Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals

1
Department of Chemistry, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece
2
Chemical Process and Energy Resources Institute, Centre for Research and Technology Hellas, 57001 Thessaloniki, Greece
*
Author to whom correspondence should be addressed.
Catalysts 2019, 9(1), 43; https://doi.org/10.3390/catal9010043
Submission received: 31 October 2018
/
Revised: 10 December 2018
/
Accepted: 10 December 2018
/
Published: 4 January 2019
(This article belongs to the Special Issue Solid Catalysts for the Upgrading of Renewable Sources)
Abstract
:Lignocellulosic biomass is an abundant renewable source of chemicals and fuels. Lignin, one of biomass main structural components being widely available as by-product in the pulp and paper industry and in the process of second generation bioethanol, can provide phenolic and aromatic compounds that can be utilized for the manufacture of a wide variety of polymers, fuels, and other high added value products. The effective depolymerisation of lignin into its primary building blocks remains a challenge with regard to conversion degree and monomers selectivity and stability. This review article focuses on the state of the art in the liquid phase reductive depolymerisation of lignin under relatively mild conditions via catalytic hydrogenolysis/hydrogenation reactions, discussing the effect of lignin type/origin, hydrogen donor solvents, and related transfer hydrogenation or reforming pathways, catalysts, and reaction conditions.

1. Introduction
The projected depletion of fossil fuels and the deterioration of environment by their intensive use has fostered research and development efforts towards utilization of alternative sources of energy. Biomass from non-edible crops and agriculture/forestry wastes or by-products is considered as a promising feedstock for the replacement of petroleum, coal, and natural gas in the production of chemicals and fuels. The EU has set the target of 10% substitution of conventional fuels by biomass-derived fuels (biofuels) by 2020, and USA of 20% substitution by 2030 [1,2,3].
Lignocellulosic biomass consists mainly of cellulose, hemicellulose and lignin, all of which can be converted into a wide variety of platform chemicals that can be further transformed to fuels, engineering polymers, pharmaceuticals, cosmetics, etc. (Figure 1). Cellulose is a linear polymer consisting of glucose molecules linked with β-1,4-glycosidic bonds and hemicellulose is branched polysaccharide composed of C5 and C6 sugars [4]. Lignin is an amorphous polymer with p-coumaryl, coniferyl, and sinapyl alcohols being its primary building units. Lignocellulosic biomass can be derived from hardwoods (beech, birch, poplar, etc.), softwoods (pine, spruce, cedar, etc.), grasses (switchgrass, miscanthus, etc.), as well as various agricultural byproducts/wastes (straws, husks, bagasse, etc.). The percentage of cellulose, hemicellulose and lignin in lignocellulosic biomass depends on the nature of the source as well as on the type of the individual member, i.e., hardwood vs. softwood and poplar vs. beech within hardwoods. A number of pretreatment methods have been proposed for the selective isolation of each biomass component. These include physical methods such milling [5,6,7], sometimes combined with H2SO4, chemical methods such as acid (H2SO4, HCl, H3PO4), alkaline (NaOH), organosolv, ozone and oxidative treatment [6,8,9,10,11] and physicochemical such as ammonia fiber, SO2 and steam explosion [6,12,13,14], wet oxidation [15] and hydrothermal methods [16,17]. The isolated fractions in the form of carbohydrate or phenolic biopolymers of varying molecular weight, functionality, particle size and other physicochemical characteristics, can be utilized as such in polymer composites, pharmaceutical formulations, etc. [18,19,20] Furthermore, the downstream selective depolymerization of these biopolymers to their primary building units, i.e., glucose, xylose, alkoxy-phenols, etc., and their consequent transformation to a wide variety of platform chemicals and eventually to final products, may offer even higher value to biomass valorization, via the “biorefinery” concept. Pyrolysis and hydrogenolysis/hydrogenation [21,22,23,24] represent probably the most studied thermochemical biomass (or its components) depolymerization processes towards the production of valuable compounds with a potential in fuels, chemicals and polymers industry [23,25,26].
The aim of this review is to focus on the heterogeneous catalytic transfer hydrogenation reactions for the depolymerization of various types of lignins, including technical lignins deriving from established industrial processes, i.e., kraft, soda or lignosulphonate lignin from the pulp and paper or related industries, as well as enzymatic/acid hydrolysis and organosolv lignins as part of the 2nd generation bioethanol production process.
2. Lignin Chemistry
2.1. Lignin Structure and Isolation
Lignin is an amorphous polymer formed by the polymerization of p-coumaryl, coniferyl and sinapyl alcohols via the phenylpropanoid pathway [28]. The structures of the three monolignols, being phenylpropene rings with one (coniferyl), two (sinapyl) or no (p-coumaryl) methoxy-substituents, are shown in Figure 2. Coniferyl alcohol (G units) is the main building block of softwood lignins with up to ca. 90% content, whereas hardwood lignins contain, in addition to coniferyl units, increased amounts of sinapyl alcohol (S units), reaching 50–75%. Grass lignin is also composed of coniferyl and sinapyl alcohol units, exhibiting also traces of p-coumaryl alcohol (H units) [29,30].
The building blocks of lignin are linked via ether or carbon-carbon bonds formed between the aliphatic chain of monolignols and the aromatic moieties. The most dominant linkage is the β-O-4 aryl ether between the β-carbon of the aliphatic chain and the O-atom from the aromatic moiety, with 45–50% abundance in softwood and 60–62% in hardwood [29,31,32]. Other linkages appearing in lignin are β-β (resinol), β-5 (phenylcoumaran), β-1 (spirodienone), α-O-4, 4-O-5 (diaryl ether), α-O-γ, 5-5 (bisphenyl) and dibenzodioxocin [29,31,32,33]. Representative schematic representations of softwood and hardwood lignin structures, as well as the dominant linkages, are shown in Figure 3.
The methods of lignin isolation can be classified into two categories based on the solubilization of lignin, as reported by Kim and co-workers [34]: the first category includes the methods in which lignin is isolated as insoluble residue after the solubilization of cellulose and hemicellulose while in the second category lignin is isolated in the process solution leaving cellulose and hemicellulose in the insoluble solids. Each isolation process may result in varying lignin yields with different molecular weight and other properties, and possible contaminations, as can be observed in Table 1.
Kraft lignin is produced by the treatment of wood feedstock with NaOH and Na2S at 170 °C for 2 h [3,33,35]. During kraft pulping, the hydroxide and hydrosulfide anions react with lignin, causing its depolymerizaiton into smaller water/alkali soluble fragments [31]. Besides the depolymerization via the cleavage of aryl ether bonds, introduction of thiol group, stilbene and carbohydrate linkages can occur [33,35]. Additionally, the isolated lignin is contaminated with carbohydrates from hemicellulose and a small amount of sulfur [4]. Kraft pulping is the dominant process and constitutes about 85% of total lignin production and is recognized as by-product in paper/pulp industry [36]. Similar to the Kraft process, soda pulping is more often used for the fractionation of non-woody biomass e.g grass, straw and sugarcane bagasse in the presence of NaOH or NaOH-anthraquinone at 140–170 °C [4,33]. Lignin is partially depolymerized during soda pulping via the cleavage of α- and β-aryl ether bonds, first in phenolic units and finally in non-phenolic units [35]. The resulting lignin is considered to be free of impurities compared to the Kraft lignin.
Another industrial process for the isolation of lignin is the sulfite pulping where the lignocellulosic biomass is digested at 140–170 °C with an aqueous solution of a sulfite or bisulfate salt of Na+, NH4+, Mg or Ca [35]. This process can be carried out in the whole range of pH scale by selecting the appropriate salt. During the sulfite pulping, the linkages between the lignocellulosic compounds as wells as the ether bonds between lignin units can be cleaved by the nucleophilic attack of the sulfite anion [4,35]. As a consequence, sulfonation of the lignin aliphatic chain can occur.
The fractionation of lignocellulosic feedstocks via the organosolv process involves the treatment of biomass in organic solvents at the temperature range of 180–200 °C [4]. In this process a wide variety of organic compounds such as alcohols, ketones, acids, ethers and their mixtures with water have been used as solvents [37,38,39,40,41]. The fractionation can be improved by the addition of inorganic acids (H3PO4, HCl, H2SO4 [29,41,42]. Luterbacher et al. suggested the formaldehyde addition in the organosolv process for the stabilization of lignin during biomass pretreatment [43]. The subsequent hydrogenolysis of the extracted lignin resulted in 47–78% monomers, in contrast to the hydrogenolysis of lignin extracted in absence of formaldehyde which led to only 7–26% monomers. The organosolv pretreatment of biomass [11,44], as well as the recently reported hybrid steam explosion/organosolv process [45], have been proven beneficial for the enzymatic saccharification of the remaining cellulose, while at the same time achieving high yields of recovered lignin of relatively low molecular weight and high purity [11,33,45].
2.2. Lignin Valorization
The chemical structure and composition of lignin offer numerous exploitation opportunities towards the production of a vast variety of valuable products. For example, lignin itself can be used either directly without modification or after chemical modification in the polymer industry. One of the main applications of lignin is the substitution of phenol in the phenol-formaldehyde resins, without modifying the properties of the final product. Furthermore, lignin can be mixed with polymers such as polyolefins, polyesters and polyurethanes in the form of blends, copolymers and composites for the production of eco-friendly plastics with improved properties [19,46,47]. After chemical modifications lignin can be also added in epoxy resins. Another possible exploitation of lignin, is the thermochemical conversion to carbon functional materials [48] and chemicals for pharmaceutical applications [49].
In addition to utilizing lignin as such, the platform chemicals/monomers, i.e., phenolics, aromatics, alkanes that derive from various depolymerization processes may lead to the production of even higher added value fuels, chemicals and products, usually via more controlled selective catalytic reaction pathways and related processes. Of course, the economics and sustainability of the integrated technology and the final products depend greatly on the effectiveness of the initial depolymerization process. The main thermochemical processes for lignin depolymerization can be divided into three groups based on the temperature/energy requirements. i.e., pyrolysis and more specifically fast pyrolysis leading mainly to the production of bio-oil (relatively high temperature/energy, ca. 400–700 °C), hydrotreatment or hydroprocessing in the absence of solvents (moderate temperatures, ca. 350–450 °C) and liquid phase depolymerization comprising various acid/base and reductive/oxidative reactions (relatively low temperatures, ca. ≤ 400 °C) [29]. The “lignin-first” process is a relatively new strategy that applies directly on the lignocellulosic biomass and provides efficient lignin solubilization and depolymerization in a single step/reactor, as described below.
In fast pyrolysis, lignin is heated up to 400–700 °C under high heating/cooling rates in the absence of oxygen, with or without catalyst [34,50]. The main products of no-catalytic, thermal fast pyrolysis of lignin are bio-oil (containing substituted alkoxyphenols and few aromatics), char and gases (mainly CO, CO2, CH4). Despite being a high temperature/energy process that could lead to uncontrolled depolymerization and breaking of C-O and C-C bonds, in a recent work of Lazaridis et al. it has been shown on the basis of 2D HSQC NMR results that the composition profile in terms of G- or S-units of the parent lignin is “transferred” to the composition of the produced thermal pyrolysis lignin bio-oil [51]. On the other hand, the catalytic fast pyrolysis of lignin where the primary thermal pyrolysis vapors/products are in situ converted to less oxygenated products via dehydration, decarbonylation, dealkoxylation, cracking and aromatization reactions, may provide bio-oils with substantially altered composition, containing mainly alkyl-phenols, mono-aromatics (BTX) and naphthalenes, depending on the physicochemical characteristics of the catalysts [30,51,52]. Gasification is also an important thermochemical process, widely studied with biomass as feedstock, showing also potential for lignin valorization via synthesis gas production or hydrogen production and utilization [29]. The ratio of the produced gases (H2, CO, CO2 and CH4) is dependent upon process parameters, i.e., temperature, pressure, presence of steam and oxygen, heating rate, and the elemental composition of feed lignin. Due to the sulfur content of technical lignins, the gasification process can also produce H2S.
With regard to the liquid phase depolymerization processes, various catalytic reaction mechanisms have been proposed including acidic, alkaline, oxidative or reductive pathways. Lignin depolymerization under acidic conditions has been mainly studied by the use of metal salts (metal acetates, metal chlorides and metal triflates) with Lewis acid properties [53]. In supercritical water at 400 °C, the yield of products, composed mainly of oxygenated mono-aromatics, was in the range of 6.2–6.9 wt.% with metal (Fe, Cu, Co, Ni, Al; max. yield with FeCl2) chlorides as acidic agent and 7.1–7.9 wt.% with metal (Fe, Cu, Co, Ni; max. yield with Co(Ac)2) acetates. The conversion was increased when the solvent changed from water to ethanol. Formic acid has also been studied as acidic catalyst, in ethanol/water mixtures with relatively low yield of monomer phenolics [54], while H2SO4 was successfully used on hydrolysis lignin (Mw > 20,000 g/mol), yielding ~ 75 wt.% of depolymerized lignin with Mw of 1660 g/mol [55]. With regard to alkaline conditions, when NaOH and KOH were used as homogeneous base catalysts, up to ~20 wt.% yield of oil was obtained, consisting of monomeric phenolic compounds, such as catechol, cresols, syringol and guaiacol. However, the relatively low oil production was attributed to substantial repolymerization reactions [56] Hulterberg and co-workers have studied the base (NaOH)—catalyzed depolymerization of pine kraft lignin in a continuous flow reactor [57]. The optimum conditions for higher production of monomeric phenolic compounds, less char formation and partial deoxygenated dimeric/oligomeric fractions, were determined to be 240 °C for residence time of 2 min, using 5 wt.% lignin loading and NaOH/lignin ratio of ~1 (w/w).
The depolymerization of lignin under oxidative conditions has been studied by the use of H2O2, O2 and nitrobenzene as oxidants and metal oxide catalysts (organometallic, single oxides and perovskites) at low temperatures. The oxidation resulted in the cleavage of lignin C-O and C-C bonds and the production of low molecular weight compounds mainly aldehydes, carboxylic acids and alcohols [30,47,58]. A well-known lignin oxidation process is the vanillin production from Borregaard Company via the catalytic oxidation of lignosulfonates with O2 as oxidizing agent. A detailed description of various lignin depolymerization/valorization processes can be found in previous reviews of Zakzeski et al. [3], Pandey and Kim [34], Li et al. [30], Sun et al. [47], Xu et al. [59] and Schutyser et al. [58]. Apart from the thermochemical depolymerization processes, enzymatic deconstruction of lignin had been also proposed by the use of oxidative enzymes, mainly laccases and peroxidase, from fungi and bacteria [59,60,61].
A more detailed analysis and overview of the reductive depolymerization processes with emphasis on the use of hydrogen donor solvents and catalytic transfer hydrogenation/hydrogenolysis methods is presented in the next sections.
3. Reductive Depolymerization
In contrary to the oxidative depolymerization, reductive depolymerization is taking place in the presence of reducing agents and redox catalysts. Sels and co-workers have reported a categorization of reductive depolymerization process based on hydrogen source and reaction temperature [50,58]. When H2 gas is used as the reducing agent, the process is called hydroprocessing and when hydrogen donor solvents are used, the process is usually called liquid phase reforming. On the other hand, there are many studies using hydrogen donor solvents which refer to transfer hydrogenation instead of reforming, without however elaborating on the possible reaction steps and mechanism. Further subcategories of hydroprocessing, in terms of the reaction temperature, are the mild (<320 °C) and the harsh hydroprocessing (>320 °C). Mild hydroprocessing is performed in liquid phase with solvents and catalysts leading to p-substituted methoxyphenols, while harsh hydroprocessing provides a wider spectrum of products including demethoxylated phenolic species, deoxygenated aromatics, alkanes, catechols and methoxy-phenols. Harsh hydroprocessing may also take place in the absence of solvents. In the solvent-free hydroprocessing of Kraft lignin by the use of NiMo/MgO-La2O3 at 350 °C, 4 h reaction time and 100 bar initial H2 pressure, the conversion was 87% with the highest total monomer yield 26.4 wt.% which included 15.7 wt.% alkyl-phenolics [62]. Similar results were obtained for Alcell lignin by the use of supported noble metals at 400 °C, for 4 h reaction time and initial H2 pressure of 100 bar, with Ru/TiO2, exhibiting the highest catalytic activity providing bio-oil yield 78.3 wt.%, and 9.1 wt.% alkylphenolics, 2.5 wt.% aromatics, and 3.5 wt.% catechols [63]. The bifunctional hydroprocessing with metals supported on acidic materials has been also identified as a separate case, leading to alkane production via additional hydrolysis and dehydration reactions due to the acidic nature of the support, at temperatures below 320 °C [50].
3.1. Reductive Depolymerization of Lignin Model Compounds
Due to the complex nature of lignin, many studies were carried out with model compounds simulating the structure and the bonds of lignin, in order to elucidate the kinetics and pathways for lignin depolymerization. As mentioned in Section 2.1, the most abundant linkage in lignin polymer is the β-O-4 ether bond. The transformation of β-O-4 and 4-O-5 model compounds shown in Figure 4, is discussed below.
Zhu et al. studied the hydrogenolysis of nine compounds containing different functional groups, i.e., benzyl alcohol, aromatic methoxyl and phenolic hydroxyl groups, in methanol and formic acid, acting as hydrogen donors, and Pd/C as the catalyst [64]. In the compounds without a benzyl alcohol group, the β-O-4 linkages were cleaved directly and quickly, in contrast to the compounds that contained benzylic alcohol group, where additional reactions, such as the dehydrogenation of the benzyl alcohol group, hindered the cleavage of β-O-4 bonds to form aromatic monomer products. The aromatic methoxyl and the phenolic hydroxyl groups had no impact on products distribution but the aromatic methoxyl group in both non-phenolic and phenolic compounds seemed to promote the cleavage of β-O-4 bonds, while the phenolic hydroxyl group had a small negative impact on the cleavage of these bonds.
The group of Zhang et al. screened a wide range of monometallic catalysts (Ru, Rh, Pd, Pt, Ir, Ag, Au, Cu, Fe, Co, Ni, Re and Sn) in the hydrogenolysis of 2-phenoxy-1-phenylethanol (130 °C, 10 bar H2) and Ni showed the highest selectivity towards monomers, i.e., 22% with 14% being cyclohexanol, but not the highest conversion (58%). The highest conversion (>99%) was observed by Ru and Rh [65]. The catalytic activity of nickel increased by the addition of Ru, Rh, Pd, Pt and Au leading to full conversion of 2-phenoxy-1-phenylethanol and the NiAu catalyst showed the highest monomer yield 71% (37% cyclohexanol). The optimum ratio Ni:Au was proved to be 7:3. The same group examined further the synergistic activity of NiM (M=Ru, Rh, Pd) and observed that Ni85Ru15 led to complete conversion of 2-phenoxy-1-phenylethanol with 58% monomer yield (32% cyclohexanol) at 130 °C, 10 bar H2 [66].
The activity of Pd-Ni bimetallic nanoparticles supported on ZrO2 was examined in the hydrotreatment of 2-phenoxy-1-phenylethanol with NaBH4 or H2 gas as hydrogen source at 80 °C. The optimum reaction system corresponded to Pd:Ni ratio of 1:8, with H2 gas as hydrogen source at 80 °C for 12 h, yielding 100% formation of cyclohexanol [67]. The presence of NaBH4 induced the initial formation of phenol which was further converted to cyclohexanol at increased NaBH4 amount. The catalytic activity of the same bimetallic Pd-Ni catalysts was also tested in the hydrogenolysis of 2-phenethyl phenyl ether in isopropanol at 210 °C. Under inert atmosphere the conversion of the substrate reached 67% with 60% aromatic yield, whereas the addition of hydrogen gas increased the conversion to 75% with 70% aromatic yield [68].
The hydrogenolysis of 2-phenoxy-1-phenylethanol with HCOOH as hydrogen donor and carbon supported metal catalysts at 80 °C, resulted in the production of acetophenone and phenol [69]. The presence of equivalent amount of base (NH3) promote the reaction and among the M/C (M=Pd, Rh, Ir, Re, Ni) catalysts tested, Pd/C exhibited the highest activity. Changing the hydrogen source from HCOOH to propanol or hydrogen gas, the reaction was not performed. On the other hand, the use of other amines, i.e., ethylamine, diethylamine and p-allylamine as bases instead of NH3, can be successfully applied resulting in >95% conversion. In another work by the same group, the effect of other H-donor solvents was examined under redox-neutral reaction conditions and the NaBH4 was proved to be the best H-donor with 100% substrate (2-phenoxy-1-phenylethanol) conversion [70].
The hydrogenolysis of 1-(2,4-dihydroxyphenyl)-2-(4-methoxyphenoxy)-ethanone (DHPMPE) model compound in water/ethanol and Pt/C as catalyst at 275 °C yielded quantitative amounts of 4-methoxypenol and a mixture of 6-hydroxy-3-coumaranone and 2,4 dihydroxyacetophenone [71]. The authors suggested that the reaction mechanism is based on β-O-4 bond cleavage and the cyclisation of 2,4 dihydroxyacetophenone (produced via hydrogenolysis of the initially formed pentacyclic ether) to 6-hydroxy-3-coumaranone favored by the presence of -OH group in α position of the aromatic ring. In the hydrogenolysis reaction of guaiacylglycerol-β-guaiacylether (GGGE) under the same conditions, the reaction products were 2-methoxyphenol and 4-propyl-2-methoxyphenol [71]. The reactant and product concentration profiles in the conversion of the DHPMPE and GGGE lignin model compounds are shown in Figure 5.
Rinaldi and co-workers examined the effect of solvent type as well as their hydrogen donor activity (protic with/without Lewis basicity and aprotic polar/non polar) in the hydrogenolysis reaction of diphenyl ether, a compound containing 4-O-5 ether bond [72]. The hydrogenolysis reaction was studied at 90 °C under hydrogen pressure (50 bar) by the use of Raney Ni catalyst. Among the protic solvents with Lewis basicity, 2-propanol exhibited the highest conversion 72.7% (83% monomers–40.5% cyclohexanol) and the use of Hex-F-2-PrOH as protic solvent without Lewis basicity led to full reactant conversion but with lower monomers yield (32.7% monomers–16.6% cyclohexanol). Among the aprotic solvents, 2-Me-THF led to 61.1% conversion (74.2% monomers–34.4% cyclohexanol) whereas full conversion was observed by methylcyclohexane (44.6% monomers–22.8% cyclohexanol), despite the fact that aprotic solvents are not hydrogen donor compounds. The ability of the above solvents to act as hydrogen donors was also tested in the absence of hydrogen gas. In the case of 2-propanol, the conversion decreased to 16.6% (100% monomers–49.8% benzene) while with the rest catalysts, the reaction was not performed.
3.2. Lignin-First Strategy
Lignin first strategy refers to the reductive catalytic fractionation of the whole biomass feedstock and not to the conversion of isolated lignins. Actually, this process is a combination of lignocellulosic biomass fractionation and simultaneous conversion of lignin to monomers [50]. The composition and the structure of the primary biomass influences significantly the obtained products. Klein et al. compared the extent of lignin depolymerization during the reaction of birch, poplar and eucalyptus wood in methanol under N2 atmosphere (2 bars), at 200 °C and Ni/C as catalyst, and pointed out that birch wood (hardwood) is a better feedstock, enabling the formation of more lignin-derived products [75]. The preferred use of hardwood in this process is also remarked by Galkin et al. who found the following ranking considering the monophenol yield: birch > poplar > spruce > pine and attributed this effect to the relatively increased abundance of β-O-4 moieties in hardwoods (Figure 6) [76]. When hardwoods were compared with softwoods and grasses, hardwoods showed higher monomer yield and delignification amount, followed by grass and softwood [77]. Furthermore, the ratio between S- and G- units of lignin in biomass feedstock was also correlated with the respective ratio in the final products [77,78].
The catalytic conversion of woody feedstocks under H2 gas pressure to lignin monomers is known from the early 1940s. The group of Hibbert studied the digestion of maple wood and woodmeal in the presence of 1,4 dioxane and Cu-CrO catalyst at 280 °C under H2 pressure and found that the major products were 4-n-propylcyclohexanol and 3-(4-hydroxycyclohexyl)-propanol-1 [79]. Recently, the reductive catalytic fractionation of lignocellulosic feedstocks has been studied by the use of noble metals Pd [80,81,82,83], Pt [80], Ru [77], Rh [80] and transition metals, mainly Ni [75,84,85] as catalysts. A comparative study of the performance of Pd/C and Ru/C catalysts in the reductive processing of birch sawdust under H2 pressure, showed that the two catalysts resulted in almost the same monomer yield and delignification efficiency but in completely different product selectivity and OH-content of lignin oil [86]. Pd/C favored the formation of para-propyl phenolics (75%) while Ru/C the formation of para-propanol phenolics (91%) and increased OH-content. Considering catalyst stability, recovery and reuse, Sels and co-workers studied the performance of Ni-Al2O3 pellets positioned in a basket within a stirred batch reactor [87].
As for the reaction medium, the most common solvents studied are H2O [80,88], alcohols [75,77,83,84,85,88] and mixture of organic solvents with H2O [80,81,82,85]. The polarity of the solvent was shown to influence delignification efficiency and formation of soluble mono-, di- and oligomer phenolics in the Pd/C-catalyzed reductive liquid processing of birch wood. The major phenolic monomers obtained were 4-propanolsyringol and 4-propanolguaiacol. The proposed ranking of solvents was: H2O > MeOH ≈ EG > EtOH > 1-Pr-OH > 1-BuOH >THF >Dioxane>Hexane, although a too polar solvent like water caused significant solubilization of carbohydrates [88]. Song et al. studied the conversion of birch sawdust into 4-propylguaiacol and 4-propylsyringol with a range of alcohols over Ni/C catalyst and the highest conversion was 50% with >90% selectivity of the above products [78]. The authors proposed that lignin is initially fragmented into smaller lignin species with a molecular weight of m/z ca. 1100 to ca. 1600 via alcoholysis reaction, followed by hydrogenolysis of the fragments into the phenolic monomers. Considering the formic acid, there is no need to be added externally in the reaction, as it can be produced in situ from the acetyl groups in lignocellulosic biomass [4,82].
In order to promote the fractionation of lignocellulose, Hensen et al. added Brønsted acid co-catalysts (HCl, H2SO4, H3PO4 and CH3COOH) for a possible replacement of the expensive Al(OTf)3 in the process [89]. In the presence of Pd/C at 180 °C, the best co-catalyst was the HCl, resulting in 44 wt.% lignin monomers from oak sawdust, being similar to the performance of Al(OTf)3 which provided 46 wt.% monomers. In a similar work, by Yan et al., H3PO4 enhanced the efficiency of lignin depolymerization from white birch wood sawdust with H2 in dioxane/water using Pt/C as the catalyst, resulting in 46.4% monomer yield [80]. Among NaOH (alkaline), H3PO4 (acidic) and neutral conditions, H3PO4 resulted in enhanced delignification (85 % with NaOH vs. 96% with H3PO4) of poplar wood in methanol (MeOH) with Pd/C as the catalyst [90]. In the presence of H3PO4, the lignin-derived oil was characterized by a narrow molecular weight distribution and a monomer yield very close to theoretical whereas in the presence of NaOH, the monomer yield was lower due to repolymerization. More detailed description of the lignin-first process can be found in the reviews published by the groups of Samec [4], Barta [47] and Sels [58,91].
3.3. Depolymerization with External Hydrogen Source
Many groups examined the catalytic reductive depolymerization of various types of lignins utilizing external hydrogen source (H2 gas). In these studies, the catalysts play a major role in the conversion and the selectivity of the final products. The most widely used catalysts are Pd/C [81,92,93,94,95,96], Ni/C [93,94,95,96,97], Pt/C [93,94,95,98], Pt/Al2O3 [99], Cu-based porous oxides [100,101], supported NiW and NiMo [102], Ru-based materials [93,94,95,96,103], bimetallic NiM (M = Ru, Rh, Pd) [66], Ni-Au [65,104], WP [105], RuxNi1-x/SBA-15 [106], modified SBA-15 and Al-SBA-15 [107], MoOx/CNT [108], S2O82−-KNO3/TiO2 [109]. Another significant parameter in the reductive depolymerization using molecular H2 is the reaction medium. The most widely used solvents are water [104], alcohols [92,93,94,95,97,98,100,101,102,103,106,107] or mixtures of water with organic compounds [81,99,105] under sub or super- critical conditions. In most cases, the beneficial effect of a hydrogen donor solvent is aimed, as discussed in the following section. The conversion activity can be also enhanced by the addition of other compounds which may facilitate lignin depolymerization and formation of phenolic monomers, such as metal chlorides [92,93] and NaOH [104].
4. Catalytic Hydrogenolysis of Lignins Using Hydrogen Donors
4.1. Kraft Lignins
Ma et al. reported the complete ethanolysis of Kraft lignin by the use of α-MoC1−x/AC (280 °C) and the main products were phenols, phenyl and C6 alcohols and C8-C10 esters [110]. The addition of H2 gas in the reaction instead of the inert atmosphere had a negative impact on the formation of liquid products, increasing the alcohols and decreasing the ester yield. Comparing ethanol, methanol, isopropanol and water, ethanol was the most effective solvent providing more liquid products. In a following paper, the effect of catalyst properties was examined in the depolymerization of lignin under supercritical ethanol [111]. The activity sequence of the catalysts was: carbide > metal > nitride > oxide and the main products were esters, alcohols and aromatics with different ratios depended on the catalyst. Kraft lignin ethanolysis was also studied by the same group using alumina-supported molybdenum catalysts at 280 °C, reduced at different temperatures [112]. The depolymerization resulted in C6 alcohols (mainly hexanol), C8-C10 esters (2-hexanoic acid ethyl ester), monophenols (2-methoxy-4-methyl phenol), benzyl alcohols (O-methyl benzyl alcohol) and arenes (xylene). The increase in the reduction temperature from 500 to 750 °C resulted in a gradual increase of total production yield reaching 1390 mg/g lignin but further increase to 800 °C, decrease the yield. The same trend was exhibited in each group of products. The inferior activity of the material reduced at 750 °C was correlated with the metallic phase of Mo and the lower activity of the material reduced at 800 °C with the collapse of porous structure due to sintering phenomena. The effect of the reaction time was also studied. When the reaction time increased from 4 to 6 h the product yield increased dramatically from 142 to 1390 mg/g lignin but when the time was prolonged to 10 h, the product yield decreased.
The conversion of Kraft lignin into monomeric alkyl phenols was achieved over Cu/Mo-ZSM-5 catalysts at 220 °C using hydrogen produced by reforming and water gas shift reactions taking place in the water/methanol solvent system and in the presence of NaOH which was used for enhanced lignin solubility [113]. If no NaOH, methanol or water was used in the reaction, low conversions were observed, despite the fact that in the presence of methanol or water high selectivity to phenol was observed but low monomeric product yield. The optimum water: methanol ratios for the production of monomeric products were determined to be 1:1 and 3:1. The first ratio (1:1) led to 95.7 wt.% conversion and 70.3% selectivity for phenol, 3-methoxy and 2,5,6 trimethyl phenol. The molecular weight of EtOAc soluble products were 286.7 g/mol. In all reaction systems, almost no char was observed, with the exception of the experiment in the absence of NaOH which resulted in high char formation (20.4 wt.%). The proposed mechanism consisted of four steps and is shown in Figure 7.
The reductive depolymerization of kraft lignin was also examined in a water–ethanol mixture 50/50 (v/v) with formic acid as an in-situ hydrogen source, by the use of Ni-based catalysts compared to 5% Ru/C [114]. The effectiveness of the formic acid is evident even in the absence of catalysts (89 wt.% yield of liquid depolymerized lignin at 200 °C). The catalysts resulted in decrease in the molecular weight of the products but also in an unfortunate increase of solid residue due to condensation reactions evoked by the acidic properties of the supports. 10% Ni/Zeolite led to a slight increase of the yield up to 93.5 wt.%, decrease of Mw to 3150 g/mol and 9.3 wt.% solid residue. The ability of formic acid to depolymerize Kraft lignin in the absence of any other catalyst was also reported by the same group in a previous publication [115]. Under the optimum operating conditions of ca. 300 °C, 1 h, 18.6 wt.% substrate concentration, 50/50 (v/v) water–ethanol medium containing formic acid (FA) with FA-to-lignin mass ratio of 0.7, lignin (Mw ~10,000 g/mol) was effectively de-polymerized towards a liquid product (DL, Mw 1270 g/mol) at a yield of ~90 wt.% and <1 wt.% yield of solid residue (SR). Higher acidity caused condensation of the intermediate products. Higher temperatures or prolonged reaction times resulted also in repolymerization. Formic acid as hydrogen source has been also used by Liguori and Barth for the depolymerization of Kraft lignin to phenols in water and with Pd-Nafion SAC-13 as catalyst at 300 °C [116]. The main products obtained were guaiacol, pyrocatechol and resorcinol. Nafion SAC-13 acted as a Brønsted acid, activating the lignin aryl ether sites and promoting the hydrogenolysis to phenols.
A combination of a supported metal catalyst (Ru/C) with various MgO based catalysts was studied in the depolymerization of kraft lignin in supercritical ethanol as hydrogen donor solvent [117]. In the absence of Ru/C, the most active catalyst proved to be MgO/ZrO2 with the highest bio-oil yield (47.7 wt.%) consisting mainly of phenolic compounds, followed by MgO/C (43.3 wt.%) and MgO/Al2O3 (42.1 wt.%), as can be observed in Table 2. The number of base/acid sites of the MgO based catalysts was found to be correlated with the catalytic activity: MgO/ZrO2 which possessed the highest number of base sites and the less acid sites, exhibited the highest catalytic activity. In contrast, the catalyst with the highest acidity, i.e., MgO/Al2O3, was the less active one. The addition of Ru/C led to higher bio-oil yield in the range of 70.9–82.7 wt.%, rich in higher alcohols and aliphatic esters, while the highest bio-oil yield (88.1 wt.%) and the lower solid residue (8.8 wt.%) was observed for Ru/C when used alone. The unexpected increase of catalytic bio-oil molecular weights, compared to the non-catalytic bio-oil, was attributed to the higher concentrations of heavy compounds. The addition of hydrogen gas in the experiment catalyzed by Ru/C + MgO/ZrO2 did not improve the bio-oil yield (76.9 wt.%). Upon replacement of Kraft lignin with organosolv lignin, the Ru/C + MgO/ZrO2 resulted in lower yield and lower molecular weight of bio-oil, with a higher yield of aromatic monomer. The higher depolymerization efficiency of organsolv lignin was attributed to the less condensed structure and the absence of catalyst-poising sulfur.
Esposito et al. synthesized two different nickel-based materials, TiN-Ni and TiO2-Ni and tested their activity in the hydrogenolysis of Kraft lignin in various alcohols, in a flow reactor system under relatively mild temperature and pressure conditions, considering mainly the effect of alcohols on lignin solubility, without discussing their potential hydrogen donating function [118]. Higher catalytic activity exhibited by the TiN-Ni, was attributed to the better dispersion of Ni in TiN phase as well as to the more favorable titanium oxidation state, i.e., being (III) in TiN compared to (IV) in TiO. Substituted phenols (3.2 wt.%) and aromatic fragments (60 wt.%) with small molecular weights were obtained for TiN-Ni.
Supercritical water/isopropanol systems were applied for the depolymerization of Kraft lignin over Fe on Rh/La2O3/CeO2-ZrO2 [119]. Different ratios of water and isopropanol were used to adjust the optimum in situ H2 production, which was correlated with the hydrogen donating capability of water and isopropanol and the relative amount of Fe in the catalyst. Gradual increase in water content led to a gradual decrease in H2 selectivity. Considering the products, the increase in water content, resulted in a progressive increase of aromatics and aliphatic acid/esters while a sharp decrease in hydrogenated cyclics was observed. The products distribution obtained at the different ratio of isopropanol/water can be seen in Figure 8. Apart from the liquid products, similar trends were observed in the gas products.
Singh et al. studied the depolymerization of Kraft lignin in methanol at 220 °C by the use of homogeneous (NaOH) and heterogeneous catalysts (HZM-5 and iron turnings from lathe machining) [120]. Compared to the non-catalytic experiment, which resulted in 73.6 wt.% depolymerization yield, the homogeneous NaOH led to 68.5 wt.% yield, 5.1 wt.% monomeric compounds and 100–1000 g/mol molecular weight distribution. The heterogeneous HZSM-5 resulted in higher depolymerization yield of 85.1 wt.%, with lower monomers amount 4.2 wt.% and the same molecular weight distribution while the iron turnings resulted in lower depolymerization (44.4 wt.%) with 1.7 wt.% monomers and 100–2000 g/mol molecular weight distribution. In all cases, the main products were alkyl substituted phenols. It was suggested that methanol, in addition to being a solvent in the reaction, acted also as a hydrogen donor. The hydrogen released due to thermal reforming of methanol-induced hydrogenolysis of the ether linkages in lignin resulting in lignin depolymerization and demethoxylation.
4.2. Soda Lignins
Hensen et al. carried out a thorough investigation of the reductive depolymerization of soda lignin by the use of CuMgAlOx catalysts. In a first article, the group examined the influence of solvent, reaction time, and catalyst [121]. The most effective combination proved to be CuMgAlOx with ethanol at 300 °C, resulting in 17 wt.% monomers yield, comprising of aromatic products with small amounts of furans, hydrogenated substituted cyclic compounds and deoxygenated aromatics, as can be observed in Figure 9. The Cu content of 20 wt.% in the CuMgAlOx mixed oxide was found to induce the highest activity for Guerbet, esterification and alkylation reactions, resulting in higher monomers yield and low repolymerization [122]. Both the non-catalytic experiment and the use of methanol resulted in low monomer yields (5 and 6 wt.%) and in the latter case the products were mainly methylated phenols and guaiacol-type compounds. Copper in MgAlOx improved the monomer production and the formation of deoxygenated aromatics compared to the MgAlOx, NiMgAlOx, and PtMgAlOx catalysts. Considering the repolymerization via the phenolic hydroxyl groups, the authors suggested that ethanol not only acts as a hydrogen-donor solvent, but also as a capping agent and formaldehyde scavenger, as shown in Figure 10 [121,123]. In a following paper, they discussed the influence of reaction temperature on the products [124]. At lower temperatures in the range of 200–250 °C, recondensation reactions are dominant whereas at higher temperatures of 380–420 °C, the char formation due to carbonization played a major role. At the intermediate range of 300–340 °C, the depolymerization of lignin is enhanced, resulting in the formation of reactive phenolic intermediates which can be protected by alkylation, Guerbet and esterification reactions. CuO in the parent CuMgAlOx catalyst favors the alkylation reactions while its progressive reduction to Cu may lead to the undesirable increased hydrogenation of the aromatic ring.
Soda lignin depolymerization was performed over metals catalysts supported on ZSM-5 zeolite in supercritical ethanol at 440 °C [125]. When comparing the transition metals Ni, Co and Cu at 10 wt.% loading on ZSM-5 with Si/Al2 of 200, the 10% Cu/ZSM-5(200) catalyst showed the highest yield of monoaromatic compounds (15.3 wt.%). Changing the metal loading from 10% to 5 and 30% a small decrease in monoaromatic compounds was observed. In order to find the optimum Si/Al2 ratio of ZSM-5, Si/Al2 was varied from 30 to 200. The highest yield 98.2 wt.% of monoaromatic compounds was obtained over 10 wt.% Cu/ZSM-5(30) due to the higher acid density. The beneficial effect of Cu in the depolymerization was confirmed by the experiment conducted in Cu-free ZSM-5 (30) which resulted in 89.4 wt.% monoaromatic compounds. The authors suggested that the in situ produced hydrogen atoms were adsorbed onto the surface of Cu leading to cleavage of ether bonds and thus promoting the depolymerization.
4.3. Alkali Lignins
In the hydrogenolysis of alkali lignin in supercritical ethanol, Zhou et al. found that CuNiAl-hydrotalcite was more active catalyst than Ni/ZSM-5 or Ru/C, resulting in 49.5 wt.% bio-oil yield at 290 °C [126]. The strong basic sites of hydrotalcite, compared to the acidic ZSM-5 and carbon, inhibited the recondensation of reactive compounds of bio-oil. The addition of phenol as co-solvent to ethanol (phenol/lignin=0.8), increased bio-oil yield to 72.3 wt.%. Phenol is suggested to promote the hydrogenolysis due to the enhanced solubilization of lignin and the capping agent action favoring the formation of mono-phenolics compounds and suppressing repolymerization. Higher amounts of phenol resulted in lower bio-oil yields and increased molecular weights, phenomena which attributed to secondary repolymerization reactions. Also, the addition of hydrogen gas did not enhanced the bio-oil yield (70.3 wt.%). The optimum temperature and time considering the bio-oil yield were determined to be 290 °C and 3 h.
The effective activity of nickel-based catalysts was also reported by Li et al., in the hydrogen transfer conversion of alkali lignin using isopropanol/water solvent [127]. Alkali lignin showed high conversion (93%) over Raney Ni catalysts at 180 °C, superior than with Pd/C catalyst which led to low conversion and liquefaction rates. Lower conversion was observed for Klason lignin due to its more condensed nature, attributed to the high acid concentrations in the Klason lignin preparation process.
The synergistic activity of formic acid and Pd/C was examined in the catalytic depolymerization of alkali lignin in subcritical water [128]. When the reaction was contacted without formic acid and Pd/C at 265 °C for 1 h, the liquid products yield was 58.2 wt.% and the solid residue 30.6 wt.%. The addition of formic acid slightly increased the liquid products to 61.6 wt.% but extremely decreased the solid residue to 0.64 wt.%. The addition of Pd/C catalyst either in the presence of formic acid or not, resulted in lower liquid products (45.8 and 41.3 wt.%) and higher solid residues (16.3 and 54.4 wt.%). The products yields from lignin depolymerization in all reaction systems are shown in Table 3. The catalyst favored the conversion of formic acid and production of H2 via reforming and water–gas shift reactions and promoted the repolymerization reactions. Significant differences are observed in the composition of liquid products. In the absence of formic acid and Pd/C, the main compound was guaiacol, while in the presence of formic acid or both formic acid and Pd/C, catechol was the main compound. Pd/C can catalyze the hydrogenolysis of the aryl–O ether bond resulting in significant yield of phenol and char formation.
4.4. Organosolv Lignins
Toledano et al. studied the hydrogenolysis of organosolv lignin from olive tree prunings under microwave irradiation, using a variety of hydrogen donor solvents without any H2 addition [129,130]. In a first paper, they examined the activity of metallic (Ni, Ru, Pd, Pt) catalysts supported on Al-SBA-15 and found that 10% Ni/Al-SBA-15 with tetralin as solvent provided improved bio-oils with 17 wt.% yield, consisting of simple phenolics, including mesitol and syningaldehyde, as well as, a small amount of esters. The product distribution obtained over Ni, Ru, Pd, Pt catalysts supported on Al-SBA-15 can be seen in Table 4. The other metals exhibited lower activity with high remaining lignin due to repolymerization phenomena [129]. In a further work, the same group carried out detailed research on the solvent effect on the hydrogenolysis of organosolv lignin from olive tree prunings with 10% Ni/Al-SBA-15 at 150 °C [130]. The most effective solvent, as shown in Figure 11, was proved to be formic acid resulting in high bio-oil yield (28.89 wt.%), no biochar and a wide variety of phenolics compounds. Formic acid was noted to exhibit additional acidolytic properties for the depolymerization of lignin. Less efficient solvents were glycerol and tetralin with the later leading mainly to phthalates. The authors highlighted the unexpected behavior of isopropanol which in the case of the catalytic experiment, it was proven less efficient compared to the blank experiment due to the dehydrogenation of isopropanol to acetone over the Ni/Al-SBA-15 catalyst.
Organosolv lignin from switchgrass had been successfully depolymerized in ethanol, 20 wt.% Pt/C and formic acid as hydrogen donor molecule at 350 °C for the production of phenol and substituted phenols with higher H/C and lower O/C molar ratios [131]. In a similar reaction system, organosolv lignin was depolymerized using isopropanol as solvent, Ru/C as catalyst and formic acid as hydrogen donor at 400 °C [132]. The catalytic experiments resulted in 71.2 wt.% lignin oil, negligible solids formation and significant amount of water (9.6%) due to the decomposition of formic acid. The activity of the catalyst was confirmed by the experiment conducted in the absence of Ru/C which led to only 18 wt.% conversion and a large amount of solids derived either from the unconverted lignin or repolymerization reactions. The effective conversion of lignin was further compared with the hydrotreatment experiment, conducted in the absence of solvent (only Ru/C + H2 gas), exhibiting lower oil yield (63.1 wt.%) compared to the use of isopropanol as solvent. Considering the chemical composition of derived oils, the major products were ketones (methyl isobutylketone) followed by aromatics, catechols and alkylphenolics. In an attempt to improve the catalytic reaction system, methanol and ethanol were also tested as solvents. Methanol gave almost similar yield (68.4 wt.%) with isopropanol but higher amounts of alkylphenolics and aromatics whereas ethanol resulted in lower yield (63.4 wt.%).
The effect of biomass feedstock and isolation method of lignin on the depolymerization in supercritical ethanol and formic acid, in absence of catalyst, at 250–350 °C was also investigated [133]. The reaction was performed in lignin derived from oak (hardwood) and pine (softwood), isolated as ethanosolv, formasolv and Klason types. Regardless the isolation method, all lignins exhibited bio-oil and conversion yields above 90 wt.% with low solid residue <2.5 wt.% at 350 °C. At this temperature, the combination of ethanol with formic acid facilitated the hydrogen production which quenched the radicals, suppressing the repolymerization. For the hardwood type biomass, at lower reaction temperature (250 °C), the ethanosolv and the formasolv lignin exhibited lower bio-oil yield, 68.0 and 77.5 wt.%, respectively, while Klason lignin bio-oil yield dramatically decreased to 19.3 wt.% due to the abundance of C-C recalcitrant bonds formed in the Klason process. With regard to bio-oil composition, at 350 °C, linear and branched short-chain oxygenated species from the decomposition of ethanol and formic acid, monoaromatic species and long chain fatty acid alkyl esters from the esterification of woody biomass with ethanol were produced. At lower reaction temperature, where the deoxygenation/hydrogenation reactions are limited, carbonyl or double bond-containing monoaromatics were formed. With regard to softwood biomass, the ethanosolv and formasolv lignins exhibited 97.1 and 99.4 wt.% conversion and 88.1 and 90.7 wt.% bio-oil yield. Again, the Klason lignin depolymerization resulted in lower conversion (95.1 wt.%) and bio-oil yield (81.7 wt.%). At the low temperatures of 250–300 °C, the most important parameter was suggested to be the relative abundance of ether linkages in the lignin structure.
The catalytic activity of Cu based porous metal oxides towards the depolymerization of organosolv lignin extracted from candlenuts was examined in supercritical methanol at 310 °C [134]. Taking into consideration lignin conversion, the following rank was determined: Cu20PMO > Cu20PMO Cu20Cr20PMO > Cu20La20PMO (PMO stands for porous metal oxide). For the best catalyst Cu20PMO, lignin conversion reached 48.3% after 1 h reaction. Due to the lower methanol reforming ability of lanthanum, the reaction was also carried out for higher times and the conversion reached 98% after 6 h. Under the same reaction conditions, the activity Cu-free porous metal oxides were also examined. [Mg/Al]PMO exhibited the highest lignin conversion of 74.6% for 5 h reaction. Comparison of CuPMOs to Cu-free analogs showed that Cu promotes higher yields of methanol-soluble products and suppresses re-condensation reactions. The Cu20La20PMO variant was suggested as the most effective catalyst in terms of limiting over-reduction of aromatic intermediates due to the lower methanol-reforming of lanthanum, thus regulating the in situ production of hydrogen.
Organosolv lignin isolated from eucalyptus was depolymerized in water to syringol monomers over β-CaP2O6 and CoP2O6 [135]. Despite that eucalyptus is a hardwood feedstock, the use of phosphate catalysts selectively produced only syringol with yield of 8.47% over β-CaP2O6 and 6.67 % over CoP2O6. The hydrogenolysis of organosolv lignin from hybrid poplar in supercritical ethanol at 320 °C, by the use of amorphous B-containing FeNi alloyed catalysts, resulted in lignin depolymerization and the production of deoxygenated aliphatic side chains [136].
4.5. Lignosulfonate
Lignosulfonate depolymerization via hydrogen transfer reactions was investigated over Raney Ni at 200 °C and a solvent mixture composed of water, isopropanol, butanol and hexane, introducing the concept of an emulsion microreactor [137]. Isopropanol was considered to be the hydrogen donor solvent for the hydrogenolysis of lignolsulfonate while the combination of the above three solvents provided higher monomer yields, compared to the more classical water, alcohol or water-alcohol mixtures, as can be seen in Table 5. The higher yield of phenolic monomers was observed for the solvent mixture water: butanol: isopropanol: hexane = 1:1:3:0.1, with 4-ethyl guaiacol being the most abundant compound. The facile phase separation after the hydrogenolysis reaction was suggested as an important advantage of the described emulsion microreactor.
4.6. Enzymatic and Acid Hydrolysis Lignins
The role of formic acid in the reductive depolymerization of lignin has been studied in the work of Oregui-Bengoechea et al. [138]. In the hydrogenolysis of enzymatic hydrolysis eucalyptus lignin over NiMo/sulfated alumina and ethanol at 320 °C, the synergistic action of formic acid and catalyst was shown by the increased oil yield (38.4 wt.%) compared to the uncatalyzed experiment (23 wt.%) and the experiment where gaseous H2 had been used instead of formic acid (19.7 wt.%). All the derived oils contained methoxy-, hydroxyl- and alkyl- substituted benzenes. The authors suggested that the formic acid is involved in lignin depolymerization via a formylation-elimination-hydrogenolysis mechanism, which includes also the catalytic decomposition of formic acid that provides molecular H2 for the hydrogenolysis reaction (Figure 12). The important role of the solvent was discussed and ethanol proved to be more effective (oil yield 38.4 wt.%) than methanol (23.8 wt.%) or isopropanol (21.7 wt.%), as suggested in similar studies [130].
Similar observations about the enhanced depolymerization in the presence of formic acid and ethanol had been reported also by Kristianto et al. for the concentrated acid hydrolysis lignin from fruit bunch palm oil [139]. From the catalytic activity screening of 5% Pd/C, 5% Ru/Al2O3, 5% Ru/C and 10% Ni/C at 300 °C it was shown that the most active catalyst was 5% Ru/C with 31 wt.% bio-oil yield and 47.1 wt.% solid residue. The addition of formic acid increased the bio-oil yield to 62.9 wt.% and decreased the solid residue to 19.8 wt.%, making formic acid better hydrogen donor than the external gas. The products obtained from the reaction were phenol and its derivatives whose amount increased with increase in formic acid/lignin ratio and reaction time. Small amounts of phenolic compounds with ethyl and ester groups were observed due to alkylation and esterification reactions of phenol intermediates with ethanol.
The synergistic effect of Raney Ni and zeolites catalysts had been investigated in the depolymerization of enzymatic hydrolysis lignin from bamboo residues [140]. In a methanol/water reaction mixture at 250 °C, when Raney Ni was combined with zeolite catalysts, the yield of mono-phenols significantly increased from 12.9 wt.% (Raney Ni) and 5 wt.% (zeolite) to 27.9 wt.% (HUSY and Raney Ni). The optimum ratio of zeolite: Raney Ni was determined to be 8:4. The authors proposed that Raney Ni could act as lignin cracking and methanol reforming catalyst and zeolite as Brønsted and/or Lewis solid acid essential for ether solvolysis and dehydration. Furthermore, zeolites can act as a blocking agent to prevent reactions between the original lignin and the unstable lignin fragments.
4.7. Lignins Extracted by Deep Eutectic Solvents
Das et al. studied the hydrogenolysis of deep eutectic solvent extracted lignin by the use of Ru/C, Pd/C and Pt/C at 270 °C with isopropanol as hydrogen donor solvent [141]. The most effective catalyst was Ru/C resulting in the highest oil yield 36.28 wt.% and the lowest char formation 46.43 wt.%. The main products were phenol, substituted phenols and long chain fatty acids derived by the reductive couple reaction of lignin. Increase in catalyst loading from 2 wt.% to 15 wt.% resulted in increase in oil yield but further increase (20 wt.%), decreased the oil yield due to side chain reactions, hindering the production of monomeric phenolic compounds. Higher reaction temperature (300 °C) did not enhance lignin depolymerization, resulting in decrease in oil yield and increase of gas products yield due to the further decomposition of lignin-derived compounds to gaseous products or condensation to char formation. Similar results was obtained by changing the reaction time. Gradual increase from 30 to 60 min, increased the oil yield but further increase to 180 min, resulted in decrease of oil yield and increase of gas products yield.
4.8. Selection and Design Criteria for an Effective Catalyst in Reductive Depolymerization of Lignin Using Hydrogen Donors
As discussed in the previous sections, the catalysts which have been widely studied in the reductive depolymerization of lignin by the use of hydrogen donors are based on noble (Pd, Ru, Pt) or transition metals (Ni, Cu) supported on carbon, zeolites and silica materials due to their known ability to catalyse the cleavage (hydrogenolysis) of C-O and C-C bonds, the hydrodeoxygenation of oxygenated compounds and the hydrogenation of aromatic double bonds. A summary of the most representative catalytic systems is shown in Table 6. However, the specific mechanisms of the in situ hydrogen production has been scarcely discussed with general reference to reforming of alcohols and related water gas shift reaction as well as decomposition of formic acid. Furthermore, while the terms “transfer hydrogenation” or “hydrogen transfer” have been used in some cases, there was no systematic effort to elucidate the reaction pathways involved in relation to the catalyst properties and the experimental conditions.
Supercritical alcohols can donate hydrogen in the form of molecular hydrogen, hydride, or protons, with the hydride deriving from α-hydrogen and the proton from the alcohol hydroxyl, forming simultaneously electron-deficient hydroxylalkylation species, alkoxide ions and aldehydes [142]. Similarly, decomposition of formic acid in supercritical water conditions leads to in situ H2 formation. However, the relatively high temperatures, e.g., >280 °C, at which most of the lignin hydrogenolysis studies in supercritical solvents have been conducted, may also induce repolymerization-condensation reactions of the initially formed monomer phenolics, especially in the presence of acidic catalysts. Thus, optimization of the overall system, i.e., type of solvent/hydrogen donor—reaction temperature—catalyst properties, is required in order to achieve high yields of liquid products enriched in monomers.
With regard to the catalyst properties, three main interactive criteria should be considered: (i) effect on the in situ hydrogen production mechanism, (ii) high hydrogenation reactivity and facilitated activation of the lignin C-O or C-C bonds, and (iii) stabilization of reactive intermediates via alkylation or other reactions. The first criterion is related with the catalyst properties that should be tailored towards enhanced reforming and WGS reaction activity and/or transfer and stabilization via surface intermediates of hydride (H:) or protons (H+) by selecting appropriate metals and supports. In this latter case, which is less discussed in the literature, an effective catalyst or catalyst support surface would comprise of Lewis acid sites and Brønsted or Lewis basic sites that can attract H: and H+ from alcohols, respectively, thus initiating the steps of hydrogen transfer and lowering the overall reaction required temperature. Materials with such properties can be various transition metal oxides or mixed oxides, such as ZrO2, TiO2, MgO, etc and their modified/doped analogues, as well as metal-modified zeolites and other aluminosilicates. The surface acid-base properties of these materials can be tuned by selecting the appropriate composition. The second criterion refers to the intrinsic (de)hydrogenation and redox activity of noble or transition metals which includes the dissociative adsorption of molecular H2, as two hydrogen atoms, which are available to participate in the various hydrogenation or hydrogenolysis pathways. The hydrogen atoms on the noble/transition metals may interact directly with the abundant ether bonds in the lignin fragments or with a double bond (at deeper hydrogenation conditions) or they can interact via spill-over phenomena with a nearby sorbed intermediate, such as an ether bond reacting with the Brønsted acid sites of the support of the hydrogenating metal. Thus, the synergistic action of the metal with the support could also lead to facile hydrogenation/hydrogenolysis reaction reducing further the required reaction temperature. The third criterion is related with the effect of the catalyst on the reactions occurring between the alcohol solvent and the formed reactive intermediates in lignin hydrogenolysis with the aim to inhibit their repolymerization, one representative example being that of favoring their alkylation with methyl or ethyl moieties (from methanol or ethanol respectively) in the presence of metal oxides such as SiO2-Al2O3, CuO, TiO2, etc. A balance between hydrogenation and alkylation may also be desirable, in order to limit repolymerization and not lose the aromatic nature of the obtained monomers, thus pointing to metal oxides (mainly being used as supports) that cannot be easily reduced in situ in the presence of hydrogen.
Along these lines and in addition to the more classical hydrogenation catalysts, e.g., noble or transition metals on carbon, zeolites, silica, etc., more “sophisticated” multifunctional catalytic formulation have been recently reported, as described in the sections above, including TiN-Ni and TiO2-Ni, FeNiB alloys, Cu based materials such as Cu20La20PMO (porous metal oxides) and CuMgAlOx, Fe on Rh/La2O3/CeO2-ZrO2, β-CaP2O6 and CoP2O6, and others. With regard to catalyst recovery from batch reactor systems, new magneticcatalytic formulations with weakly acidic Brønsted-type centers, such as Fe3O4@SiO2@Re and Co@Nb2O5@Fe3O4 which exhibited promising behavior in the reductive depolymerization of lignin using gaseous H2, could be also effectively used in catalytic transfer hdyrogenolysis reactions [143,144].
5. Conclusions and Outlook
The valorization of lignin, being the most abundant natural phenolic/aromatic polymer, has tremendous potential provided that efficient depolymerization processes will be developed. The reductive depolymerization via catalytic hydrogenation/hydrogenolysis reactions utilizing hydrogen donors offers a possible route with minimum requirements of molecular (gaseous) hydrogen. Small alcohols and water may act both as solvents and hydrogen donors, under sub- or supercritical conditions, providing the necessary hydrogen for breaking the primary ether bonds of the lignin macromolecule and leading to deeper C-C bond hydrogenolysis and hydrogenation of the derived alkoxy-phenols depending on process parameters and catalysts used. Methanol, ethanol, isopropanol or mixtures of these alcohols with water, can lead to high degrees of lignin depolymerization and monomers yield, with ethanol being the most promising candidate. Apart from the alcohol-based solvents, formic acid can also act as hydrogen donor, in cooperation with alcohols or water, thus leading to enhanced depolymerization and increased bio-oil production.
Although the selection of the most appropriate solvent is crucial for inducing transfer hydrogenolysis/hydrogenation reactions in lignin depolymerization, still the major role is being played by the catalyst type and its properties, especially when relatively low/moderate temperatures are aimed that do not favor undesirable hydrogenation of the aromatic ring and repolymerization/condensation reactions. As discussed in Section 4.8, an effective catalyst in reductive depolymerization of lignin using hydrogen donors should promote the in situ generation of hydrogen via alcohol reforming, WGS or dehydrogenation (via H: and H+ abstraction) reactions or formic acid decomposition, should exhibit high hydrogenation reactivity and favor activation of the lignin aryl-ether bonds, and should be able to contribute to stabilization of reactive intermediates, for example, via alkylation reactions leading to increased bio-oil yield and minimum solid residues.
Finally, towards the potential upscaling of lignin depolymerization via catalytic transfer hydrogenolysis using hydrogen donors, continuous flow reactors and processes need to be developed by considering important parameters such as the effective solubilization of lignin as feedstock, increased catalyst reactivity to balance the relatively short contact times and low rate of catalyst deactivation.
The lignin bio-oils rich in alkoxy- or alkyl-phenols, derived either from reductive depolymerization or from thermal/catalytic fast pyrolysis, could be used without or after further upgrading (e.g., glycidilation) in the production of epoxy or phenol-formaldehyde resins and related polymers and composites, while specific components such as vanillin will find application in the food industry, cosmetics, and pharmaceuticals. Furthermore, the lignin bio-oil can be upgraded via hydrodeoxygation (HDO) reactions to hydrocarbon (alkanes) fuels.
Acknowledgements
We acknowledge support of this work by the project “INVALOR: Research Infrastructure for Waste Valorization and Sustainable Management” (MIS 5002495) which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund). We would also like to acknowledge COST Action CA17128 (LignoCOST) for promoting exchange and dissemination of knowledge and expertise in the field of lignin valorization.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- European Parliament and the Council, Directive 2009/28/EC. 2009. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=celex%3A32009L0028 (accessed on 21 December 2018).
- Perlack, R.D.; Wright, L.L.; Turhollow, A.F.; Graham, R.L.; Stokes, B.J.; Erbach, D.C. Biomass as Feedstock for a Bioenergy and Bioproducts Industry: The Technical Feasibility of a Billion-Ton Annual Supply; U.S. Department of Energy: Oak Ridge National Laboratory, Oak Ridge, TN, USA, 2005.
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Galkin, M.V.; Samec, J.S. Lignin Valorization through Catalytic Lignocellulose Fractionation: A Fundamental Platform for the Future Biorefinery. ChemSusChem 2016, 9, 1544–1558. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.S.; Inoue, H.; Endo, T.; Yano, S.; Bon, E.P. Milling pretreatment of sugarcane bagasse and straw for enzymatic hydrolysis and ethanol fermentation. Bioresour. Technol. 2010, 101, 7402–7409. [Google Scholar] [CrossRef]
- Agbor, V.B.; Cicek, N.; Sparling, R.; Berlin, A.; Levin, D.B. Biomass pretreatment: Fundamentals toward application. Biotechnol. Adv. 2011, 29, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Mayer-Laigle, C.; Solhy, A.; Arancon, R.A.D.; de Vries, H.; Luque, R. Mechanical pretreatments of lignocellulosic biomass: Towards facile and environmentally sound technologies for biofuels production. RSC Adv. 2014, 4, 48109–48127. [Google Scholar] [CrossRef]
- Gonzales, R.R.; Sivagurunathan, P.; Kim, S.H. Effect of severity on dilute acid pretreatment of lignocellulosic biomass and the following hydrogen fermentation. Int. J. Hydrogen Energy 2016, 41, 21678–21684. [Google Scholar] [CrossRef]
- Xu, J.K.; Sun, R.C. Chapter 19—Recent Advances in Alkaline Pretreatment of Lignocellulosic Biomass. In Biomass Fractionation Technologies for a Lignocellulosic Feedstock Based Biorefinery; Mussatto, S.I., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 431–459. [Google Scholar]
- Garcia-Cubero, M.A.; Gonzalez-Benito, G.; Indacoechea, I.; Coca, M.; Bolado, S. Effect of ozonolysis pretreatment on enzymatic digestibility of wheat and rye straw. Bioresour. Technol. 2009, 100, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Nitsos, C.; Stoklosa, R.; Karnaouri, A.; Vörös, D.; Lange, H.; Hodge, D.; Crestini, C.; Rova, U.; Christakopoulos, P. Isolation and Characterization of Organosolv and Alkaline Lignins from Hardwood and Softwood Biomass. ACS Sustain. Chem. Eng. 2016, 4, 5181–5193. [Google Scholar] [CrossRef]
- Dale, B.E.; Leong, C.K.; Pham, T.K.; Esquivel, V.M.; Rios, I.; Latimer, V.M. Hydrolysis of lignocellulosics at low enzyme levels: Application of the AFEX process. Bioresour. Technol. 1996, 56, 111–116. [Google Scholar] [CrossRef]
- Duque, A.; Manzanares, P.; Ballesteros, I.; Ballesteros, M. Chapter 15—Steam Explosion as Lignocellulosic Biomass Pretreatment. In Biomass Fractionation Technologies for a Lignocellulosic Feedstock Based Biorefinery; Mussatto, S.I., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 349–368. [Google Scholar]
- Tang, Y.; Chandra, R.P.; Sokhansanj, S.; Saddler, J.N. Influence of steam explosion processes on the durability and enzymatic digestibility of wood pellets. Fuel 2018, 211, 87–94. [Google Scholar] [CrossRef]
- Schmidt, A.S.; Thomsen, A.B. Optimization of wet oxidation pretreatment of wheat straw. Bioresour. Technol. 1998, 64, 139–151. [Google Scholar] [CrossRef]
- Nitsos, C.K.; Matis, K.A.; Triantafyllidis, K.S. Optimization of hydrothermal pretreatment of lignocellulosic biomass in the bioethanol production process. ChemSusChem 2013, 6, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Foston, M.; Ragauskas, A.J. Comparative studies on hydrothermal pretreatment and enzymatic saccharification of leaves and internodes of alamo switchgrass. Bioresour. Technol. 2011, 102, 7224–7228. [Google Scholar] [CrossRef] [PubMed]
- Oksman, K.; Aitomäki, Y.; Mathew, A.P.; Siqueira, G.; Zhou, Q.; Butylina, S.; Tanpichai, S.; Zhou, X.; Hooshmand, S. Review of the recent developments in cellulose nanocomposite processing. Compos. Part A Appl. Sci. Manuf. 2016, 83, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Patil, S.; Argyropoulos, D.S. Thermal properties of lignin in copolymers, blends, and composites: A review. Green Chem. 2015, 17, 4862–4887. [Google Scholar] [CrossRef]
- Liu, X.; Lin, Q.; Yan, Y.; Peng, F.; Sun, R.; Ren, J. Hemicellulose from Plant Biomass in Medical and Pharmaceutical Application: A Critical Review. Curr. Med. Chem. 2017, 24, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Espro, C.; Gumina, B.; Szumelda, T.; Paone, E.; Mauriello, F. Catalytic Transfer Hydrogenolysis as an Effective Tool for the Reductive Upgrading of Cellulose, Hemicellulose, Lignin, and Their Derived Molecules. Catalysts 2018, 8, 313. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef]
- Zhou, C.H.; Xia, X.; Lin, C.X.; Tong, D.S.; Beltramini, J. Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels. Chem. Soc. Rev. 2011, 40, 5588–5617. [Google Scholar] [CrossRef]
- Lappas, A.A.; Kalogiannis, K.G.; Iliopoulou, E.F.; Triantafyllidis, K.S.; Stefanidis, S.D. Catalytic pyrolysis of biomass for transportation fuels. Wires Energy Environ. 2012, 1, 285–297. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; Pineda, A.; Balu, A.M.; Luque, R.; Campelo, J.M.; Romero, A.A.; Ramos-Fernandez, J.M. Catalytic transformations of biomass-derived acids into advanced biofuels. Catal. Today 2012, 195, 162–168. [Google Scholar] [CrossRef]
- Espro, C.; Gumina, B.; Paone, E.; Mauriello, F. Upgrading Lignocellulosic Biomasses: Hydrogenolysis of Platform Derived Molecules Promoted by Heterogeneous Pd-Fe Catalysts. Catalysts 2017, 7, 78. [Google Scholar] [CrossRef]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. 2016, 55, 8164–8215. [Google Scholar] [CrossRef] [Green Version]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Chakar, F.S.; Ragauskas, A.J. Review of current and future softwood kraft lignin process chemistry. Ind. Crop. Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Dorrestijn, E.; Laarhoven, L.J.J.; Arends, I.W.C.E.; Mulder, P. The occurrence and reactivity of phenoxyl linkages in lignin and low rank coal. J. Anal. Appl. Pyrol. 2000, 54, 153–192. [Google Scholar] [CrossRef]
- Gillet, S.; Aguedo, M.; Petitjean, L.; Morais, A.R.C.; Lopes, A.M.D.; Lukasik, R.M.; Anastas, P.T. Lignin transformations for high value applications: Towards targeted modifications using green chemistry. Green Chem. 2017, 19, 4200–4233. [Google Scholar] [CrossRef]
- Pandey, M.P.; Kim, C.S. Lignin Depolymerization and Conversion: A Review of Thermochemical Methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Lora, J. Chapter 10—Industrial Commercial Lignins: Sources, Properties and Applications. In Monomers, Polymers and Composites from Renewable Resources; Belgacem, M.N., Gandini, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 225–241. [Google Scholar] [CrossRef]
- Tejado, A.; Pena, C.; Labidi, J.; Echeverria, J.M.; Mondragon, I. Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis. Bioresour. Technol. 2007, 98, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lei, F.; Li, P.; Jiang, J. Lignocellulosic biomass to biofuels and biochemicals: A comprehensive review with a focus on ethanol organosolv pretreatment technology. Biotechnol. Bioeng. 2018, 115, 2683–2702. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Harrison, M.D.; Rackemann, D.W.; Doherty, W.O.S.; O’Hara, I.M. Organosolv pretreatment of plant biomass for enhanced enzymatic saccharification. Green Chem. 2016, 18, 360–381. [Google Scholar] [CrossRef] [Green Version]
- Villaverde, J.; Ligero, P.; Vega, A. Formic and acetic acid as agents for a cleaner fractionation of Miscanthus x giganteus. J. Clean. Prod. 2010, 18, 395–401. [Google Scholar] [CrossRef]
- Katahira, R.; Mittal, A.; McKinney, K.; Ciesielski, P.N.; Donohoe, B.S.; Black, S.K.; Johnson, D.K.; Biddy, M.J.; Beckham, G.T. Evaluation of Clean Fractionation Pretreatment for the Production of Renewable Fuels and Chemicals from Corn Stover. ACS Sustain. Chem. Eng. 2014, 2, 1364–1376. [Google Scholar] [CrossRef]
- Smit, A.; Huijgen, W. Effective fractionation of lignocellulose in herbaceous biomass and hardwood using a mild acetone organosolv process. Green Chem. 2017, 19, 5505–5514. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Gilkes, N.; Kadla, J.; Pye, K.; Saka, S.; Gregg, D.; Ehara, K.; Xie, D.; Lam, D.; Saddler, J. Bioconversion of hybrid poplar to ethanol and co-products using an organosolv fractionation process: Optimization of process yields. Biotechnol. Bioeng. 2006, 94, 851–861. [Google Scholar] [CrossRef]
- Shuai, L.; Amiri, M.T.; Questell-Santiago, Y.M.; Heroguel, F.; Li, Y.; Kim, H.; Meilan, R.; Chapple, C.; Ralph, J.; Luterbacher, J.S. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 2016, 354, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Raghavendran, V.; Nitsos, C.; Matsakas, L.; Rova, U.; Christakopoulos, P.; Olsson, L. A comparative study of the enzymatic hydrolysis of batch organosolv-pretreated birch and spruce biomass. AMB Express 2018, 8, 114. [Google Scholar] [CrossRef]
- Matsakas, L.; Nitsos, C.; Raghavendran, V.; Yakimenko, O.; Persson, G.; Olsson, E.; Rova, U.; Olsson, L.; Christakopoulos, P. A novel hybrid organosolv: Steam explosion method for the efficient fractionation and pretreatment of birch biomass. Biotechnol. Biofuels 2018, 11, 160. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef]
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Jiang, H.; Yu, H.Q. Thermochemical conversion of lignin to functional materials: A review and future directions. Green Chem. 2015, 17, 4888–4907. [Google Scholar] [CrossRef]
- Bjørsvik, H.-R.; Liguori, L. Organic Processes to Pharmaceutical Chemicals Based on Fine Chemicals from Lignosulfonates. Org. Process. Res. Dev. 2002, 6, 279–290. [Google Scholar] [CrossRef]
- Van den Bosch, S.; Koelewijn, S.F.; Renders, T.; Van den Bossche, G.; Vangeel, T.; Schutyser, W.; Sels, B.F. Catalytic Strategies Towards Lignin-Derived Chemicals. Top. Curr. Chem. 2018, 376, 36. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, P.A.; Fotopoulos, A.P.; Karakoulia, S.A.; Triantafyllidis, K.S. Catalytic Fast Pyrolysis of Kraft Lignin With Conventional, Mesoporous and Nanosized ZSM-5 Zeolite for the Production of Alkyl-Phenols and Aromatics. Front. Chem. 2018, 6, 295. [Google Scholar] [CrossRef] [PubMed]
- Custodis, V.B.F.; Karakoulia, S.A.; Triantafyllidis, K.S.; van Bokhoven, J.A. Catalytic Fast Pyrolysis of Lignin over High-Surface-Area Mesoporous Aluminosilicates: Effect of Porosity and Acidity. ChemSusChem 2016, 9, 1134–1145. [Google Scholar] [CrossRef]
- Guvenatam, B.; Heeres, E.H.J.; Pidko, E.A.; Hensen, E.J.M. Lewis-acid catalyzed depolymerization of Protobind lignin in supercritical water and ethanol. Catal. Today 2016, 259, 460–466. [Google Scholar] [CrossRef]
- Wang, H.; Tucker, M.; Ji, Y. Recent Development in Chemical Depolymerization of Lignin: A Review. J. Appl. Chem. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C.C. Hydrolytic depolymerization of hydrolysis lignin: Effects of catalysts and solvents. Bioresour. Technol. 2015, 190, 416–419. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Labidi, J. Organosolv lignin depolymerization with different base catalysts. J. Chem. Technol. Biotechol. 2012, 87, 1593–1599. [Google Scholar] [CrossRef]
- Abdelaziz, O.Y.; Li, K.; Tunå, P.; Hulteberg, C.P. Continuous catalytic depolymerisation and conversion of industrial kraft lignin into low-molecular-weight aromatics. Biomass Convers. Biorefinery 2017, 8, 455–470. [Google Scholar] [CrossRef] [Green Version]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef]
- Xu, C.; Arancon, R.A.; Labidi, J.; Luque, R. Lignin depolymerisation strategies: Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.; Ahmad, M.; Hardiman, E.M.; Rahmanpour, R. Pathways for degradation of lignin in bacteria and fungi. Nat. Prod. Rep. 2011, 28, 1883–1896. [Google Scholar] [CrossRef] [PubMed]
- Sena-Martins, G.; Almeida-Vara, E.; Duarte, J.C. Eco-friendly new products from enzymatically modified industrial lignins. Ind. Crops Prod. 2008, 27, 189–195. [Google Scholar] [CrossRef]
- Kumar, C.R.; Anand, N.; Kloekhorst, A.; Cannilla, C.; Bonura, G.; Frusteri, F.; Barta, K.; Heeres, H.J. Solvent free depolymerization of Kraft lignin to alkyl-phenolics using supported NiMo and CoMo catalysts. Green Chem. 2015, 17, 4921–4930. [Google Scholar] [CrossRef]
- Kloekhorst, A.; Heeres, H.J. Catalytic Hydrotreatment of Alcell Lignin Using Supported Ru, Pd, and Cu Catalysts. ACS Sustain. Chem. Eng. 2015, 3, 1905–1914. [Google Scholar] [CrossRef]
- Zhu, G.D.; Ouyang, X.P.; Jiang, L.F.; Zhu, Y.; Jin, D.X.; Pang, Y.X.; Qiu, X.Q. Effect of functional groups on hydrogenolysis of lignin model compounds. Fuel Process. Technol. 2016, 154, 132–138. [Google Scholar] [CrossRef]
- Zhang, J.G.; Asakura, H.; van Rijn, J.; Yang, J.; Duchesne, P.; Zhang, B.; Chen, X.; Zhang, P.; Saeys, M.; Yan, N. Highly efficient, NiAu-catalyzed hydrogenolysis of lignin into phenolic chemicals. Green Chem. 2014, 16, 2432–2437. [Google Scholar] [CrossRef]
- Zhang, J.; Teo, J.; Chen, X.; Asakura, H.; Tanaka, T.; Teramura, K.; Yan, N. A Series of NiM (M = Ru, Rh, and Pd) Bimetallic Catalysts for Effective Lignin Hydrogenolysis in Water. ACS Catal. 2014, 4, 1574–1583. [Google Scholar] [CrossRef]
- Zhang, J.-W.; Cai, Y.; Lu, G.-P.; Cai, C. Facile and selective hydrogenolysis of β-O-4 linkages in lignin catalyzed by Pd–Ni bimetallic nanoparticles supported on ZrO2. Green Chem. 2016, 18, 6229–6235. [Google Scholar] [CrossRef]
- Mauriello, F.; Paone, E.; Pietropaolo, R.; Balu, A.M.; Luque, R. Catalytic Transfer Hydrogenolysis of Lignin-Derived Aromatic Ethers Promoted by Bimetallic Pd/Ni Systems. ACS Sustain. Chem. Eng. 2018, 6, 9269–9276. [Google Scholar] [CrossRef]
- Galkin, M.V.; Sawadjoon, S.; Rohde, V.; Dawange, M.; Samec, J.S.M. Mild Heterogeneous Palladium-Catalyzed Cleavage of β-O-4′-Ether Linkages of Lignin Model Compounds and Native Lignin in Air. ChemCatChem 2014, 6, 179–184. [Google Scholar] [CrossRef]
- Galkin, M.V.; Dahlstrand, C.; Samec, J.S. Mild and Robust Redox-Neutral Pd/C-Catalyzed Lignol beta-O-4’ Bond Cleavage Through a Low-Energy-Barrier Pathway. ChemSusChem 2015, 8, 2187–2192. [Google Scholar] [CrossRef] [PubMed]
- Besse, X.; Schuurman, Y.; Guilhaume, N. Reactivity of lignin model compounds through hydrogen transfer catalysis in ethanol/water mixtures. Appl. Catal. B Environ. 2017, 209, 265–272. [Google Scholar] [CrossRef]
- Wang, X.; Rinaldi, R. Solvent effects on the hydrogenolysis of diphenyl ether with Raney nickel and their implications for the conversion of lignin. ChemSusChem 2012, 5, 1455–1466. [Google Scholar] [CrossRef]
- Macala, G.S.; Matson, T.D.; Johnson, C.L.; Lewis, R.S.; Iretskii, A.V.; Ford, P.C. Hydrogen transfer from supercritical methanol over a solid base catalyst: A model for lignin depolymerization. ChemSusChem 2009, 2, 215–217. [Google Scholar] [CrossRef]
- Subbotina, E.; Galkin, M.V.; Samec, J.S.M. Pd/C-Catalyzed Hydrogenolysis of Dibenzodioxocin Lignin Model Compounds Using Silanes and Water as Hydrogen Source. ACS Sustain. Chem. Eng. 2017, 5, 3726–3731. [Google Scholar] [CrossRef]
- Klein, I.; Saha, B.; Abu-Omar, M.M. Lignin depolymerization over Ni/C catalyst in methanol, a continuation: Effect of substrate and catalyst loading. Catal. Sci. Technol. 2015, 5, 3242–3245. [Google Scholar] [CrossRef]
- Galkin, M.V.; Smit, A.T.; Subbotina, E.; Artemenko, K.A.; Bergquist, J.; Huijgen, W.J.; Samec, J.S. Hydrogen-free catalytic fractionation of woody biomass. ChemSusChem 2016, 9, 3280–3287. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, S.; Schutyser, W.; Vanholme, R.; Driessen, T.; Koelewijn, S.F.; Renders, T.; De Meester, B.; Huijgen, W.J.J.; Dehaen, W.; Courtin, C.M.; et al. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ. Sci. 2015, 8, 1748–1763. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Wang, F.; Cai, J.; Wang, Y.; Zhang, J.; Yu, W.; Xu, J. Lignin depolymerization (LDP) in alcohol over nickel-based catalysts via a fragmentation–hydrogenolysis process. Energy Environ. Sci. 2013, 6, 994. [Google Scholar] [CrossRef]
- Godard, H.P.; McCarthy, J.L.; Hibbert, H. Hydrogenation of wood. J. Am. Chem. Soc. 1940, 62, 988. [Google Scholar] [CrossRef]
- Yan, N.; Zhao, C.; Dyson, P.J.; Wang, C.; Liu, L.T.; Kou, Y. Selective degradation of wood lignin over noble-metal catalysts in a two-step process. ChemSusChem 2008, 1, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Torr, K.M.; van de Pas, D.J.; Cazeils, E.; Suckling, I.D. Mild hydrogenolysis of in-situ and isolated Pinus radiata lignins. Bioresour. Technol. 2011, 102, 7608–7611. [Google Scholar] [CrossRef] [PubMed]
- Galkin, M.V.; Samec, J.S. Selective route to 2-propenyl aryls directly from wood by a tandem organosolv and palladium-catalysed transfer hydrogenolysis. ChemSusChem 2014, 7, 2154–2158. [Google Scholar] [CrossRef] [PubMed]
- Parsell, T.; Yohe, S.; Degenstein, J.; Jarrell, T.; Klein, I.; Gencer, E.; Hewetson, B.; Hurt, M.; Kim, J.I.; Choudhari, H.; et al. A synergistic biorefinery based on catalytic conversion of lignin prior to cellulose starting from lignocellulosic biomass. Green Chem. 2015, 17, 1492–1499. [Google Scholar] [CrossRef]
- Luo, H.; Klein, I.M.; Jiang, Y.; Zhu, H.Y.; Liu, B.Y.; Kenttamaa, H.I.; Abu-Omar, M.M. Total Utilization of Miscanthus Biomass, Lignin and Carbohydrates, Using Earth Abundant Nickel Catalyst. ACS Sustain. Chem. Eng. 2016, 4, 2316–2322. [Google Scholar] [CrossRef]
- Ferrini, P.; Rinaldi, R. Catalytic biorefining of plant biomass to non-pyrolytic lignin bio-oil and carbohydrates through hydrogen transfer reactions. Angew. Chem. 2014, 53, 8634–8639. [Google Scholar] [CrossRef]
- Van den Bosch, S.; Schutyser, W.; Koelewijn, S.F.; Renders, T.; Courtin, C.M.; Sels, B.F. Tuning the lignin oil OH-content with Ru and Pd catalysts during lignin hydrogenolysis on birch wood. Chem. Commun. 2015, 51, 13158–13161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bosch, S.; Renders, T.; Kennis, S.; Koelewijn, S.F.; Van den Bossche, G.; Vangeel, T.; Deneyer, A.; Depuydt, D.; Courtin, C.M.; Thevelein, J.M.; et al. Integrating lignin valorization and bio-ethanol production: On the role of Ni-Al2O3 catalyst pellets during lignin-first fractionation. Green Chem. 2017, 19, 3313–3326. [Google Scholar] [CrossRef]
- Schutyser, W.; Van den Bosch, S.; Renders, T.; De Boe, T.; Koelewijn, S.F.; Dewaele, A.; Ennaert, T.; Verkinderen, O.; Goderis, B.; Courtin, C.M.; et al. Influence of bio-based solvents on the catalytic reductive fractionation of birch wood. Green Chem. 2015, 17, 5035–5045. [Google Scholar] [CrossRef]
- Huang, X.; Ouyang, X.; Hendriks, B.M.S.; Gonzalez, O.M.M.; Zhu, J.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J.M. Selective production of mono-aromatics from lignocellulose over Pd/C catalyst: The influence of acid co-catalysts. Faraday Discuss. 2017, 202, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Renders, T.; Schutyser, W.; Van den Bosch, S.; Koelewijn, S.F.; Vangeel, T.; Courtin, C.M.; Sels, B.F. Influence of Acidic (H3PO4) and Alkaline (NaOH) Additives on the Catalytic Reductive Fractionation of Lignocellulose. ACS Catal. 2016, 6, 2055–2066. [Google Scholar] [CrossRef]
- Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Schutyser, W.; Sels, B.F. Lignin-first biomass fractionation: The advent of active stabilisation strategies. Energy Environ. Sci. 2017, 10, 1551–1557. [Google Scholar] [CrossRef]
- Shu, R.; Long, J.; Xu, Y.; Ma, L.; Zhang, Q.; Wang, T.; Wang, C.; Yuan, Z.; Wu, Q. Investigation on the structural effect of lignin during the hydrogenolysis process. Bioresour. Technol. 2016, 200, 14–22. [Google Scholar] [CrossRef]
- Shu, R.Y.; Xu, Y.; Ma, L.L.; Zhang, Q.; Wang, T.J.; Chen, P.R.; Wu, Q.Y. Hydrogenolysis process for lignosulfonate depolymerization using synergistic catalysts of noble metal and metal chloride. RSC Adv. 2016, 6, 88788–88796. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, J.; Hwang, H.; Kim, J.K.; Song, I.K.; Choi, J.W. Catalytic depolymerization of lignin macromolecule to alkylated phenols over various metal catalysts in supercritical tert-butanol. J. Anal. Appl. Pyrol. 2015, 113, 99–106. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, J.; Kim, U.J.; Choi, J.W. Conversion of Lignin to Phenol-Rich Oil Fraction under Supercritical Alcohols in the Presence of Metal Catalysts. Energy Fuels 2015, 29, 5154–5163. [Google Scholar] [CrossRef]
- Yuan, Z.S.; Tymchyshyn, M.; Xu, C.B. Reductive Depolymerization of Kraft and Organosolv Lignin in Supercritical Acetone for Chemicals and Materials. ChemCatChem 2016, 8, 1968–1976. [Google Scholar] [CrossRef]
- Lama, S.M.G.; Pampel, J.; Fellinger, T.P.; Beskoski, V.P.; Slavkovic-Beskoski, L.; Antonietti, M.; Molinari, V. Efficiency of Ni Nanoparticles Supported on Hierarchical Porous Nitrogen-Doped Carbon for Hydrogenolysis of Kraft Lignin in Flow and Batch Systems. ACS Sustain. Chem. Eng. 2017, 5, 2415–2420. [Google Scholar] [CrossRef]
- Park, J.; Oh, S.; Kim, J.Y.; Park, S.Y.; Song, I.K.; Choi, J.W. Comparison of degradation features of lignin to phenols over Pt catalysts prepared with various forms of carbon supports. RSC Adv. 2016, 6, 16917–16924. [Google Scholar] [CrossRef]
- Bouxin, F.P.; McVeigh, A.; Tran, F.; Westwood, N.J.; Jarvis, M.C.; Jackson, S.D. Catalytic depolymerisation of isolated lignins to fine chemicals using a Pt/alumina catalyst: Part 1—Impact of the lignin structure. Green Chem. 2015, 17, 1235–1242. [Google Scholar] [CrossRef]
- Barta, K.; Warner, G.R.; Beach, E.S.; Anastas, P.T. Depolymerization of organosolv lignin to aromatic compounds over Cu-doped porous metal oxides. Green Chem. 2014, 16, 191–196. [Google Scholar] [CrossRef]
- Barta, K.; Matson, T.D.; Fettig, M.L.; Scott, S.L.; Iretskii, A.V.; Ford, P.C. Catalytic disassembly of an organosolv lignin via hydrogen transfer from supercritical methanol. Green Chem. 2010, 12, 1640–1647. [Google Scholar] [CrossRef]
- Narani, A.; Chowdari, R.K.; Cannilla, C.; Bonura, G.; Frusteri, F.; Heeres, H.J.; Barta, K. Efficient catalytic hydrotreatment of Kraft lignin to alkylphenolics using supported NiW and NiMo catalysts in supercritical methanol. Green Chem. 2015, 17, 5046–5057. [Google Scholar] [CrossRef] [Green Version]
- Verziu, M.; Tirsoaga, A.; Cojocaru, B.; Bucur, C.; Tudora, B.; Richel, A.; Aguedo, M.; Samikannu, A.; Mikkola, J.P. Hydrogenolysis of lignin over Ru-based catalysts: The role of the ruthenium in a lignin fragmentation process. Mol. Catal. 2018, 450, 65–76. [Google Scholar] [CrossRef]
- Konnerth, H.; Zhang, J.G.; Ma, D.; Prechtl, M.H.G.; Yan, N. Base promoted hydrogenolysis of lignin model compounds and organosolv lignin over metal catalysts in water. Chem. Eng. Sci. 2015, 123, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.L.; Tian, Y.; Hao, W.Y.; Ma, R.; Li, Y.D. Production of phenols from catalytic conversion of lignin over a tungsten phosphide catalyst. Appl. Catal. A Gen. 2014, 481, 64–70. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, S.Y.; Choi, I.G.; Choi, J.W. Evaluation of RuxNi1-x/SBA-15 catalysts for depolymerization features of lignin macromolecule into monomeric phenols. Chem. Eng. J. 2018, 336, 640–648. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, Q.; Shu, R.; Xu, Y.; Ma, L.; Wang, T. Catalytic depolymerization of the hydrolyzed lignin over mesoporous catalysts. Bioresour. Technol. 2017, 226, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.P.; Wang, S.Z.; Li, H.L.; Li, Z.W.; Shi, Z.J.; Xiao, L.; Sun, R.G.; Fang, Y.M.; Song, G.Y. Catalytic Hydrogenolysis of Lignins into Phenolic Compounds over Carbon Nanotube Supported Molybdenum Oxide. ACS Catal. 2017, 7, 7535–7542. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Wang, H.; Ma, Q.; Li, S.; Chang, H.M.; Jameel, H. Liquefaction of kraft lignin by hydrocracking with simultaneous use of a novel dual acid-base catalyst and a hydrogenation catalyst. Bioresour. Technol. 2017, 243, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Hao, W.; Ma, X.; Tian, Y.; Li, Y. Catalytic ethanolysis of Kraft lignin into high-value small-molecular chemicals over a nanostructured alpha-molybdenum carbide catalyst. Angew. Chem. Int. Ed. 2014, 53, 7310–7315. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.L.; Ma, R.; Hao, W.Y.; Chen, M.M.; Iran, F.; Cui, K.; Tian, Y.; Li, Y.D. Common Pathways in Ethanolysis of Kraft Lignin to Platform Chemicals over Molybdenum-Based Catalysts. ACS Catal. 2015, 5, 4803–4813. [Google Scholar] [CrossRef]
- Ma, X.; Cui, K.; Hao, W.; Ma, R.; Tian, Y.; Li, Y. Alumina supported molybdenum catalyst for lignin valorization: Effect of reduction temperature. Bioresour. Technol. 2015, 192, 17–22. [Google Scholar] [CrossRef]
- Singh, S.K.; Ekhe, J.D. Cu–Mo doped zeolite ZSM-5 catalyzed conversion of lignin to alkyl phenols with high selectivity. Catal. Sci. Technol. 2015, 5, 2117–2124. [Google Scholar] [CrossRef]
- Huang, S.H.; Mahmood, N.; Zhang, Y.S.; Tymchyshyn, M.; Yuan, Z.S.; Xu, C.B. Reductive de-polymerization of kraft lignin with formic acid at low temperatures using inexpensive supported Ni-based catalysts. Fuel 2017, 209, 579–586. [Google Scholar] [CrossRef]
- Huang, S.; Mahmood, N.; Tymchyshyn, M.; Yuan, Z.; Xu, C.C. Reductive de-polymerization of kraft lignin for chemicals and fuels using formic acid as an in-situ hydrogen source. Bioresour. Technol. 2014, 171, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Liguori, L.; Barth, T. Palladium-Nafion SAC-13 catalysed depolymerisation of lignin to phenols in formic acid and water. J. Anal. Appl. Pyrol. 2011, 92, 477–484. [Google Scholar] [CrossRef]
- Limarta, S.O.; Ha, J.M.; Park, Y.K.; Lee, H.; Suh, D.J.; Jae, J. Efficient depolymerization of lignin in supercritical ethanol by a combination of metal and base catalysts. J. Ind. Eng. Chem. 2018, 57, 45–54. [Google Scholar] [CrossRef]
- Molinari, V.; Clavel, G.; Graglia, M.; Antonietti, M.; Esposito, D. Mild Continuous Hydrogenolysis of Kraft Lignin over Titanium Nitride–Nickel Catalyst. ACS Catal. 2016, 6, 1663–1670. [Google Scholar] [CrossRef]
- Luo, L.; Yang, J.; Yao, G.; Jin, F. Controlling the selectivity to chemicals from catalytic depolymerization of kraft lignin with in-situ H2. Bioresour. Technol. 2018, 264, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Nandeshwar, K.; Ekhe, J.D. Thermochemical lignin depolymerization and conversion to aromatics in subcritical methanol: Effects of catalytic conditions. New J. Chem. 2016, 40, 3677–3685. [Google Scholar] [CrossRef]
- Huang, X.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J. Catalytic depolymerization of lignin in supercritical ethanol. ChemSusChem 2014, 7, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atay, C.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Role of Cu–Mg–Al Mixed Oxide Catalysts in Lignin Depolymerization in Supercritical Ethanol. ACS Catal. 2015, 5, 7359–7370. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.M.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J.M. Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics. Green Chem. 2015, 17, 4941–4950. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Atay, C.; Zhu, J.; Palstra, S.W.L.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J.M. Catalytic Depolymerization of Lignin and Woody Biomass in Supercritical Ethanol: Influence of Reaction Temperature and Feedstock. ACS Sustain. Chem. Eng. 2017, 5, 10864–10874. [Google Scholar] [CrossRef]
- Jeong, S.; Yang, S.; Kim, D.H. Depolymerization of Protobind lignin to produce monoaromatic compounds over Cu/ZSM-5 catalyst in supercritical ethanol. Mol. Catal. 2017, 442, 140–146. [Google Scholar] [CrossRef]
- Zhou, M.H.; Sharma, B.K.; Liu, P.; Ye, J.; Xu, J.M.; Jiang, J.C. Catalytic in Situ Hydrogenolysis of Lignin in Supercritical Ethanol: Effect of Phenol, Catalysts, and Reaction Temperature. ACS Sustain. Chem. Eng. 2018, 6, 6867–6875. [Google Scholar] [CrossRef]
- Li, Z.; Bi, Z.; Yan, L. Two-step hydrogen transfer catalysis conversion of lignin to valuable small molecular compounds. Green Process. Synth. 2017, 6, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Onwudili, J.A.; Williams, P.T. Catalytic depolymerization of alkali lignin in subcritical water: Influence of formic acid and Pd/C catalyst on the yields of liquid monomeric aromatic products. Green Chem. 2014, 16, 4740–4748. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Pineda, A.; Romero, A.A.; Luque, R.; Labidi, J. Microwave-assisted depolymerisation of organosolv lignin via mild hydrogen-free hydrogenolysis: Catalyst screening. Appl. Catal. B Envrion. 2014, 145, 43–55. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Labidi, J.; Pineda, A.; Balu, A.M.; Luque, R. Heterogeneously Catalysed Mild Hydrogenolytic Depolymerisation of Lignin Under Microwave Irradiation with Hydrogen-Donating Solvents. ChemCatChem 2013, 5, 977–985. [Google Scholar] [CrossRef]
- Xu, W.; Miller, S.J.; Agrawal, P.K.; Jones, C.W. Depolymerization and hydrodeoxygenation of switchgrass lignin with formic acid. ChemSusChem 2012, 5, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Kloekhorst, A.; Shen, Y.; Yie, Y.; Fang, M.; Heeres, H.J. Catalytic hydrodeoxygenation and hydrocracking of Alcell® lignin in alcohol/formic acid mixtures using a Ru/C catalyst. Biomass Bioenergy 2015, 80, 147–161. [Google Scholar] [CrossRef]
- Park, J.; Riaz, A.; Insyani, R.; Kim, J. Understanding the relationship between the structure and depolymerization behavior of lignin. Fuel 2018, 217, 202–210. [Google Scholar] [CrossRef]
- Warner, G.; Hansen, T.S.; Riisager, A.; Beach, E.S.; Barta, K.; Anastas, P.T. Depolymerization of organosolv lignin using doped porous metal oxides in supercritical methanol. Bioresour. Technol. 2014, 161, 78–83. [Google Scholar] [CrossRef]
- Klamrassamee, T.; Laosiripojana, N.; Faungnawakij, K.; Moghaddam, L.; Zhang, Z.Y.; Doherty, W.O.S. Co- and Ca-phosphate-based catalysts for the depolymerization of organosolv eucalyptus lignin. RSC Adv. 2015, 5, 45618–45621. [Google Scholar] [CrossRef] [Green Version]
- Regmi, Y.N.; Mann, J.K.; McBride, J.R.; Tao, J.M.; Barnes, C.E.; Labbe, N.; Chmely, S.C. Catalytic transfer hydrogenolysis of organosolv lignin using B-containing FeNi alloyed catalysts. Catal. Today 2018, 302, 190–195. [Google Scholar] [CrossRef]
- Liu, S.; Lin, Z.; Cai, Z.; Long, J.; Li, Z.; Li, X. Selective depolymerization of lignosulfonate via hydrogen transfer enhanced in an emulsion microreactor. Bioresour. Technol. 2018, 264, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Oregui-Bengoechea, M.; Gandarias, I.; Arias, P.L.; Barth, T. Unraveling the Role of Formic Acid and the Type of Solvent in the Catalytic Conversion of Lignin: A Holistic Approach. ChemSusChem 2017, 10, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Kristianto, I.; Limarta, S.O.; Lee, H.; Ha, J.M.; Suh, D.J.; Jae, J. Effective depolymerization of concentrated acid hydrolysis lignin using a carbon-supported ruthenium catalyst in ethanol/formic acid media. Bioresour. Technol. 2017, 234, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.T.; Li, Z.; Tang, X.; Sun, Y.; Zeng, X.H.; Liu, S.J.; Lin, L. Depolymerization of Cellulolytic Enzyme Lignin for the Production of Monomeric Phenols over Raney Ni and Acidic Zeolite Catalysts. Energy Fuels 2015, 29, 1662–1668. [Google Scholar] [CrossRef]
- Das, L.; Li, M.; Stevens, J.; Li, W.Q.; Pu, Y.Q.; Ragauskas, A.J.; Shi, J. Characterization and Catalytic Transfer Hydrogenolysis of Deep Eutectic Solvent Extracted Sorghum Lignin to Phenolic Compounds. ACS Sustain. Chem. Eng. 2018, 6, 10408–10420. [Google Scholar] [CrossRef]
- Seo, M.; Yoon, D.; Hwang, K.S.; Kang, J.W.; Kim, J. Supercritical alcohols as solvents and reducing agents for the synthesis of reduced graphene oxide. Carbon 2013, 64, 207–218. [Google Scholar] [CrossRef]
- Tudorache, M.; Opris, C.; Cojocaru, B.; Apostol, N.G.; Tirsoaga, A.; Coman, S.M.; Parvulescu, V.I.; Duraki, B.; Krumeich, F.; van Bokhoven, J.A. Highly Efficient, Easily Recoverable, and Recyclable Re–SiO2–Fe3O4 Catalyst for the Fragmentation of Lignin. ACS Sustain. Chem. Eng. 2018, 6, 9606–9618. [Google Scholar] [CrossRef]
- Opris, C.; Cojocaru, B.; Gheorghe, N.; Tudorache, M.; Coman, S.M.; Parvulescu, V.I.; Duraki, B.; Krumeich, F.; van Bokhoven, J.A. Lignin fragmentation over magnetically recyclable composite Co@Nb2O5@Fe3O4 catalysts. J. Catal. 2016, 339, 209–227. [Google Scholar] [CrossRef]
Figure 1.
Chemicals derived by the valorization of lignocellulosic biomass. Reproduced from reference [27] with permission from MDPI.
Figure 1.
Chemicals derived by the valorization of lignocellulosic biomass. Reproduced from reference [27] with permission from MDPI.

Figure 2.
Building blocks of lignin.

Figure 3.
Schematic representations of (a) softwood and (b) hardwood lignin structures. Reprinted with permission from reference [3]. Copyright 2010, American Chemical Society.
Figure 3.
Schematic representations of (a) softwood and (b) hardwood lignin structures. Reprinted with permission from reference [3]. Copyright 2010, American Chemical Society.


Figure 4.
Lignin model compounds studied in the catalytic reductive depolymerization.

Figure 5.
Reaction profiles of (a) 1-(2,4-dihydroxyphenyl)-2-(4-methoxyphenoxy)-ethanone (DHPMPE) and (b) guaiacylglycerol-β-guaiacylether (GGGE) lignin model compounds in 50 vol.% ethanol + 50 vol.% water, Pt/C, 275 °C, 80 bar. Reproduced from reference [71] with permission from Elsevier.
Figure 5.
Reaction profiles of (a) 1-(2,4-dihydroxyphenyl)-2-(4-methoxyphenoxy)-ethanone (DHPMPE) and (b) guaiacylglycerol-β-guaiacylether (GGGE) lignin model compounds in 50 vol.% ethanol + 50 vol.% water, Pt/C, 275 °C, 80 bar. Reproduced from reference [71] with permission from Elsevier.

Figure 6.
Yield of phenolic compounds produced by the lignin-first approach as a function of β-O-4′ moiety in the native lignin of different lignocellulose substrates. Reproduced from reference [76] with permission from John Wiley and Sons.
Figure 6.
Yield of phenolic compounds produced by the lignin-first approach as a function of β-O-4′ moiety in the native lignin of different lignocellulose substrates. Reproduced from reference [76] with permission from John Wiley and Sons.

Figure 7.
Possible mechanism for the selective formation of phenol, 3-methoxy, 2,5,6-trimethyl (PMT). Reproduced from reference [113] by permission of The Royal Society of Chemistry.
Figure 7.
Possible mechanism for the selective formation of phenol, 3-methoxy, 2,5,6-trimethyl (PMT). Reproduced from reference [113] by permission of The Royal Society of Chemistry.

Figure 8.
Qualitative liquid products distribution from Kraft lignin depolymerization under the different ratio of isopropanol to water (0.1 g kraft lignin, 40 wt.% Rh/La2O3/CeO2-ZrO2 catalyst, 373 °C reaction temperature, 2 h reaction time and 2 mmol Fe). Reproduced from reference [119] with permission from Elsevier.
Figure 8.
Qualitative liquid products distribution from Kraft lignin depolymerization under the different ratio of isopropanol to water (0.1 g kraft lignin, 40 wt.% Rh/La2O3/CeO2-ZrO2 catalyst, 373 °C reaction temperature, 2 h reaction time and 2 mmol Fe). Reproduced from reference [119] with permission from Elsevier.

Figure 9.
Product distribution following reaction: (a) blank reaction at 300 °C for 4 h in ethanol, (b) CuMgAlOx at 300 °C for 4 h in ethanol, (c) CuMgAlOx at 300 °C for 4 h in methanol, (d) CuMgAlOx at 300 °C for 8 h in ethanol. The numbers in the molecules correspond to the molecular weight, whereas colors are used to facilitate the reading of the figure. Reproduced from reference [121] with permission from John Wiley and Sons.
Figure 9.
Product distribution following reaction: (a) blank reaction at 300 °C for 4 h in ethanol, (b) CuMgAlOx at 300 °C for 4 h in ethanol, (c) CuMgAlOx at 300 °C for 4 h in methanol, (d) CuMgAlOx at 300 °C for 8 h in ethanol. The numbers in the molecules correspond to the molecular weight, whereas colors are used to facilitate the reading of the figure. Reproduced from reference [121] with permission from John Wiley and Sons.

Figure 10.
Possible reaction routes during lignin depolymerization in ethanol in the presence of catalyst. Reproduced from reference [121] with permission from John Wiley and sons.
Figure 10.
Possible reaction routes during lignin depolymerization in ethanol in the presence of catalyst. Reproduced from reference [121] with permission from John Wiley and sons.

Figure 11.
Products yields (%, w/w) referenced to initial lignin weight of bio-oil (black), biochar (white) and residual lignin (grey) derived by hydrogenolysis of lignin under mild, microwave-assisted, H2 free conditions, using various solvents: tetralin (TL), glycerol (GLY), formic acid (FA) and isopropanol (IP). Reaction conditions: 0.5 g lignin, 0.5 g catalyst, 12.5 mL solvent, microwaves, Taverage = 423 K, t = 30 min. Reproduced from reference [130] with permission from John Wiley and sons.
Figure 11.
Products yields (%, w/w) referenced to initial lignin weight of bio-oil (black), biochar (white) and residual lignin (grey) derived by hydrogenolysis of lignin under mild, microwave-assisted, H2 free conditions, using various solvents: tetralin (TL), glycerol (GLY), formic acid (FA) and isopropanol (IP). Reaction conditions: 0.5 g lignin, 0.5 g catalyst, 12.5 mL solvent, microwaves, Taverage = 423 K, t = 30 min. Reproduced from reference [130] with permission from John Wiley and sons.

Figure 12.
Proposed mechanism for formic acid-aided depolymerization of lignin through formylation, elimination, and hydrogenolysis. Reproduced from reference [138] with permission from John Wiley and sons.
Figure 12.
Proposed mechanism for formic acid-aided depolymerization of lignin through formylation, elimination, and hydrogenolysis. Reproduced from reference [138] with permission from John Wiley and sons.


{kind=link}
{kind=link}
{kind=link}
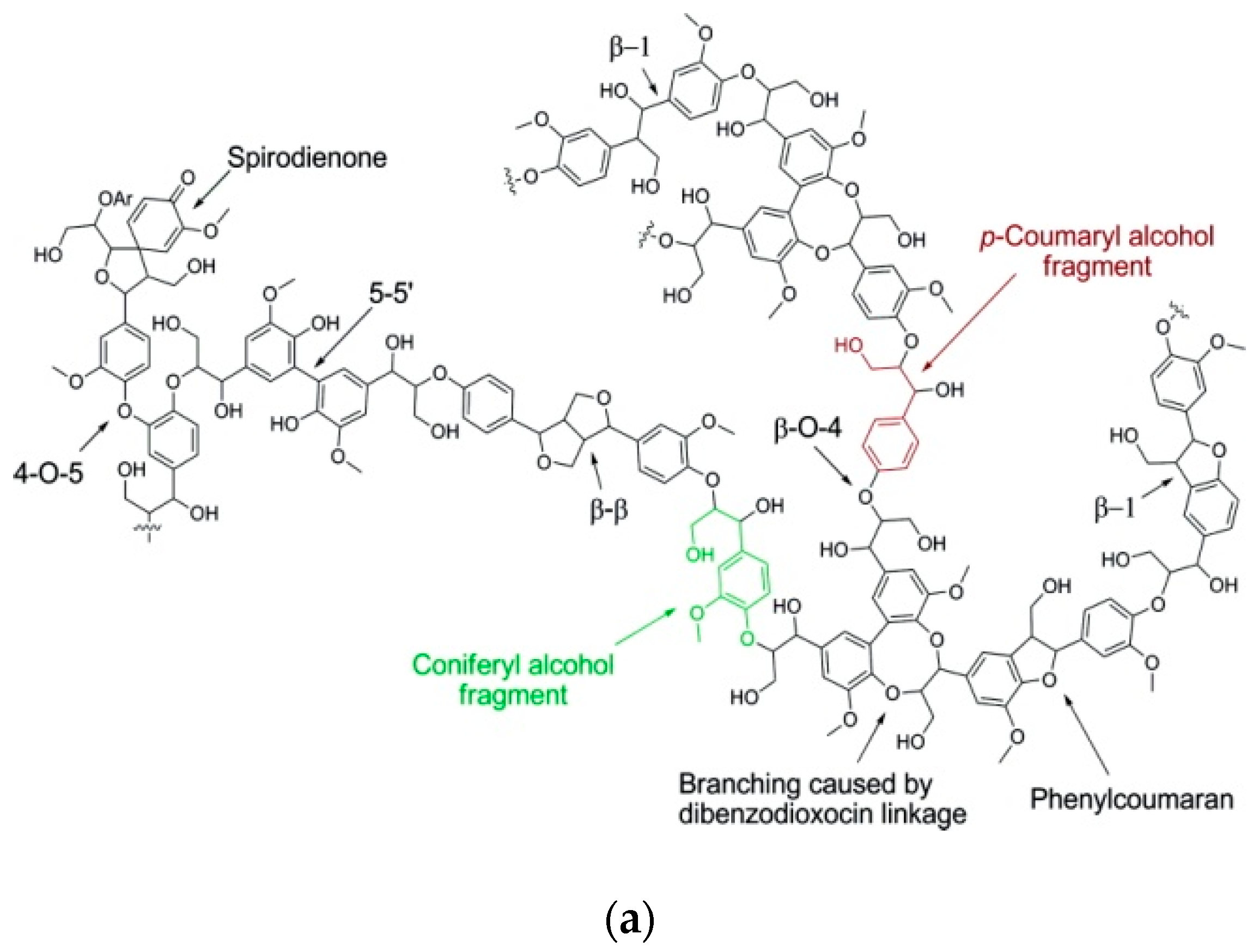{kind=link}
{kind=link}
{kind=link}
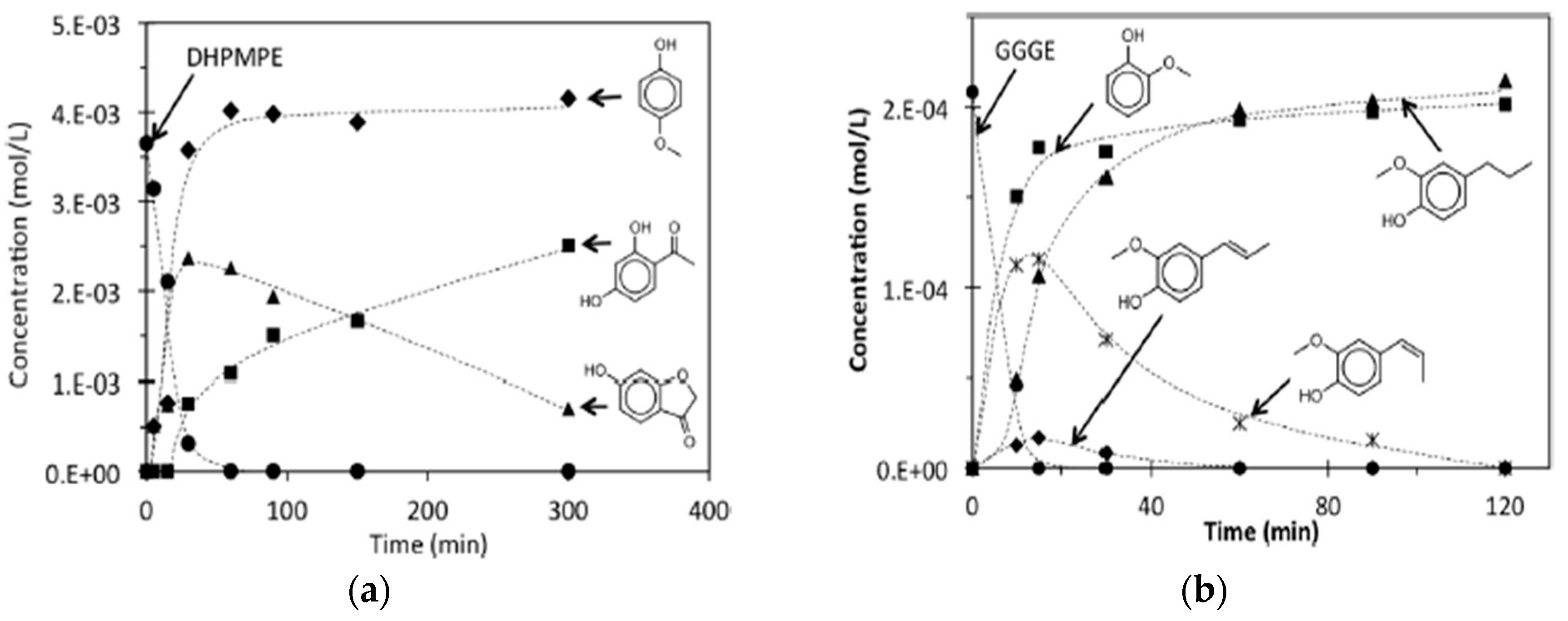{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
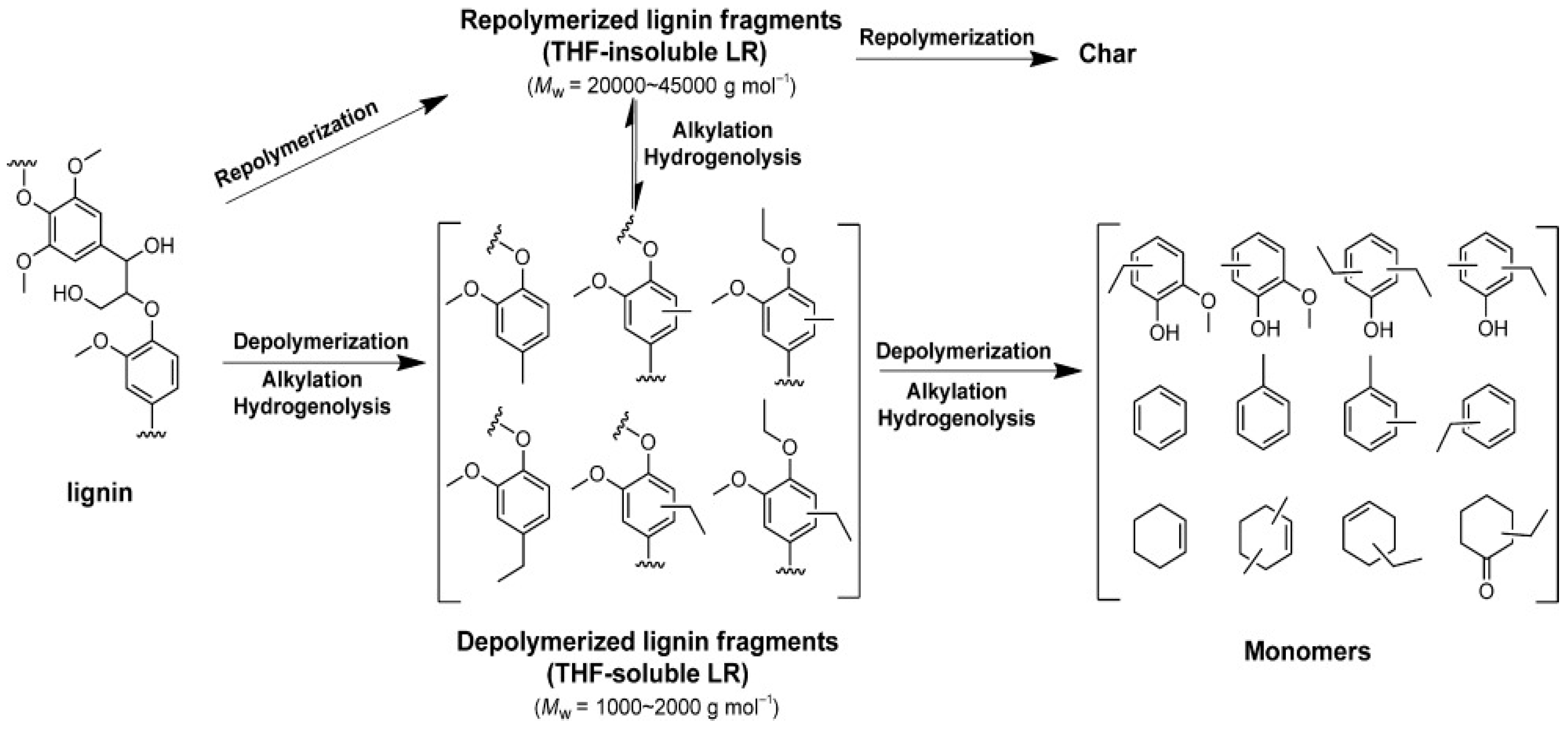{kind=link}
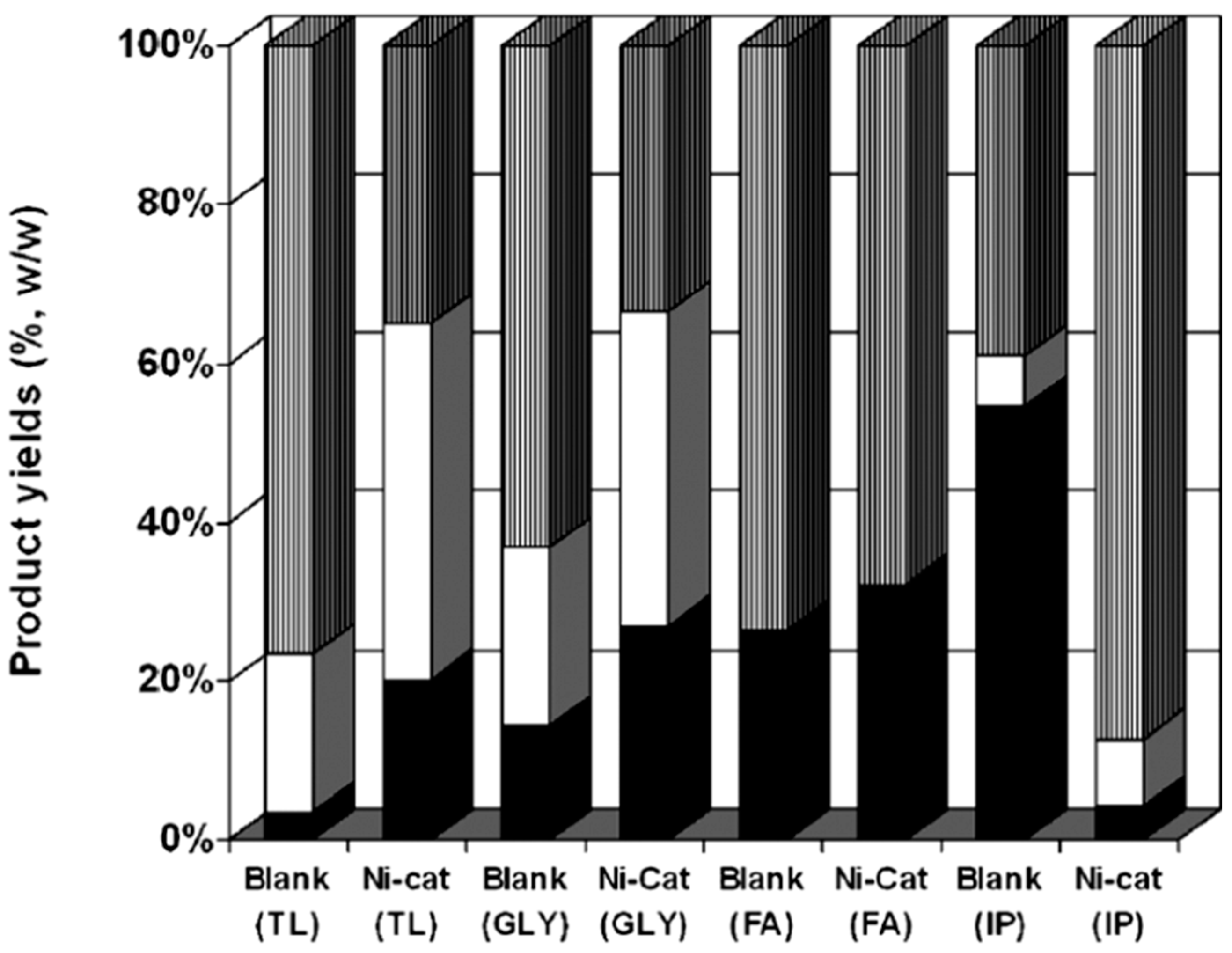{kind=link}
{kind=link}
Table 1.
Major lignin isolation processes and the properties of the obtained lignin [4].
Table 1.
Major lignin isolation processes and the properties of the obtained lignin [4].
| Process | Agent | T (°C) | MW (Da) | Polydispersity | Contamination |
|---|---|---|---|---|---|
| Kraft | NaOH + Na2S | 170 | 1000–3000 | 2.5–3.5 | Sulfur |
| Soda | NaOH + anthraquinone | 140–170 | 1000–3000 | 2.5–3.5 | Sulfur |
| Sulfite | sulfite salts | 140–170 | 1000–50,000 | 4.2–7.0 | Sulfur |
| Organosolv | organic solvents | 180–200 | 500–5000 | 1.5 | Sulfur free |
Table 2.
Product yields, molecular weight averages of bio-oils, and monomer yields obtained from the depolymerization of kraft lignin over MgO loaded on different supports (i.e., carbon, Al2O3, and ZrO2), Ru/C, and physical mixtures of Ru/C and each MgO catalyst at 350 °C after a reaction time of 60 min in the 40 mL batch reactor. Reproduced from reference [117] with permission from Elsevier.
Table 2.
Product yields, molecular weight averages of bio-oils, and monomer yields obtained from the depolymerization of kraft lignin over MgO loaded on different supports (i.e., carbon, Al2O3, and ZrO2), Ru/C, and physical mixtures of Ru/C and each MgO catalyst at 350 °C after a reaction time of 60 min in the 40 mL batch reactor. Reproduced from reference [117] with permission from Elsevier.
| Catalyst | Product Yield (wt.%) | Molecular Weight of Bio-Oil (g/mol) | Monomer Yield (wt.%) | ||||
|---|---|---|---|---|---|---|---|
| Bio-Oil | SR | Mn (g/mol) | Mw (g/mol) | Total | Phenolic Monomers | Aliphatic Esters | |
| No catalyst | 36.2 | 47.3 | 347 | 737 | 1.75 | 1.70 | 0.04 |
| MgO/C 10% a | 43.3 | 37.3 | 370 | 834 | 2.10 | 1.83 | 0.26 |
| MgO/Al2O3 10% | 42.1 | 37.5 | 383 | 844 | 1.98 | 1.76 | 0.23 |
| MgO/ZrO2 10% | 47.5 | 32.7 | 365 | 776 | 4.22 | 3.90 | 0.32 |
| Ru/C 10% | 88.1 | 8.8 | 398 | 906 | 5.05 | 4.54 | 0.51 |
| Ru/C 10% + MgO/C 10% | 79.2 | 10.0 | 371 | 944 | 5.73 | 5.15 | 0.58 |
| Ru/C 10% + MgO/Al2O3 10% | 70.9 | 21.0 | 434 | 1064 | 4.52 | 3.76 | 0.77 |
| Ru/C 10% + MgO/ZrO2 10% | 82.7 | 11.3 | 323 | 832 | 6.10 | 5.16 | 0.94 |
a Percent value indicates the catalyst loading amount with respect to lignin weight.
Table 3.
Product yields from lignin depolymerization with additives at 265 °C, 6.5 MPa from 1–6 h hold time. Reproduced from reference [128] by permission of The Royal Society of Chemistry.
Table 3.
Product yields from lignin depolymerization with additives at 265 °C, 6.5 MPa from 1–6 h hold time. Reproduced from reference [128] by permission of The Royal Society of Chemistry.
| Sample | Hold Time (h) | Liquid (wt.%) | Solid Residue (wt.%) | Gas (wt.%) | % Balance |
|---|---|---|---|---|---|
| Lignin | 1.0 | 58.2 | 30.6 | 5.56 | 94.4 |
| Lignin | 3.0 | 51.6 | 35.9 | 9.12 | 96.6 |
| Lignin | 6.0 | 33.3 | 46.0 | 17.0 | 96.3 |
| Lignin/FA | 1.0 | 61.6 | 0.64 | 36.5 | 98.7 |
| Lignin/FA | 3.0 | 52.3 | 0.19 | 46.1 | 98.6 |
| Lignin/FA | 6.0 | 41.1 | 1.12 | 58.3 | 101 |
| Lignin/Pd/C | 1.0 | 41.3 | 54.4 | 0.84 | 96.5 |
| Lignin/Pd/C | 3.0 | 36.1 | 57.3 | 1.25 | 94.7 |
| Lignin/Pd/C | 6.0 | 35.6 | 59.1 | 2.25 | 97.0 |
| Lignin/FA-Pd/C | 1.0 | 45.8 | 16.3 | 38.7 | 101 |
| Lignin/FA-Pd/C | 3.0 | 38.5 | 18.3 | 49.8 | 107 |
| Lignin/FA-Pd/C | 6.0 | 31.6 | 22.9 | 53.4 | 108 |
| Pd/C | 6.0 | - | 98.5 | - | 98.5 |
Table 4.
Compounds identified in appreciable quantities in lignin bio-oils obtained by the hydrogenolysis of organsolv lignin from olive tree prunings under microwave irradiation using Ni, Pd, Pt, Ru catalysts supported on Al-SBA-15. Compounds yields expressed as mg of each compound per gram of lignin. Reproduced from ref. [129] with permission from Elsevier.
Table 4.
Compounds identified in appreciable quantities in lignin bio-oils obtained by the hydrogenolysis of organsolv lignin from olive tree prunings under microwave irradiation using Ni, Pd, Pt, Ru catalysts supported on Al-SBA-15. Compounds yields expressed as mg of each compound per gram of lignin. Reproduced from ref. [129] with permission from Elsevier.
| Blank | Ni2% AlSBA | Ni5% AlSBA | Ni10% AlSBA | Pd2% AlSBA | Pt2% AlSBA | Ru2% AlSBA | |
|---|---|---|---|---|---|---|---|
| Mesitol (2) | 0.56 | 2.26 | 1.19 | 1.13 | 0.49 | 1.67 | 1.40 |
| 2,3,6-Trimehtylphenol (3) | 0.19 | 0.76 | 0.22 | 0.28 | 0.18 | 0.17 | 0.45 |
| 6-Ethyl-o-cresol (5) | 0.13 | 0.14 | 0.04 | - | - | 0.32 | 0.19 |
| 4-Ethyl-m-cresol (6) | - | 0.11 | 0.04 | - | - | 0.08 | - |
| Vanillin (7) | 0.20 | 0.20 | 0.19 | 0.20 | - | 0.13 | 0.11 |
| Diethylphthalate (10) | 2.34 | 10.10 | 1.67 | 1.69 | 10.93 | 10.66 | 7.44 |
| 3,4-Dimethoxyphenol (11) | 0.05 | 0.12 | 0.05 | 0.15 | 0.24 | 0.09 | 0.09 |
| Syringaldehyde (12) | 0.47 | 0.68 | 0.57 | 0.56 | 0.15 | 0.43 | 0.43 |
| Butyl-octyl ester phthalic acid (16) | 0.21 | 0.36 | 0.30 | 0.19 | 0.34 | 0.36 | 0.37 |
a Reaction conditions: 0.5 g lignin, 0.5 g catalyst, 12.5 mL tetralin, microwaves, 400 W, average temperature 140 °C, 30 min reaction.
Table 5.
Catalytic depolymerization of sodium lignosulfonate in emulsion microreactor. Reproduced from reference [137] with permission from Elsevier.
Table 5.
Catalytic depolymerization of sodium lignosulfonate in emulsion microreactor. Reproduced from reference [137] with permission from Elsevier.
| Solvent (mL) | Yield of Phenolic Monomers (mg∙g−1) | ||||
|---|---|---|---|---|---|
| H2O | n-BuOH | i-PrOH | 4-Ethyl Guaiacol | Others | Total |
| 25 | / | / | 18.8 ± 0.3 | 37.3 ± 0.5 | 56.1 ± 0.7 |
| 20 | 5.0 | / | 22.0 ± 0.4 | 39.8 ± 0.6 | 61.8 ± 0.8 |
| 15 | 10 | / | 23.5 ± 0.2 | 41.9 ± 0.5 | 65.4 ± 0.7 |
| 10 | 15 | / | 22.3 ± 0.3 | 40.2 ± 0.6 | 62.5 ± 0.8 |
| 20 | / | 5.0 | 21.3 ± 0.5 | 32.1 ± 0.6 | 53.4 ± 1.1 |
| 15 | 5.0 | 5.0 | 24.8 ± 0.4 | 50.2 ± 1.9 | 75.0 ± 2.2 |
| 10 | 5.0 | 10 | 33.8 ± 0.3 | 50.3 ± 2.5 | 84.1 ± 2.8 |
| 10 | 10 | 5.0 | 27.7 ± 0.1 | 52.8 ± 3.0 | 80.5 ± 3.0 |
| 5.0 | 15 | 5.0 | 27.2 ± 0.5 | 55.5 ± 2.4 | 82.7 ± 2.8 |
| 5.0 | 10 | 10 | 29.9 ± 0.6 | 65.5 ± 3.1 | 95.4 ± 3.5 |
| 5.0 | 5.0 | 15 | 39.3 ± 0.7 | 76.8 ± 2.3 | 116.1 ± 2.8 |
Condition: sodium lignosulfonate 0.5 g, Raney Ni 2.0 g, hexane 0.5 mL, 473 K for 2.0 h.
Table 6.
Representative optimum results on catalytic hydrogenolysis of various lignins using small alcohols or formic acid as hydrogen donors.
Table 6.
Representative optimum results on catalytic hydrogenolysis of various lignins using small alcohols or formic acid as hydrogen donors.
| Lignin | Catalyst & Reaction Conditions | Conversion/Yields a (wt.%) | Main Products | Reference |
|---|---|---|---|---|
| Kraft | α-MoC1−x/AC, EtOH, 280 °C | 100/-/- | Esters, alcohols, aromatics | [110] |
| Kraft | Cu/Mo-ZSM-5, H2O:MeOH=1:1, 220 °C, NaOH | 95.7/-/19.9 | Phenolics | [113] |
| Kraft | 10% Ni/Zeolite, H2O:EtOH=1:1, HCOOH, 200 °C | 93.5 b | Aromatics | [114] |
| Kraft | Pd-Nafion SAC-13, H2O, HCOOH, 300 °C | -/-/10.5 | Phenolics | [116] |
| Kraft | MgO/ZrO2, EtOH, 350 °C | -/47.5/4.22 | Phenolics | [117] |
| Kraft | Ru/C+ MgO/ZrO2, EtOH, 350 °C | -/82.7/6.10 | Phenolics, higher alcohols, aliphatic esters | [117] |
| Kraft | HZSM-5, MeOH, 220 °C | 85.1/-/4.2 | Phenolics | [120] |
| Kraft | Iron turnings, MeOH, 220 °C | 44.4/-/1.7 | Phenolics | [120] |
| Soda | CuMgAlOx, EtOH, 300 °C | -/-/17 | Aromatics, furans, hydrogenated cyclics | [121,122,123,124] |
| Soda | 10 wt.% Cu/ZSM-5(30), EtOH, 440 °C | -/-/98.2 | Aromatics | [125] |
| Alkali | CuNiAl-HT, EtOH, Phenol:lignin=0.8, 290 °C | -/72.3/- | - | [126] |
| Alkali | Raney Ni, i-PrOH/H2O, 180 °C | 93.5/-/- | Phenolics-aromatics | [127] |
| Alkali | Pd/C, H2O, HCOOH, 265 °C | -/45.8/- | Phenolics | [128] |
| Organosolv | 10% Ni/Al-SBA-15, Tetralin, 140 °C c | -/16.94/- | Phenolics, esters | [129] |
| Organosolv | 10% Ni/Al-SBA-15, HCOOH, 150 °C c | -/28.89/- | Phenolics | [130] |
| Organosolv | 20 wt.% Pt/C, EtOH, HCOOH, 350 °C | -/-/21 | Phenolics | [131] |
| Organosolv | Ru/C, i-PrOH, HCOOH, 400 °C | -/71.2/- | ketones aromatics, phenolics | [132] |
| Organosolv | Cu20PMO, MeOH, 310 °C | 48.3/-/- | Phenolics, aromatics | [134] |
| Organosolv | FeNiB alloys, EtOH, 320 °C | -/-/- | Phenolics | [136] |
| Enzymatic hydrolysis | NiMo/sulfated alumina, EtOH, HCOOH, 320 °C | -/38.4/- | methoxy, hydroxyl, alkyl benzenes | [138] |
| Acid hydrolysis | 5% Ru/C, EtOH, HCOOH, 300 °C | -/62.9/- | Phenolics, esters | [139] |
| Enzymatic hydrolysis | HUSY+Raney Ni, MeOH/H2O, 250 °C | -/-/27.9 | Phenolics | [140] |
| DES Extracted | Ru/C, i-PrOH, 270 °C | -/36.28/- | Phenolics, acids | [141] |
a Conversion/Bio-oil/ Monomers, b depolymerized lignin, c Microwave irradiation.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Margellou, A.; Triantafyllidis, K.S. Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals. Catalysts 2019, 9, 43. https://doi.org/10.3390/catal9010043
AMA Style
Margellou A, Triantafyllidis KS. Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals. Catalysts. 2019; 9(1):43. https://doi.org/10.3390/catal9010043
Chicago/Turabian StyleMargellou, Antigoni, and Konstantinos S. Triantafyllidis. 2019. "Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals" Catalysts 9, no. 1: 43. https://doi.org/10.3390/catal9010043
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.