Performance and Stability of Metal (Co, Mn, Cu)-Promoted La2O2SO4 Oxygen Carrier for Chemical Looping Combustion of Methane


Istituto Ricerche sulla Combustione—CNR, 80125 Napoli, Italy
*
Authors to whom correspondence should be addressed.
Catalysts 2019, 9(2), 147; https://doi.org/10.3390/catal9020147
Submission received: 18 December 2018
/
Revised: 16 January 2019
/
Accepted: 23 January 2019
/
Published: 2 February 2019
(This article belongs to the Special Issue Catalysts Deactivation, Poisoning and Regeneration)
Abstract
:Oxygen carrier materials based on La2O2SO4 and promoted by small amounts (1% wt.) of transition metals, namely Co, Mn and Cu, have been synthesized and characterized by means of X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET), Temperature-programmed reduction/oxidation (TPR/TPO) and thermogravimetry-mass-Fourier transform infrared spectrometry (TG-MS-FTIR) experiments under alternating feeds in order to investigate their potential use for the Chemical Looping Combustion process using either hydrogen or methane as the fuel. The chemical looping reactivity is based on the reversible redox cycle of sulfur from S6+ in La2O2SO4 to S2− in La2O2S and entails a large oxygen storage capacity, but it generally requires high temperatures to proceed, challenging material stability and durability. Herein we demonstrate a remarkable improvement of lattice oxygen availability and activity during the reduction step obtained by cost-effective metal doping in the order Co > Mn > Cu. Notably, the addition of Co or Mn has shown a significant beneficial effect to prevent the decomposition of the oxysulfate releasing SO2, which is identified as the main cause of progressive deactivation for the unpromoted La2O2SO4.
1. Introduction
Chemical Looping Combustion (CLC) represents one of the most investigated approaches for clean combustion of fossil fuels because it provides an easy CO2 capture [1,2,3] by splitting the process into two cyclic half-steps. The same two-step technology has been also proposed for Chemical Looping Reforming [1,2,3].
Large oxygen storage capacity (OSC), fast oxidation and reduction kinetics and high stability of the oxygen carrier (OC) are crucial parameters for process development [4]. A large variety of OCs have been proposed, most of them based on transition metals (Cu, Ni, Fe, Mn, Co) as bulk or supported simple oxides or as more complex oxides such as perovskite-like materials [3,5,6]. Perovskites show a better thermal stability and adjustable properties related to the partial or total substitution of A and B metal cations in their ABO3 structure [6].
Recently, Miccio et al. [7,8] have dispersed iron and iron/manganese oxides into a geopolymer matrix to provide low density materials with good resistance to mechanical stresses that can be suitable for fluidized bed reactors.
Additionally, CaSO4 has been investigated due to its potentially higher oxygen transport capacity compared to transition metal oxides, deriving from the redox cycle of sulfur [3,9,10]. Nevertheless, a significant sulfur release due to decomposition at high temperature represents an unsolved issue for CaSO4, in addition to agglomeration and sintering that are typical of metal oxides (MeOx). An attempt to face the loss of efficiency has been proposed by adding elemental iron [9].
Machida and co-workers [11,12,13,14,15,16] have deeply investigated rare earths based oxysulfates (Ln2O2SO4) as stable, durable, and high performing oxygen storage materials in three ways catalysts for automotive emission control. Sulfur can undergo a complete reduction from S6+ in the oxysulfate to S2− in the oxysulfide structure by reaction with a reducing agent and it can be reversibly re-oxidized to S6+ by reaction with molecular oxygen, provided that a suitable temperature is chosen for the two processes. Indeed, both the Ln2O2SO4 and its corresponding reduced compound Ln2O2S crystallize in a similar structure consisting of alternating layers of Ln2O22+ and SO42− or S2− anions. The phase transformation is therefore regarded as the removal or insertion of O2− surrounding sulfur [13]. The thermal stability of Ln2O2SO4 decreases with increasing the atomic number of Ln [11,17]. Accordingly, it has been reported that Lu2O2SO4 cannot be reduced to the corresponding oxysulfide, since it transforms directly into Lu2O3 and SO2 under reducing atmosphere [16]. Furthermore, Machida et al. [12,13,14] found that among other Ln-oxysulfates, Pr2O2SO4 is able to operate the redox cycles at the lowest temperatures, due to the beneficial effect of the existence of Pr3+/Pr4+ surface species on the redox of sulfur. They also reported that sulfate reduction becomes easier with increasing distortion of the tetragonal SO4 unit in the oxysulfate, as evidenced in Pr2O2SO4 structure [13]. However, due to the high cost and scarce availability of praseodymium, substitution of the more common lanthanum is difficult to pursue [13]. In conclusion, the oxygen storage properties of Ln2O2SO4 can be enhanced (i) by introducing redox species and/or (ii) by increasing distortion of the SO4 unit as in the case of the partial substitution of Ce for La in (La1−xCex)2O2SO4 [14] and/or (iii) by the addition of noble metals on the surface (1% wt. Pd or Pt), likely promoting the redox cycle of Ln2O2SO4 by hydrogen/oxygen spillover effect [11,12,13,14].
The cost-effective addition of small quantities of Cu [10] or Na and Fe [18] to La2O2SO4, as well as Ni to Pr2O2SO4 [19] has been recently reported to enhance oxygen storage capability (OSC) and storage/release reaction rates, in agreement with the distortion of the SO4 unit [13,15], thus allowing operation at lower temperatures than with parent oxysulfates. It can be argued that cations with different dimensions can alter the lattice to a different extent, thus affecting the OSC and stability of materials.
In this paper, we set out to investigate the effect of the addition of a low amount (1% wt.) of three different transition metals (Cu, Mn, Co) on the OSC as well as on stability/durability of a La2O2SO4 oxygen carrier. Chemical looping reaction experiments have been performed under alternating feed conditions at a fixed temperature in the range 700–900 °C, using either hydrogen or methane as the fuel (i.e., reducing agent). The emission of sulfur compounds under repeated redox cycles has been investigated to provide a detailed analysis of conditions promoting the undesired decomposition of the oxygen carrier.
2. Results
In Table 1 the list of metal-promoted OCs is reported with the corresponding values of Brunauer–Emmett–Teller (BET) surface area. The original surface area of the as-prepared La2O2SO4 is rather low (2.7 m2/g), in agreement with previous results reported for the simple preparation method based on the thermal decomposition of bulk lanthanum sulfate at 1027 °C under inert flow [20,21]. The further addition of 1% wt. of transition metals followed by heat treatment at 900 °C does not significantly alter the specific surface area of the materials.
In Figure 1a X-ray diffraction (XRD) patterns of fresh La2O2SO4 and transition metal-promoted La2O2SO4 are reported. All spectra show the characteristic lines of monoclinic lanthanum oxysulfate (JCPDS 085-1535). The presence of the transition metals causes a decrease of crystallinity, as shown by the lower intensity of XRD peaks, which is particularly evident for the case of Co-La2O2SO4. As mentioned in the Introduction, the structure of La2O2SO4 consists of alternating stacks of La2O22+ layers and layers of anions SO42− [12,15], therefore the possible inclusion of the transition metal into the lattice can generate a distortion since the radius of the transition metal ion differs from that of La3+ (1.03 Å). Notably, for the same oxidation state (including the metallic state), cobalt shows the smallest dimensions. In particular, the ionic radius of Me3+, supposed to substitute La3+ in the lattice, is 0.60 Å for cobalt and 0.70 Å for manganese. Copper, which is generally in the +2 oxidation state, has a ionic radius as Cu2+ similar to Mn3+ (0.73Å). That could explain the lowest crystallinity of the Co-La2O2SO4 material.
The effect of metal doping on the thermal stability of La2O2SO4 has been studied by thermogravimetry-mass spectrometry (TG-MS) experiments ramping up to 1300 °C at 10 °C/min under N2 flow: the corresponding derivative weight loss plots (Figure 2a) are superimposed and flat for all materials, indicating an almost constant weight up to ca. 1050 °C. Above this temperature, which corresponds well to the decomposition temperature previously reported for La oxysulfate [21], all materials experience a significant weight loss, at similar increasing rates up to 1300 °C, where the process is not yet completed (during 10 min under isothermal conditions). Accordingly, the MS traces for SO2 (m/z = 64), O2 (m/z = 32) and CO2 (m/z = 48) in the evolved gas display similar trends for undoped La2O2SO4 (Figure 2b) as well as for metal- doped materials (exemplified in Figure 2c for the case of Mn-La2O2SO4). In particular, a limited CO2 release can be observed in the temperature range of 250–500 °C corresponding to the decomposition of same superficial La- carbonates. A rather small SO2 peak in the range of 800–950 °C suggests the decomposition of some residual La sulfates. Eventually, a large SO2 release begins at temperatures around 1050 °C, which is accompanied by the evolution of O2, thus suggesting the occurrence of La oxysulfate decomposition to La2O3. Poston et al. [21] detected (by XRD analysis) the simultaneous formation of La2O3 and La2O2S phases during thermal treatment of La2O2SO4 at T ≥ 1050 °C in air. According to our results, the oxygen release never occurs independently from SO2. Therefore, it can be concluded that under N2 flow none of the doped or undoped La oxysulfates is able to release oxygen and turns spontaneously into the corresponding oxysulfide without decomposing.
Sequential H2-TPR/O2-TPO analysis have been performed to compare the redox behavior of the carriers and results are reported in Figure 3a,b.
The main H2 consumption event starts for all materials (promoted or not) at ca. 630 °C, and the total H2 uptake accounts for the complete transformation of La2O2SO4 into the La2O2S [10]. However, Co-La2O2SO4 is most easily reduced among others, as testified by the clear shift towards lower temperatures of its H2 consumption peak showing a maximum at ca. 810 °C. Notably, Cu- and Mn-promoted La2O2SO4 samples show their peaks respectively at 855 °C and 890 °C, whereas the undoped La oxysulfate requires as much as 925 °C. Furthermore, Cu- and Co-promoted OCs display additional small peaks in the low temperature region that can be assigned respectively to the reduction of some segregated/supported CuO and Co3O4 [22]. In the case of Mn-La2O2SO4 a very small peak at ca. 560 °C could be related to the reduction of some Mn2O3 [23], whereas the eventual presence of Mn3O4 could not be detected since its reduction requires higher temperatures [23] and would, therefore, be masked by the onset of reduction of La2O2SO4.
The same trend is confirmed by the results of the TG-MS experiments carried out in the thermo-balance under a 2%H2/N2 flow: Figure 4a and Table 2 show all samples experience a similar weight loss (ca. 15.2%), which approaches the theoretical value (15.8%) estimated from the reduction reaction:
La2O2SO4 + 4H2 → La2O2S + 4H2O
Small differences are due to some impurities, such as carbonates, which are present in the original samples (Figure 2b,c). In contrast, the complete transformation of La2O2SO4 into La2O3 would account for a weight loss as large as 19.8%. The onset temperature of weight loss associated with the reduction of La2O2SO4 (ca.630 °C) is not affected by doping with Cu or Mn, whereas Co addition lowers it by 30 °C (Table 2). However, all transition metal (oxides) dopants act as a catalyst to facilitate the surface reaction with the reducing gas, confirming the order of activity is Cu < Mn < Co.
In particular, as shown in Table 2, Co-doping lowers the temperature required to complete the transformation of La2O2SO4 into La2O2S by as much as 200 °C. In the case of Pd- and Pt-promoted La2O2SO4 or Pr2O2SO4 [11,14] it was inferred that the easier reducibility at lower temperatures is caused by the higher reactivity of atomic hydrogen produced via H2 spillover from noble metal nanoparticles with respect to molecular H2. On the contrary, Me-promoted La2O2SO4 materials show similar onset temperatures but, thereafter, reduction proceeds at a significantly faster rate, suggesting this effect could be rather a consequence of distortion of the oxysulfate lattice.
A serious issue for the durability of La oxysulfates comes from their irreversible decomposition [14] while reacting under a reducing atmosphere. Figure 4b presents the MS profiles for SO2 emission recorded during TG experiments under H2/N2 flow. In fact, besides the H2 consumption and the corresponding emission of water deriving from its oxidation (not shown), a small SO2 emission is observed for each material, peaking at the same temperature of the inflection point of the corresponding TG curve (Table 2). Osseni et al. [17] observed by XRD the bulk formation of Lu2O3 during Lu2O2SO4 reduction with H2 at 650, 690 and 800 °C via reaction (2), which possibly occurs to a limited extent also when Ln = La:
Ln2O2SO4 + H2 → Ln2O3 + SO2 + H2O
In the present case the amount of SO2 emitted, estimated by peak area after specific calibration, is always rather small: it accounts for ca. 0.06% of the original weight for the case of the undoped and Cu-doped La2O2SO4, whereas it drops down to 0.009% for Co- and Mn-doped materials, possibly due to their faster oxygen mobility and lower temperature level required to complete reduction.
Moreover, a second SO2 emission starts from 925–945 °C for all OCs. In fact, the two SO2 emission peaks are partly overlapped for the unpromoted La2O2SO4. It can be argued that the second SO2 emission corresponds to the decomposition of some residual lanthanum oxysulfate [19]; however, the reaction of La-oxysulfide with residual water [19] present in the chamber cannot be ruled out
Therefore, present results suggest that a faster oxygen mobility in the OC enables operation under conditions far from those leading to a significant and irreversible decomposition.
La2O2S + 2H2O → La2O3 + SO2 + 2H2
As shown in Figure 3b, during TPO the undoped La2O2S starts to be reoxidized at ca. 700 °C displaying a broad peak centered at 850 °C, with an estimated oxygen consumption corresponding to the theoretical value needed for the restoration of La2O2SO4 [10]. All metal promoted OCs in their oxysulfide form display similar onset temperatures for reoxidation, although the associated O2 consumption peak is somehow broader than for the undoped material and its maximum occurs at around 900 °C. Unresolved signals also appear in the low-mid temperature region of the TPO profiles of metal-doped OC materials that are related to the reoxidation of Cu or MeOx phases [10].
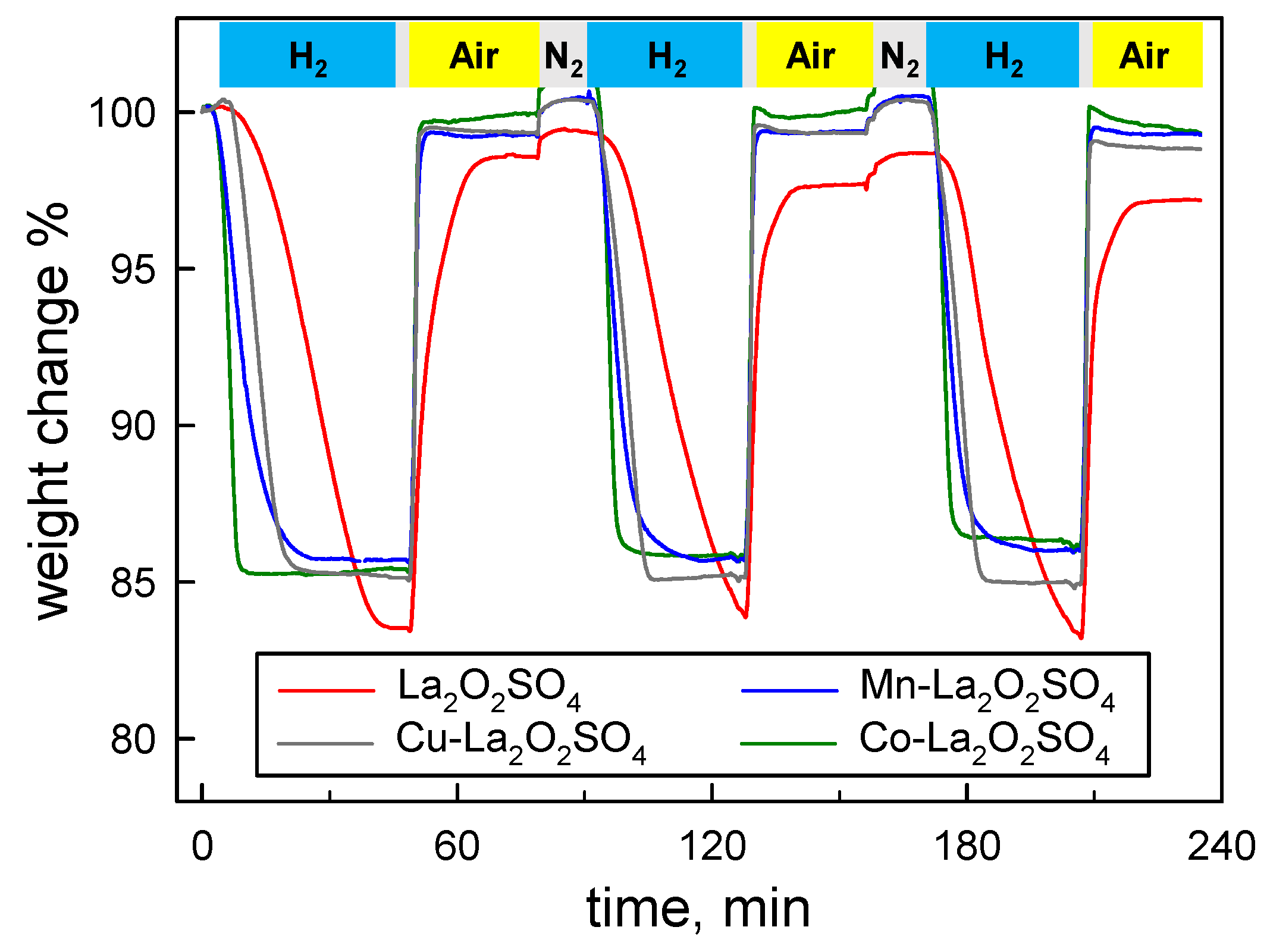
The oxygen storage performance has been next evaluated at constant temperature under alternating feed stream conditions, where reducing (fuel) and oxidizing (air) gases are cycled. Figure 5 shows the weight changes recorded for doped and undoped OC materials at 800 °C during three cycles obtained alternating 5% H2/N2 and air flows. During the first reduction step, all metal-promoted materials undergo a weight loss of ca. 15%, which is compatible with an almost complete reduction of La2O2SO4 to La2O2S. However, the unpromoted La2O2SO4 material shows a larger weight loss that slightly exceeds the value expected for the transformation into the corresponding oxysulfide. Moreover, La2O2SO4 needs ca. 40 min to complete its weight loss, whereas all metal promoted samples require a shorter time: the rate of reaction increases in the order Cu < Mn < Co.
Re-oxidation to lanthanum oxysulfate occurs immediately after the exposure to air flow for the three metal promoted materials (showing superimposed TG traces in Figure 5), whilst it takes about 20 min for the unpromoted sample. In fact, the larger weight loss displayed by La2O2SO4 during its first reduction step is irreversible, as confirmed by the incomplete recovery of its original weight after reoxidation. The second and the third cycles show similar qualitative features, Co-La2O2SO4 quickly turning into Co-La2O2S and Cu- and Mn-promoted carriers taking few more minutes. On the contrary, the reduction of pure La2O2SO4 appears even harder than in the first cycle, since the weight loss is not completed within the time allowed for this step. Slowing down of the reduction rate of La2O2SO4 after the first reduction/reoxidation cycle was already observed [10] during cyclic H2-TPR/O2-TPO experiments with maximum temperatures of 1027 °C. Therefore, a progressive loss of the initial redox properties of La2O2SO4 occurs also when the chemical looping process is operated at a fixed and relatively moderate temperature level.
Lowering the reaction temperature mainly affects the time required to complete the reduction step with H2, which indeed represents the limiting step, whereas reoxidation in air results always very fast. In fact, due to its intrinsically easier reducibility, only Co-La2O2SO4 is able to complete full redox cycles at 700 °C (not shown) within the given time (45 min).
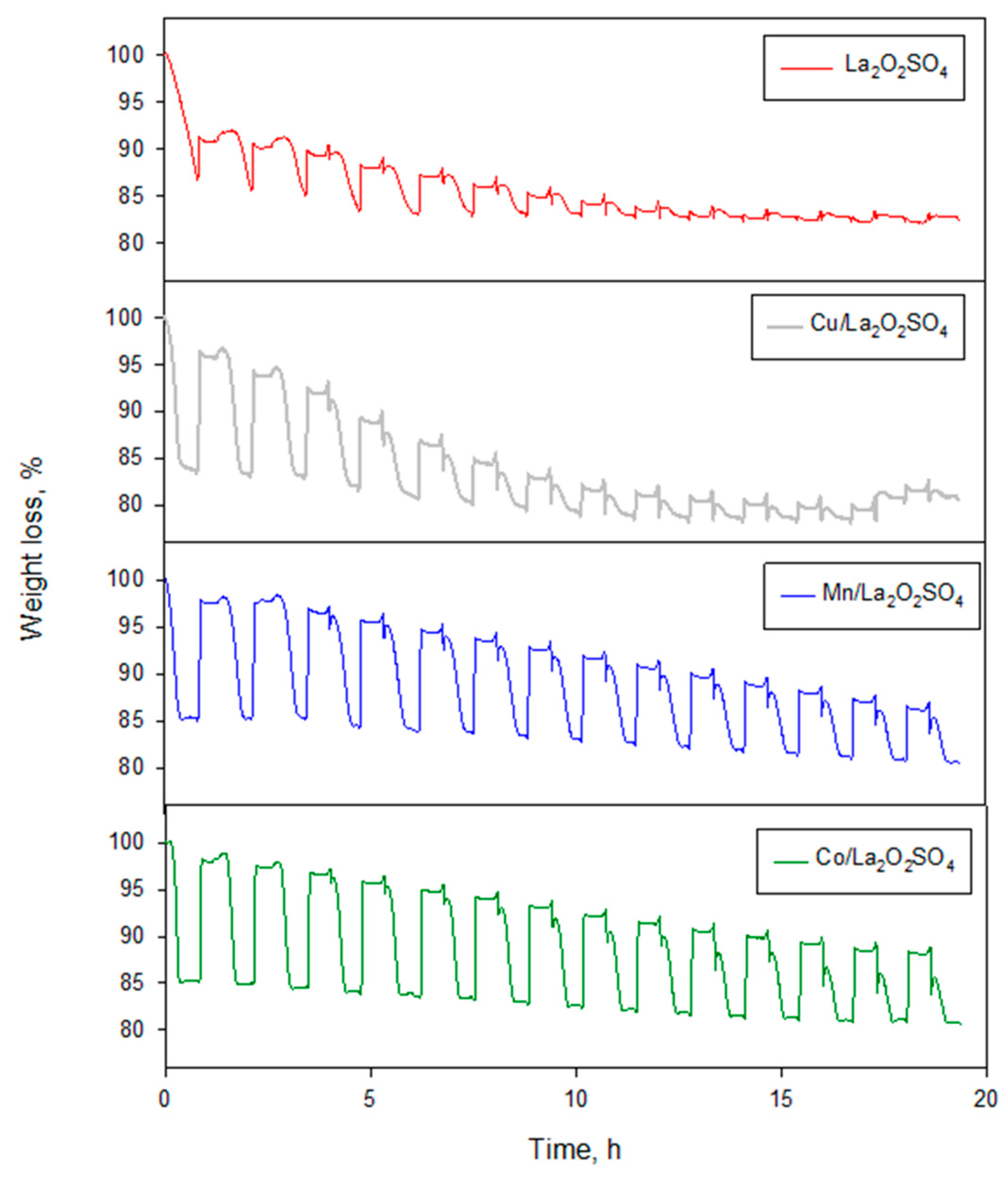
In order to extend this investigation to a more common fuel, chemical looping combustion in the flow microbalance has been performed using methane (5% vol. in N2) as the reducing agent. Due to its much lower reactivity with respect to H2, the operating temperature has been preliminarily set at 900 °C. The oxygen storage performance of the carriers has been verified during 15 cycles (Figure 6), which also allows testing their thermo-chemical stability.
All OCs, except the unpromoted one, initially display the typical weight loss of ca. 15%, suggesting the transformation of La2O2SO4 into La2O2S can occur also by reaction with methane. Judging by the slope of the weight loss profile, the reaction rate increases in the order Cu < Mn < Co, thus following the same trend of reactivity found with H2 as fuel. On the contrary, the weight loss of La2O2SO4 is not yet completed after 45 min under CH4 flow. Thereafter, reoxidation by air does not completely restore the original weight of samples, particularly for those less reactive materials. In fact, the OSC of the undoped La2O2SO4 is progressively reduced in the subsequent cycles and vanishes after the 10th cycle.
Doping by copper has a modest beneficial effect on the residual OSC, but the weight of the sample progressively approaches a stable level compatible with the irreversible transformation into La2O3. On the other hand, the promoting effect of manganese and particularly that of cobalt is evident both in terms of reaction rates as well as residual oxygen storage capacity, which is still ca. 45% of its initial value for Co-La2O2SO4 during the last cycle.
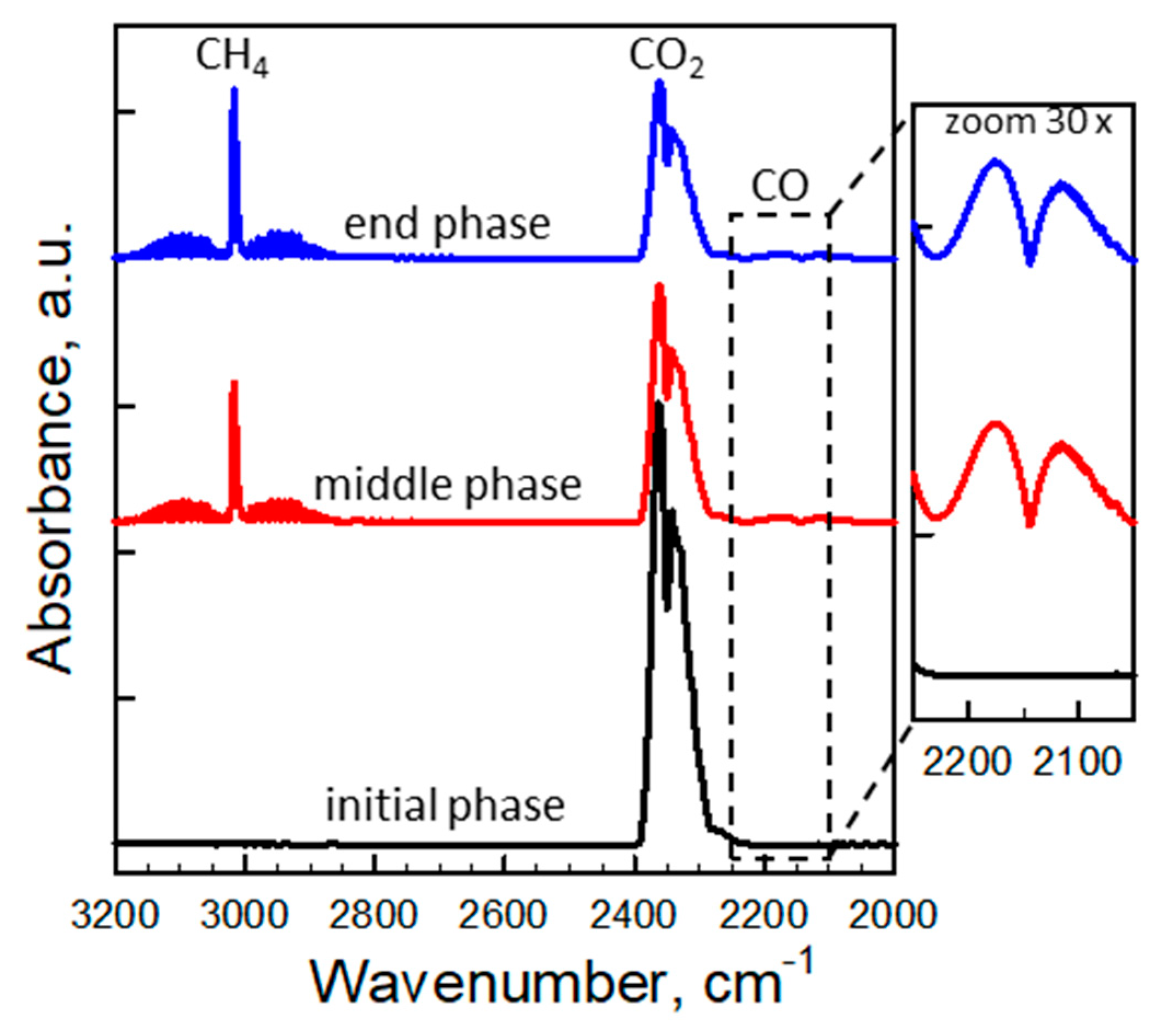
Figure 7 shows the infrared (IR) spectra of evolved gas collected at three times (initial, middle, end) during the 7th reducing step for Co-La2O2SO4. The band around 2340 cm−1 is assigned to CO2 formed by the reaction between CH4 and lattice oxygen from the material (Equation (4)). CO2 is the only product detected during the initial phase of reaction over Co- and Mn-doped materials, which promote the deep oxidation of methane with 100% selectivity.
Thereafter, the intensity of the CO2 signal declines, as a consequence of a reduction in the lattice oxygen mobility/availability and, therefore, in the overall reaction rate. Notably, the appearance of a weak doublet band at 2170–2115 cm−1 (insert of Figure 7) indicates the formation of only small amounts of CO in addition to CO2 via the partial oxidation of methane (Equation (5)), while the characteristic band centered at 3020 cm−1 is associated to increasing amounts of unconverted methane. As expected, a comparison of IR spectra of all OCs (not shown) highlights that the order of deep oxidation rate of methane follows the same trend of lattice oxygen mobility/availability in the OC materials.
La2O2SO4 + CH4 → La2OsS + CO2 + 2H2O
La2O2SO4 + 2CH4 → La2OsS + 2CO + 4H2O
XRD patterns of La2O2SO4 and metal-promoted La2O2SO4 at the end of 15 cycles are presented in Figure 1b. The low intensity of signals compared to the corresponding ones in Figure 1a is due to the small amount of each material (12–16 mg) available after the experiments in the flow microbalance. All samples show quite complex patterns suggesting the presence of two or more crystalline phases. In particular they all show a well detectable peak at ca. 13° that can be assigned to lanthanum hydroxide (JCPDS 36–1481) produced by hydration of La2O3. This confirms the loss of oxygen storage capacity is caused by the irreversible decomposition of La2O2SO4 with formation of lanthanum oxide. While most of the signals in the XRD patters of La2O2SO4 and Cu-La2O2SO4 can be attributed to La(OH)3, Mn- and Co-promoted carriers still show well detectable peaks at 25°, 28°, 36.9°, 44.9° characteristic of La2O2S2 phase. Therefore, the addition of Mn and Co can prevent to some extent the decomposition of lanthanum oxysulfate/oxysulfide. This represents an interesting result considering that the enhancement of oxygen mobility is probably associated with a higher distortion of SO4 units caused by inclusion of cobalt or manganese ions into the oxysulfate/oxysulfide structure. Therefore, the better crystallized, un-promoted La2O2SO4 is more prone to loose sulfate units than the more amorphous Co-La2O2SO4.
In order to get further insights into the possible reactions occurring during cycled anaerobic oxidation of methane over the best performing Co-La2O2SO4, further TG experiments have been carried out analyzing evolved gases with the mass spectrometer. Figure 8 shows the weight change and the corresponding mass signals for the main gaseous products (m/z = 2, 18, 44, 64) as recorded at 900 °C by alternating reducing and oxidizing flows (40min), separated by a N2 purge (30 min) in between. As soon as exposed to methane, Co-La2O2SO4 is quickly and completely reduced according to equation (4), giving H2O and CO2 as primary products, as confirmed by the sharp and simultaneous MS peaks recorded in the effluent gas. After the expected weight loss (ca. 15.5%) is completed, a small weight increase is observed, that is probably related to some coke deposition on the sample deriving from CH4 decomposition catalyzed by Co [24]. Accordingly, a sudden emission of H2 shows up after the production of CO2 and water has stopped, and it continues during the purge phase, as long as some methane is still available in the reacting chamber. While the lattice oxygen from the OC material is consumed to oxidize methane, an evident peak of SO2 emission appears that ends when the OC is fully reduced. Notably, the maximum SO2 concentration is ca. two orders of magnitude lower than CO2. This phenomenon resembles what observed during H2-TPR experiments (Figure 4), and indicates a limited decomposition of La2O2SO4 can occur also by reaction with methane leading to SO2 and the products of methane partial oxidation (Equation (6)):
However, the formation of CO (m/z = 28) cannot be easily detected because of the use of N2 as the carrier gas.
La2O2SO4 + CH4 → La2O3 + SO2 + 2 H2 + CO
Due to the formation of some La2O3, the OC material cannot fully recover its original weight during the fast oxidation occurring as soon as it is exposed to air flow. During this phase, a small CO2 peak appears, caused by the oxidation of coke deposits previously formed on the material. The stepwise increase in the signal of H2O is assigned to humidity contained in the air flow. Moreover, a new but rather small emission of SO2 is detected during the reoxidation, therefore the occurrence of reaction (7) should be also considered:
During the subsequent cycles, the temporal profiles of the gaseous species are repeatable.
La2O2S + 3/2 O2 → La2O3 + SO2
Considering the high oxygen mobility in Co-La2O2SO4, the cyclic anaerobic oxidation of methane has been tested also at 800 °C and 750 °C in order to verify if the adverse impact of decomposition reactions (Equations (6) and (7)) could be limited. Results are shown in Figure 9 and Figure 10, respectively. As expected, at 800 °C the reduction of the oxysulfate takes place at a slower rate but the corresponding weight loss is completed within ca. 30 min under CH4 flow. Accordingly, water and CO2 are produced during the whole reduction phase, resulting in wider emission peaks than at 900 °C. Some H2 appears only at the end of this step, but is not accompanied by any detectable weight increase. It is inferred that CH4 decomposition does not significantly proceed at 800 °C, whereas some methane partial oxidation can occur when lattice oxygen from the OC is almost completely exploited. The SO2 emission during the reduction step at 800 °C is at least one order of magnitude lower than at 900 °C, confirming the hypothesis on the beneficial effect of a lower reaction temperature to limit the decomposition of the oxysulfate into the corresponding oxide. Re-oxidation under air at 800 °C is still very rapid, and the weight recovery is almost complete. However, the second (and third) reduction step is slower than the first one so that the transformation into the oxysulfide phase ends only during the purging step. As a consequence, CO2 and H2O profiles show broader peaks with similar area but lower maxima with respect to the first cycle and shifted towards the end of the period. The SO2 emission during reoxidation remains quite low. The results of the third cycle are repeatable.
As shown on the top of Figure 10, when operating at 750 °C the duration of the reduction step has been prolonged up to 90 min. The slower rate of reduction of the OC material limits its final weight loss to 15.3%, indicating that the transformation into the oxysulfide phase is not yet completed and ca. 96.5% of the total OSC has been consumed. The corresponding production of water and CO2 by methane oxidation with lattice oxygen is characterized by the presence of two emission peaks: the first one is sharp and located at the beginning of the exposure to CH4, whereas the second peak is broad and centered at ca. 45 min. It can be argued that the initial formation of an outer shell of La2O2S may slow down the reaction due to the increase of diffusional limitations (in the solid state). H2 formation is initially low and increases towards the end of the period, testifying an increase of process selectivity towards the products of partial oxidation of methane due to the slow oxygen availability from the OC material. However, no SO2 emission can be detected (sub-ppm level) during operation at 750 °C. The subsequent reoxidation by air is still fast. As already observed at 800 °C, the second reduction step is slower than the first one: based on the final weight of the OC, ca. 80% of its total OSC has been utilized. Moreover, the fraction of La2O2S displays a lower attitude to be fully re-oxidized, since the sample does not completely recover its previous weight. The results of the third cycle are repeatable, confirming that the partial reduction of the OS performance is not related to material stability issues (decomposition) but rather to an incomplete transformation between La2O2SO4 and La2O2S phases due to the reduced oxygen mobility.
Overall the results indicate that the Co-promoted La2O2SO4/La2O2S couple can be favorably operated at 800 °C in the chemical looping combustion of methane: it shows a high oxygen mobility that promotes complete oxidation of methane and full exploitation of its outstanding OS capacity, and a remarkable stability/durability during cyclic operation. Notably, the stability of the La2O2SO4/La2O2S system could be strongly enhanced in the presence of even small contents of S-bearing compounds [14], commonly found in the fuel stream. On the other hand, further studies should address the possibility to enhance lattice oxygen mobility and overall reactivity at lower temperatures, by increasing the specific surface area of the OC [18], and by varying the content of transition metal (oxide) dopant which could effectively work as an intermediate oxygen gateway [16] apart from its role as structural promoter.
3. Materials and Methods
3.1. Preparation of Oxygen Carriers
Lanthanum oxysulfate (La2O2SO4) was obtained starting from La2(SO4)3 (Sigma-Aldrich, 99.99% purity, St. Louis, MO, USA) by heating it at 1027 °C for 5 h under helium flow [10,20]. Different fractions of La2O2SO4 powder were impregnated with water solutions of Cu(NO3)2·2.5H2O (Sigma-Aldrich, purity ≥ 98%, St. Louis, MO, USA), Mn(NO3)2·4H2O (Sigma-Aldrich, purity ≥ 97%, St. Louis, MO, USA) and Co(NO3)2·6H2O (Sigma-Aldrich, purity ≥ 98%, St. Louis, MO, USA) to obtain samples with a nominal 1wt% metal loading. After impregnation, samples were dried for 12 h at 120 °C in a stove and eventually heat treated at 900 °C for 2 h under He.
3.2. Physical and Chemical Characterization
The surface area was evaluated according to the BET method by N2 adsorption at 77K with a Quantachrome Autosorb 1-C analyzer (Quantachrome Instruments, Boynton Beach, FL, USA).
Powder X-ray diffraction (XRD) analysis was performed with a Bruker D2 Phaser diffractometer (Bruker, Billerica, MA, USA) operated at diffraction angles ranging between 10 and 80° 2θ with a scan rate of 0.02° 2θ s−1.
Temperature Programmed Reduction (TPR) and Oxidation (TPO) experiments were carried out with a Micromeritics Autochem II TPD/TPR instrument (Micromeritics, Norcross, GA, USA) equipped with a TC detector. The samples (20–30 mg) were first reduced under a flow of 2% H2 in Ar (50 Ncm3/min) by heating up to 1027 °C at 10 °C/min; thereafter, they were reoxidized under a flow of 0.5% O2 in He (50 Ncm3/min) using the same temperature ramp.
TG analysis were performed in a Setaram Labsys Evo TGA-DTA-DSC 1600 (Setaram Instrumentation, Caluire, France) flow microbalance loading 15–25 mg of sample in an alumina crucible and ramping the temperature at 10 °C/min up to 1300 °C or 1150 °C, respectively under a flow (100 cc/min) of pure N2 or 2% H2 in N2.
3.3. Chemical Looping Reaction
Cyclic isothermal reduction/oxidation tests were carried out in the same Setaram Labsys Evo flow microbalance at 700, 750, 800 and 900 °C, by switching alternatively the flow between 5% H2 (or CH4) in N2 and air, at a fixed total flow rate of 100 cc/min. If not otherwise stated, the duration of the reduction phase was 45 min, whilst that of oxidation phase was 30 min, with an intermediate purge by pure N2 (2–30 min). The evolved gases, leaving the flow microbalance through two independent heated capillaries, were analyzed by a Pfeiffer Thermostar G mass spectrometer and a Perkin Elmer Spectrum GX spectrometer in order to detect all IR-active gaseous species. In this last case, IR spectra were rationed against a background spectrum of pure N2 and were collected every 1-10 min depending on the duration of the experiment.
4. Conclusions
Oxygen carrier materials based on La2O2SO4 promoted by small amounts (1% wt.) of transition metals such as Co, Mn and Cu have been synthesized and characterized by means of XRD, BET, TPR/TPO and TG-MS-FTIR experiments in order to investigate their potential use in the Chemical Looping Combustion process with either hydrogen or methane as fuel. Results demonstrate that doping the parent La2O2SO4 with transition metals can promote lattice oxygen mobility in the temperature range 700–900 °C following the order Cu < Mn < Co, catalyzing the complete reduction of La2O2SO4 to form La2O2S by reaction with either H2 and CH4. On the other hand, the reoxidation of La2O2S by molecular oxygen to restore the La2O2SO4 phase requires temperatures ≥700 °C to proceed quickly and is poorly affected by the addition of those transition metals.
Notably, the addition of Co or Mn has shown a marked beneficial effect to limit or inhibit the decomposition of the oxysulfate that releases some SO2. Such undesired side reaction proceeds to some extent together with the main reduction of the La2O2SO4 by either H2 or CH4 and it causes a progressive and irreversible loss of the original oxygen storage capacity through the formation of the stable La2O3 phase. Eventually, metal doping does not affect the thermal decomposition of La2O2SO4 occurring above ca. 1030 °C under inert flow, nor it promotes any spontaneous reduction of the lanthanum oxysulfate to oxysulfide.
The enhanced performance of the Co-promoted carrier can be assigned to H2/CH4 activation on metal sites and to the higher distortion of SO4 units in the oxysulfate lattice caused by the partial inclusion of Co3+ ion, much smaller than La3+, which enhances oxygen mobility.
TG-MS-FTIR experiments of CLC with alternating feeds have shown that Co-La2O2SO4 is able to oxidize CH4 at 800 °C producing CO2 and water with reasonable rates and high selectivity, avoiding the deterioration of performance related to a progressive decomposition. At higher temperatures, the oxygen storage performance is progressively lost due to the irreversible formation of some La2O3, that occurs as a side reaction during the reducing step of the OC material by the fuel and is accompanied by the release of SO2. At lower temperatures, the mobility of lattice oxygen becomes the limiting factor that can preclude the complete exploitation of the OSC within a reasonable contact time with the fuel stream.
Author Contributions
Data curation, G.M.; Investigation, S.C., G.M. and L.L.; Supervision, S.C. and L.L.; Writing—original draft, S.C. and L.L.; Writing—review & editing, S.C.
Funding
This research received no external funding.
Acknowledgments
Luciano Cortese is kindly acknowledged for XRD analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tang, M.; Xu, L.; Fan, M. Progress in oxygen carrier development of methane-based chemical-looping reforming: A review. Appl. Energy 2015, 151, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Bhavsar, S.; Najera, M.; Solunke, R.; Veser, G. Chemical looping: To combustion and beyond. Catal. Today 2014, 228, 96–105. [Google Scholar] [CrossRef]
- Adanez, J.; Abad, A.; Garcia-Labiano, F.; Gayan, P.; de Diego, L.F. Progress in Chemical-Looping Combustion and Reforming technologies. Prog. Energy Combust. Sci. 2012, 38, 215–282. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Che, L.; Hao, Y.; Su, Q. Cycle performance of Cu-based oxygen carrier based on a chemical-looping combustion process. J. Energy Chem. 2016, 25, 101–109. [Google Scholar] [CrossRef]
- Imanieh, M.H.; Rad, M.H.; Nadarajah, A.; González-Platas, J.; Rivera-López, F.; Martín, I.R. Novel perovskite ceramics for chemical looping combustion application. J. CO2 Util. 2016, 13, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Taylor, D.D.; Rodriguez, E.E.; Zachariah, M.R. Influence of transition metal electronegativity on the oxygen storage capacity of perovskite oxides. Chem. Commun. 2016, 52, 10369–10372. [Google Scholar] [CrossRef] [PubMed]
- Miccio, F.; Bendoni, R.; Piancastelli, A.; Medri, V.; Landi, E. Geopolymer composites for chemical looping combustion. Fuel 2018, 225, 436–442. [Google Scholar] [CrossRef]
- Bendoni, R.; Miccio, F.; Medri, V.; Landi, E. Chemical looping combustion using geopolymer-based oxygen carriers. Chem. Eng. J. 2018, 341, 187–197. [Google Scholar] [CrossRef]
- Bi, W.; Chen, T.; Zhao, R.; Wang, Z.; Wu, J.; Wu, J. Characteristics of a CaSO4 oxygen carrier for chemical-looping combustion: Reaction with polyvinylchloride pyrolysis gases in a two-stage reactor. RSC Adv. 2015, 5, 34913–34920. [Google Scholar] [CrossRef]
- Lisi, L.; Mancino, G.; Cimino, S. Chemical looping oxygen transfer properties of Cu-doped lanthanum oxysulphate. Int. J. Hydrogen Energy 2015, 40, 2047–2054. [Google Scholar] [CrossRef]
- Machida, M.; Kawamura, K.; Ito, K. Novel oxygen storage mechanism based on redox of sulfur in lanthanum oxysulfate/oxysulfide. Chem. Commun. (Camb.) 2004, 2, 662–663. [Google Scholar] [CrossRef] [PubMed]
- Machida, M.; Kawamura, K.; Ito, K.; Ikeue, K. Large-Capacity Oxygen Storage by Lanthanide Oxysulfate/Oxysulfide Systems. Chem. Mater. 2005, 17, 1487–1492. [Google Scholar] [CrossRef]
- Machida, M.; Kawano, T.; Eto, M.; Zhang, D.; Ikeue, K. Ln Dependence of the Large-Capacity Oxygen Storage/Release Property of Ln Oxysulfate/Oxysulfide Systems. Chem. Mater. 2007, 19, 954–960. [Google Scholar] [CrossRef]
- Ikeue, K.; Eto, M.; Zhang, D.J.; Kawano, T.; Machida, M. Large-capacity oxygen storage of Pd-loaded Pr2O2SO4 applied to anaerobic catalytic CO oxidation. J. Catal. 2007, 248, 46–52. [Google Scholar] [CrossRef]
- Zhang, D.; Yoshioka, F.; Ikeue, K.; Machida, M. Synthesis and Oxygen Release/Storage Properties of Ce-Substituted La-Oxysulfates, (La1−xCex)2O2SO4. Chem. Mater. 2008, 20, 6697–6703. [Google Scholar] [CrossRef]
- Zhang, D.; Kawada, T.; Yoshioka, F.; Machida, M. Oxygen Gateway Effect of CeO2/La2O2SO4 Composite Oxygen Storage Materials. ACS Omega 2016, 1, 789–798. [Google Scholar] [CrossRef]
- Osseni, S.A.; Denisenko, Y.G.; Fatombi, J.K.; Sal’nikova, E.I.; Andreev, O.V. Synthesis and characterization of Ln2O2SO4 (Ln = Gd, Ho, Dy and Lu) nanoparticles obtained by coprecipitation method and study of their reduction reaction under H2 flow. J. Nanostruct. Chem. 2017, 7, 337–343. [Google Scholar] [CrossRef]
- Zhang, W.; Arends, I.W.C.E.; Djanashvili, K. Nanoparticles of lanthanide oxysulfate/oxysulfide for improved oxygen storage/release. Dalt. Trans. 2016, 45, 14019–14022. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Li, D. Enhancing Oxygen Storage Capability and Catalytic Activity of Lanthanum Oxysulfide (La2O2S) Nanocatalysts by Sodium and Iron/Sodium Doping. ChemCatChem 2018, 10, 550–558. [Google Scholar] [CrossRef]
- Sal’nikova, E.I.; Kaliev, D.I.; Andreev, P.O. Kinetics of phase formation upon the treatment of La2(SO4)3 and La2O2SO4 in a hydrogen flow. Russ. J. Phys. Chem. A 2011, 85, 2121–2125. [Google Scholar] [CrossRef]
- Poston, J.A.; Siriwardane, R.V.; Fisher, E.P.; Miltz, A.L. Thermal decomposition of the rare earth sulfates of cerium(III), cerium(IV), lanthanum(III) and samarium(III). Appl. Surf. Sci. 2003, 214, 83–102. [Google Scholar] [CrossRef]
- Arnone, S.; Bagnasco, G.; Busca, G.; Lisi, L.; Russo, G.; Turco, M. Catalytic combustion of methane over transition metal oxides. Stud. Surf. Sci. Catal. 1998, 119, 65–70. [Google Scholar]
- Cimino, S.; Lisi, L.; Tortorelli, M. Low temperature SCR on supported MnOx catalysts for marine exhaust gas cleaning: Effect of KCl poisoning. Chem. Eng. J. 2016, 283, 223–230. [Google Scholar] [CrossRef]
- Zhang, Y.; Smith, K.J. CH4 decomposition on Co catalysts: Effect of temperature, dispersion, and the presence of H2 or CO in the feed. Catal. Today 2002, 77, 257–268. [Google Scholar] [CrossRef]
Figure 1.
X-ray diffraction (XRD) patterns of (a) as prepared and (b) used (15 cycles at 900°C) La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4.
Figure 1.
X-ray diffraction (XRD) patterns of (a) as prepared and (b) used (15 cycles at 900°C) La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4.

Figure 2.
(a) dTG curves for La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4 during heating under a N2 flow. Heating rate: 10 °C min−1. Corresponding MS temporal profiles for O2 (m/z = 32), CO2 (m/z = 44), and SO2 (m/z = 64) concentration in the evolved gas relevant to (b) La2O2SO4 and (c) Mn-La2O2SO4.
Figure 2.
(a) dTG curves for La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4 during heating under a N2 flow. Heating rate: 10 °C min−1. Corresponding MS temporal profiles for O2 (m/z = 32), CO2 (m/z = 44), and SO2 (m/z = 64) concentration in the evolved gas relevant to (b) La2O2SO4 and (c) Mn-La2O2SO4.

Figure 3.
(a) H2-TPR profiles of La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4; (b) O2-TPO profiles recorded during the subsequent re-oxidation of OC materials. Heating 10°C/min from room temperature up to 1030 °C.
Figure 3.
(a) H2-TPR profiles of La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4; (b) O2-TPO profiles recorded during the subsequent re-oxidation of OC materials. Heating 10°C/min from room temperature up to 1030 °C.

Figure 4.
(a) TG curves for La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4 during reduction under a flow of 2% H2/N2. Heating rate: 10 °C min−1; (b) Corresponding MS profiles for SO2 (m/z = 64) in the evolved gas.
Figure 4.
(a) TG curves for La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4 during reduction under a flow of 2% H2/N2. Heating rate: 10 °C min−1; (b) Corresponding MS profiles for SO2 (m/z = 64) in the evolved gas.

Figure 5.
Weight changes during chemical looping reactions under cycled feed stream of 5% CH4/N2 and air at 800 °C over La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4.
Figure 5.
Weight changes during chemical looping reactions under cycled feed stream of 5% CH4/N2 and air at 800 °C over La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4.

Figure 6.
Reducing (under CH4)/oxidation cycles of La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4 carried out at 900 °C (15 cycles).
Figure 6.
Reducing (under CH4)/oxidation cycles of La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4 carried out at 900 °C (15 cycles).

Figure 7.
Fourier transform infrared (FTIR) spectra of the evolved gas leaving the TG at three times during the reduction step (under CH4 flow) of Co-La2O2SO4 at 900 °C (7th cycle of Figure 6). The inset shows a magnification (30x) of the region of typical CO absorption bands.
Figure 7.
Fourier transform infrared (FTIR) spectra of the evolved gas leaving the TG at three times during the reduction step (under CH4 flow) of Co-La2O2SO4 at 900 °C (7th cycle of Figure 6). The inset shows a magnification (30x) of the region of typical CO absorption bands.

Figure 8.
Weight change and gas concentration profiles during chemical looping reactions under cycled feed stream of 5% CH4/N2 and air at 900 °C over Co-La2O2SO4.
Figure 8.
Weight change and gas concentration profiles during chemical looping reactions under cycled feed stream of 5% CH4/N2 and air at 900 °C over Co-La2O2SO4.

Figure 9.
Weight change and gas concentration profiles during chemical looping reactions under cycled feed stream of 5% CH4/N2 and air at 800 °C over Co-La2O2SO4.
Figure 9.
Weight change and gas concentration profiles during chemical looping reactions under cycled feed stream of 5% CH4/N2 and air at 800 °C over Co-La2O2SO4.

Figure 10.
Weight change and gas concentration profiles during chemical looping reactions under cycled feed stream of 5% CH4/N2 and air at 750 °C over Co-La2O2SO4.
Figure 10.
Weight change and gas concentration profiles during chemical looping reactions under cycled feed stream of 5% CH4/N2 and air at 750 °C over Co-La2O2SO4.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
List of oxygen carriers and corresponding values of Brunauer–Emmett–Teller (BET) surface area.
Table 1.
List of oxygen carriers and corresponding values of Brunauer–Emmett–Teller (BET) surface area.
| Oxygen Carrier | BET Area (m2/g) |
|---|---|
| La2O2SO4 | 2.7 |
| Cu-La2O2SO4 | 2.7 |
| Mn-La2O2SO4 | 2.4 |
| Co-La2O2SO4 | 2.4 |
Table 2.
Weight loss with onset, inflection, and final temperatures for the reduction of La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4 under a flow of 2% H2/N2; amount of SO2 emitted below 950 °C.
Table 2.
Weight loss with onset, inflection, and final temperatures for the reduction of La2O2SO4 and metal (Cu, Mn, Co) promoted La2O2SO4 under a flow of 2% H2/N2; amount of SO2 emitted below 950 °C.
| Δ m | Tin | Tinfl | Tf | SO2 Emitted | |
|---|---|---|---|---|---|
| % wt. | °C | % wt. | |||
| La2O2SO4 | 15.2 | 635 | 874 | 972 | 0.060 |
| Cu-La2O2SO4 | 15.1 | 625 | 817 | 910 | 0.065 |
| Mn-La2O2SO4 | 15.2 | 625 | 784 | 862 | 0.009 |
| Co-La2O2SO4 | 15.1 | 600 | 690 | 770 | 0.009 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cimino, S.; Mancino, G.; Lisi, L. Performance and Stability of Metal (Co, Mn, Cu)-Promoted La2O2SO4 Oxygen Carrier for Chemical Looping Combustion of Methane. Catalysts 2019, 9, 147. https://doi.org/10.3390/catal9020147
AMA Style
Cimino S, Mancino G, Lisi L. Performance and Stability of Metal (Co, Mn, Cu)-Promoted La2O2SO4 Oxygen Carrier for Chemical Looping Combustion of Methane. Catalysts. 2019; 9(2):147. https://doi.org/10.3390/catal9020147
Chicago/Turabian StyleCimino, Stefano, Gabriella Mancino, and Luciana Lisi. 2019. "Performance and Stability of Metal (Co, Mn, Cu)-Promoted La2O2SO4 Oxygen Carrier for Chemical Looping Combustion of Methane" Catalysts 9, no. 2: 147. https://doi.org/10.3390/catal9020147
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.