydrogenation of substrate. e 120 °C.
Table 2.
Hydroaminomethylation of eugenol (1, Figure 14) with di-n-butylamine: effect of added acid a. Adapted from [79].
| Ligand | P/Rh b | Acid | T (°C) | Con. (%) | Product Distribution (%) | Regioselectivity (%) c | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | Aldehydes | Enamines | Amines | 9 | 10 | 11 | |||||
| PPh3 | 2 | None | 120 | 100 d | 0 | 17 | 0 | 83 | 96 | 4 | 0 |
| PPh3 | 2 | H2SO4 d | 120 | 86 | 13 | 76 | 9 | 2 | 94 | 6 | 0 |
| PPh3 | 2 | HOTs d | 120 | 100 | 0 | 23 | 0 | 75 | 89 | 11 | 0 |
| PPh3 | 2 | HOTf d | 120 | 100 | 1 | 11 | 0 | 88 | 84 | 14 | 2 |
| NAPHOS | 2 | HOTf e | 100 | 100 | 5 | 9 | 0 | 86 | 99 | 1 | 0 |
| NAPHOS | 20 | HOTf e | 100 | 100 | 2 | 5 | 0 | 93 | 99 | 1 | 0 |
| NAPHOS | 40 | HOTf e | 100 | 100 | 2 | 3 | 0 | 95 | 98 | 2 | 0 |
a Conditions: 1 (10 mmol); di-n-butylamine (10 mmol); [Rh(cod)(μ-OMe)]2 (5.0 × 10−3 mmol), toluene (30 mL), 4.0 MPa (CO:H2 = 1:3), 24 h. For products, the value “zero” means not observed or <0.5%. b Phosphorus/Rhodium atomic ratio. c Related to the sum of amines (9 + 10 + 11). d 1.0 mmol. e 2.0 mmol.
Table 3.
Overview of hydroamination investigations *. Adapted from [33].
Table 3.
Overview of hydroamination investigations *. Adapted from [33].
| Catalyst/Conditions a | Amine | Geranylamine Selectivity (%) | Amine Yield (%) | TON c | TOF d (h−1) |
|---|---|---|---|---|---|
| 20.0 mol% BuLi, 50 °C, 4 h | HNEt2 | 95 | 77–83 | 4 | 1.04 |
| 33.3 mol% Na/16.7 mol% naphthalene, 20 °C, 1 h | HNEt2 | 80 | 53 | 2 | 1.60 |
| 36.0 mol% Li, 50 °C, 20 h | HNiPr2 | n.m. | 80 | 2 | 0.11 |
| 100 mol% Li, 50 mol% naphthalene, | HNEt2, morpholine, pyrrolidine, piperidine, HNnBu2 | geranylamine | 72 b | 1 b | 0.18 b |
| 100 mol% TMDAP, 20 °C, 4 h | 88 b | ||||
| 10 mol% BuLi, 50 °C, 2.5 h | HNMe2 | 88 | 79 | 8 | 3.18 |
| 1.7 mol% Na, 50 °C, 4 h | HNMe2, HNEt2, HNiPr2, HNnBu2 | 90 b | 83 b | 49 b | 12.21 b |
| 3.6 mol% Li, 25 °C, 72 h | HNMe2, HNEt2, HNiPr2, HNnBu2, piperidine, 2-methylpi- | 96 b | 87 b | 24 b | 0.34 b |
| peridine, 2,6-dimethyl-piperidine | |||||
| 36.0 mol% Li, 55 °C, 5 h | HNEt2 | 92 | 74–77 | 2 | 0.43 |
| 15.0 mol% BuLi, 55 °C, 12 h | HNEt2 | 95 | 85 | 6 | 0.47 |
| 0.1 mol% [RhCl(cod)]2, 2.3 mol% TPPTS, 100 °C, 21.5 h (biphasic) | morpholine | 53 | 59 | 590 | 27.44 |
| 0.2 mol% Pd(CF3CO2)2, 1.6 mol% DPPB, 100 °C, 4 h | morpholine | 57 | 98 | 490 | 123.50 |
a TMDAP: N,N,N′,N″-tetramethyldiaminopropane, TPPTS: triphenylphosphine trisulfonate. b According to diethylamine. c TON: turn over number. d TOF: turn over frequency. * References to original papers are provided in [33].
Table 4.
Secondary terpene alcohol amination with [Ru3(CO)12]/L9 a. Adapted from [125].
Table 4.
Secondary terpene alcohol amination with [Ru3(CO)12]/L9 a. Adapted from [125].
| Substrate | Time (h) | T (°C) | Conversion b (%) | Selectivity c,d (%) | Ketones (%) | Product |
|---|---|---|---|---|---|---|
Menthol | 21 | 150 | 25.3 | 18.0 | 82 | Menthylamine |
| 21 | 170 | 31.6 | 70.8 | 29.2 | ||
| 63 | 150 | 32.7 | 24.3 | 75.7 | ||
Carveol | 21 | 150 | 67.3 | 74.7 | Carvylamine | |
| 21 | 170 | 99.1 | 55.2 | |||
| 65.5 | 150 | 82.0 | 56 | |||
| 42 | 170 | 99.3 | 50.4 | |||
Verbenol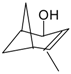 | 21 | 150 | 93.5 | 39.7 | 44.2 | Verbenylamine |
| 21 | 170 | 96.4 | 27.7 | 62.2 | ||
| 63 | 170 | 98.8 | 25.0 | 47.7 | ||
Borneol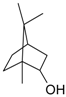 | 21 | 150 | 58.5 | 73.2 | Bornylamine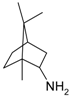 | |
| 21 | 170 | 76.8 | 83.9 | |||
| 65.5 | 150 | 72.2 | 78.7 | |||
| 63 | 170 | 79.1 | 81.2 | |||
Fenchol | 21 | 150 | 50.6 | 0 | 34.8 | Fenchylamine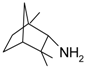 |
| 21 | 170 | 59.2 | 0 | 46.0 | ||
| 63 | 150 | 54.5 | 0 | 38.2 | ||
| 117 | 170 | 63.9 | 0 | 44.3 |
a [Ru3(CO)12] (0.066 mmol), L9 (2 mmol), substrate (10 mmol), tert-amyl alcohol (13.3 mL), NH3 (6 mL, 234 mmol), 150 and 170 °C. b Conversion determined from GC analysis on the basis of alcohol consumption and amine production. c Total of all primary amines. d Main byproducts are primary imines, amides, and ketones. ![Catalysts 08 00365 i011]() .
.
 .
.
Table 5.
Primary terpene alcohol amination with [Ru3(CO)12]/L9 a. Adapted from [125].
Table 5.
Primary terpene alcohol amination with [Ru3(CO)12]/L9 a. Adapted from [125].
| Substrate | Time (h) | T (°C) | Conversion b (%) | Selectivity c,d (%) | Product |
|---|---|---|---|---|---|
Citronellol | 21 | 150 | 33.8 | 64.8 | Citronellylamine |
| 21 | 170 | 47.4 | 61.2 | ||
| 63 | 150 | 47.9 | 71.4 | ||
| 152 | 170 | 82.9 | 65.9 | ||
Geraniol | 21 | 150 | 98.9 | 79.9 | Geranylamine |
| 21 | 170 | 99.3 | 71 | ||
| 63 | 150 | 99.7 | 70.3 | ||
| 42 | 170 | 99.4 | 67.4 | ||
Nerol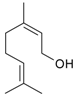 | 21 | 150 | 66.7 | 92.9 | Nerylamine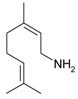 |
| 21 | 170 | 85.5 | 75.1 | ||
| 120 | 150 | 85.8 | 87 | ||
| 49 | 170 | 96 | 69.8 | ||
Farnesol | 21 | 150 | 83.8 | 77.4 | Farnesylamine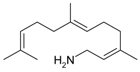 |
| 21 | 170 | 94.8 | 73.8 | ||
| 95 | 150 | 93,2 | 72.5 | ||
| 45 | 170 | 99.4 | 76.1 |
a [Ru3(CO)12] (0.066 mmol), L9 (0.2 mmol), substrate (10 mmol), tert-amyl alcohol (13.3 mL), NH3 (6 mL, 234 mmol), 150 and 170 °C. b Conversion determined from GC analysis on the basis of alcohol consumption and amine production. c Total of all primary amines. d Main byproducts are primary imines, amides, and ketones.
Table 6.
Pd(0)-catalyzed amination of nerolidyl acetate (3). Adapted from [91].
Table 6.
Pd(0)-catalyzed amination of nerolidyl acetate (3). Adapted from [91].
| Amine | Solvent | Ligand | T (°C) | Molar Ratio Amine/(3) | Yield of (4) (%) a | E:Z |
|---|---|---|---|---|---|---|
| (C2H5)2NH | THF | PPh3 | 20 | 2.0 | 40 | 60:40 |
| (C2H5)2NH | THF | PPh3 | 50 | 2.0 | 14 | 62:38 |
| (C2H5)2NH | IPE | PPh3 | 20 | 2.0 | 48 | 60:40 |
| (C2H5)2NH | IPE | PPh3 | 50 | 2.0 | 8 | 62:38 |
| (C2H5)2NH | Benzene | PPh3 | 20 | 2.0 | 35 | 76:24 |
| (C2H5)2NH | DMF | PPh3 | 20 | 2.0 | 70 | 52:48 |
| (C2H5)2NH | CH2Cl2 | PPh3 | 20 | 2.0 | 52 | 54:46 |
| (C2H5)2NH | (C2H5)2NH | PPh3 | 20 | Excess | 57 | 59:41 |
Reaction conditions: nerolidyl acetate (3) 4 mmol; catalyst 0.02 mmol (5 mol%); solvent 12 mL. a Yields of (4) were isolated yields.
Table 7.
Pd(0)-catalyzed amination of linalyl methylcarbonate (5). Adapted from [91].
Table 7.
Pd(0)-catalyzed amination of linalyl methylcarbonate (5). Adapted from [91].
| Amine | Solvent | Ligand | T (°C) | Molar Ratio Amine/(5) | Yield of (2) (%) a | E:Z |
|---|---|---|---|---|---|---|
| (C2H5)2NH | THF | PPh3 | 20 | 2.0 | 20 | 91:9 |
| (C2H5)2NH | IPE | PPh3 | 20 | 2.0 | 21 | 92:8 |
| (C2H5)2NH | Benzene | PPh3 | 20 | 2.0 | 15 | 94:6 |
| (C2H5)2NH | DMF | PPh3 | 20 | 2.0 | 42 | 86:14 |
| (C2H5)2NH | 1,2-C2H4Cl2 | PPh3 | 20 | 2.0 | 30 | 86:14 |
| (C2H5)2NH | (C2H5)2NH | PPh3 | 20 | Excess | 48 | 91:9 |
| Aniline | THF | PPh3 | 20 | 2.0 | 52 | 100:0 b |
| Aniline | IPE | PPh3 | 20 | 2.0 | 60 | 100:0 |
| N-Me-Aniline | THF | PPh3 | 20 | 2.0 | 50 | 100:0 c |
| N-Me-Aniline | IPE | PPh3 | 20 | 2.0 | 59 | 100:0 |
| Morpholine | THF | PPh3 | 20 | 2.0 | 47 | 100:0 d |
| Morpholine | IPE | PPh3 | 20 | 2.0 | 45 | 100:0 |
| Pyrrolidine | THF | PPh3 | 20 | 2.0 | 32 | 85:15 e |
| Pyrrolidine | IPE | PPh3 | 20 | 2.0 | 49 | 94:6 |
Reaction conditions: linalyl merhylcarbonate (5) 5 mmol, catalyst 0.25 mmol (5 mol%), solvent 12 mL. a Yields of (2) were isolated yields. b The product is N-geranyl aniline. c The product is N-methyl-N-geranyl aniline. d The product is geranyl morpholine. e The product is geranyl pyrrolidine.
Table 8.
Catalytic amination of myrtenyl acetate a (1). Adapted from [124].
Table 8.
Catalytic amination of myrtenyl acetate a (1). Adapted from [124].
| Secondary Amine | Product b | [a]D c | With 2.5 Equiv of Et3N | Without Et3N | ||
|---|---|---|---|---|---|---|
| Yield d (%) | Conv. (%) | Yield d (%) | Conv. e (%) | |||
| Pyrrolidine | 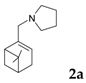 | −4.0 | 86 | 94 | 76 | 93 |
| Morpholine | 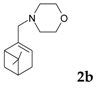 | −18.4 | 90 | 96 | 91 | 95 |
| Me2NH |  | −3.5 | 94 | 100 | 92 | 96 |
a The reaction performed in THF with 2.5 mol % of Pd(dba)2, 5 mol% PPh-and 1.3 equivalents of nucleophile under nitrogen atmosphere at room temperature. b Experiments monitored until no further evolution was encountered. c [a]D determined at: c = 2, CHCl3 for (2a); c = 1.9, CHCl3 for (2b); c = 2, CHCl3 for (2c). d Isolated yield. e Conversions were determined by GC and based on (1).
Table 9.
Ligand influence on the amination of myrtenyl acetate a (1) with morpholine in the presence of Et3N. Adapted from [124].
Table 9.
Ligand influence on the amination of myrtenyl acetate a (1) with morpholine in the presence of Et3N. Adapted from [124].
| Ligand b | Yield c (%) | Conversion d (%) |
|---|---|---|
| None | - | 2 |
| PPh3 | 90 | 96 |
| dppe | 88 | 94 |
| P(o-tolyl)3 | 13 | 22 |
| 2,2′-dipyridyl | 5 | 14 |
a The reaction mixture in THF with 2.5 mol% of Pd(dba)2, 5 mol% of ligand and 1.2 equivalents of morpholine under nitrogen atmosphere at room temperature. b Experiments monitored by GC until no further evolution was encountered. c Isolated yield. d Conversions were determined by GC and based on (1).
Table 10.
Catalytic amination of terpenic allylic esters a. Adapted from [124].
Table 10.
Catalytic amination of terpenic allylic esters a. Adapted from [124].
| Terpenic Allylic Esters | Secondary Amine | Product | Yield b (%) | Conv c (%) |
|---|---|---|---|---|
| Allyl acetate | ||||
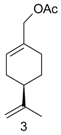 | Pyrrolidine | 4a | 48 | 60 |
| Morpholine | 4b | 48 | 56 | |
| Me2NH | 4c | 54 | 62 | |
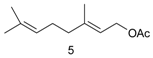 | Pyrrolidine | 6a | 39 | 58 |
| Morpholine | 6b | 40 | 56 | |
| Me2NH | 6c | 42 | 62 | |
| Allyl carbonate | ||||
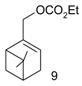 | Pyrrolidine | 2a | 94 | 96 |
| Morpholine | 2b | 94 | 96 | |
| Me2NH | 2c | 96 | 98 | |
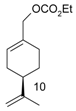 | Pyrrolidine | 4a | 50 | 68 |
| Morpholine | 4b | 52 | 62 | |
| Me2NH | 4c | 62 | 68 | |
 | Pyrrolidine | 6a | 43 | 64 |
| Morpholine | 6b | 44 | 62 | |
| Me2NH | 6c | 47 | 66 | |
a Reaction performed in THF with 2.5 mmol% of Pd(dba)2, 5 mol% dppe and 1.2 equivalents of nucleophile under nitrogen atmosphere at room temperature. b Isolated yield. c Conversions were determined by GC.
Table 11.
Effect of reaction conditions on the content of amine (4) diastereomers and diastereomeric excess value at complete myrtenol conversion. Adapted from [138].
Table 11.
Effect of reaction conditions on the content of amine (4) diastereomers and diastereomeric excess value at complete myrtenol conversion. Adapted from [138].
| Conditions | trans-(%) | cis-(%) | de(%) |
|---|---|---|---|
| 1 bar H2, 8 bar N2, 180 °C | 65 | 35 | 30 |
| 2 bar H2, 7 bar N2, 180 °C | 62 | 38 | 24 |
| 9 bar N2, 180 °C/1 bar H2, 8 bar N2, 180 °C | 80 | 20 | 60 |
Table 12.
Effect of the monoterpene alcohol structure on catalytic behavior of one-pot alcohol amination with aniline. The reaction conditions: T = 180 °C, monoterpene alcohol 1 mmol, aniline 1 mmol, toluene 10 mL, Au/ZrO2 catalyst 92 mg, 9 bar N2. Adapted from [140].
Table 12.
Effect of the monoterpene alcohol structure on catalytic behavior of one-pot alcohol amination with aniline. The reaction conditions: T = 180 °C, monoterpene alcohol 1 mmol, aniline 1 mmol, toluene 10 mL, Au/ZrO2 catalyst 92 mg, 9 bar N2. Adapted from [140].
| Alcohol | R a (mol∙L−1∙h−1) | Time (h) | Alcohol Conversion (%) | Selectivity (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Sec. Amine | Imine | Aldehyde b | ||||||
| 1 c | 2 d | 1 c | 2 d | |||||
| Myrtenol | 2.1 × 10−2 | 2 | 44 | 39 | 5 | 50 | 3 | 0 |
| 8 | 87 | 52 | 6 | 34 | 2 | 0 | ||
| 16 | 98 | 53 | 7 | 33 | 2 | 0 | ||
| Nopol | 2.4 × 10−3 | 8 | 10 | 74 | 0 | 22 | 0 | 0 |
| 16 | 40 | 76 | 0 | 19 | 0 | 0 | ||
| Perillyl alcohol | 3.1 × 10−2 | 2 | 70 | Complicated mixture of the products | ||||
| 8 | 99 | |||||||
| Perillyl alcohol e | 1.3 × 10−2 | 8 | 98 | 1 | 19 | 28 | 11 | 2 |
a Initial reaction rate of alcohol transformation, calculated within the linear part of the kinetic curves. b Selectivity to intermediate aldehyde formed from the corresponding primary or secondary alcohol, respectively. c Selectivity to the corresponding product with unsaturated C=C bond in alcohol structure (bold). d Selectivity to the corresponding product with saturated C–C bond in alcohol structure (italic). e The reaction temperature is 160 °C.
Table 13.
Effect of amine structure on catalytic behavior of one-pot myrtenol amination. The reaction conditions: T = 180 °C, myrtenol 1 mmol, amine 1 mmol, toluene 10 mL, Au/ZrO2 catalyst 92 mg, 9 bar N2. Adapted from [140].
Table 13.
Effect of amine structure on catalytic behavior of one-pot myrtenol amination. The reaction conditions: T = 180 °C, myrtenol 1 mmol, amine 1 mmol, toluene 10 mL, Au/ZrO2 catalyst 92 mg, 9 bar N2. Adapted from [140].
| Amine | R a (mol∙L−1∙h−1) | Time (h) | Alcohol Conversion (%) | Selectivity (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Sec. Amine | Imine | Myrtenal/Myrtanal | ||||||
| 1 b | 2 c | 1 b | 2 c | |||||
| Aniline | 2.1 × 10−2 | 2 | 44 | 39 | 5 | 50 | 3 | 0/0 |
| 8 | 87 | 52 | 6 | 34 | 2 | 0/0 | ||
| 16 | 98 | 53 | 7 | 33 | 2 | 0/0 | ||
| Aniline d | 2.0 × 10−2 | 16 | 98 | 69 | 9 | 10 | 2 | 5/2 |
| 4-Methylaniline | 1.6 × 10−2 | 2 | 34 | 19 | 1 | 73 | 0 | 4/9 × 10−1 |
| 8 | 89 | 37 | 1 | 53 | 0 | 8/2 | ||
| 16 | 98 | 43 | 2 | 49 | 0 | 5/1 | ||
| 4-Methylaniline e | 1.4 × 10−2 | 16 | 95 | 51 | 4 | 30 | 2 | 7/2 |
| 4-Bromoaniline | 3.4 × 10−2 | 2 | 60 | 0 | 14 | 0 | 43 | 14/0 |
| 8 | 99 | 0 | 22 | 0 | 49 | 13/0 | ||
| Benzylamine | 2.3 × 10−2 | 2 | 48 | 36 | 13 | 26 | 14 | 1 × 10−1/0 |
| 8 | 98 | 3 | 35 | 0 | 49 | 0/0 | ||
| Phenethylamine | 3.3 × 10−3 | 8 | 40 | 63 | 0 | 36 | 0 | 0/0 |
| 16 | 53 | 51 | 0 | 47 | 0 | 0/0 | ||
| 24 | 70 | 46 | 1 | 52 | 0 | 0/0 | ||
| Phenethylamine e | 3.0 × 10−3 | 16 | 48 | 69 | 3 | 20 | 0 | 3/2 |
| 3,4-Dimethoxy-phenethylamine | 7.0 × 10−4 | 16 | 13 | 72 | 0 | 27 | 0 | 0/0 |
| 3,4-Dimethoxy-phenethylamine e | 6.9 × 10−4 | 16 | 12 | 80 | 0 | 15 | 0 | 2/1 |
| 3-Aminopyridine | 1.9 × 10−3 | 8 | 19 | 21 | 0 | 74 | 0 | 2/1 |
| 16 | 31 | 20 | 1 | 60 | 0 | 11/3 | ||
| 24 | 44 | 23 | 2 | 58 | 1 | 12/3 | ||
| 3-Aminopyridine e | 2.0 × 10−3 | 16 | 33 | 30 | 2 | 49 | 0 | 14/2 |
a Initial reaction rate of alcohol transformation, calculated within the linear part of the kinetic curves. b Selectivity to the corresponding product with unsaturated C=C bond in myrtenol structure (bold). c Selectivity to the corresponding product with saturated C–C bond in myrtenol structure (italic). d The result obtained in [140] adding 0.5 mmol of 2-propanol into initial reaction mixture. e 0.5 mmol of 2-propanol was added to initial reaction mixture.
Table 14.
Allylic amination of terpenes with sulfonimidamide S*NH2 (1 in Figure 28) a. Adapted from [143].

| Product | Yield | d.r. b | Product | Yield | d.r. b |
|---|---|---|---|---|---|
 | 91 | 39:1 | 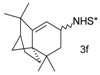 | 85 | |
 | 71 | 49:1 | 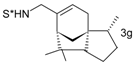 | 34(63) c | |
 | 73 | 49:1 | 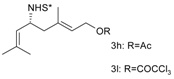 | 90 (3h) 89 (3I) | 19:1 >20:1 |
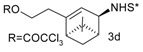 | 98 | >20:1 | 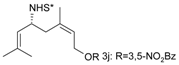 | 81 | 5:1 |
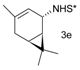 | 27 | 10:1 |
a Reaction conditions: terpene (0.2 mmol) in a 3:1 mixture of 1,1,2,2-tetrachloroethane/MeOH at −35 °C. b The diastereomeric ratios have been determined by 1H NMR or HPLC. c Yield in parentheses obtained using 5 equiv of substrate. (3a, 3b)—enantiomers of α-pinene, (3c)—limonene, (3d)—nopol trichloroacetate, (3e)—carene.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
L. Simakova, I.; V. Simakov, A.; Yu. Murzin, D. Valorization of Biomass Derived Terpene Compounds by Catalytic Amination. Catalysts 2018, 8, 365. https://doi.org/10.3390/catal8090365
AMA Style
L. Simakova I, V. Simakov A, Yu. Murzin D. Valorization of Biomass Derived Terpene Compounds by Catalytic Amination. Catalysts. 2018; 8(9):365. https://doi.org/10.3390/catal8090365
Chicago/Turabian StyleL. Simakova, Irina, Andrey V. Simakov, and Dmitry Yu. Murzin. 2018. "Valorization of Biomass Derived Terpene Compounds by Catalytic Amination" Catalysts 8, no. 9: 365. https://doi.org/10.3390/catal8090365
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.