An Enzyme Cascade Synthesis of Vanillin


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Screening of the Small Focused CYP102A1 Library
2.2. Screening of Additional CYP102A1 Variants
2.3. Molecular Dynamics Simulation
2.4. Screening of Quadruple CYP102A1 Variants for Substrate Conversion
2.5. In Vitro One-Pot Cascade Reactions
2.6. In Vivo Cascade Reactions
3. Materials and Methods
3.1. Materials
3.2. Plasmids and Strains
3.3. Molecular Biology Techniques and Enzyme Expression
3.4. Determination of Protein Concentration
3.5. In Vitro Biotransformations
3.6. In Vivo Biotransformations
3.7. Sample Treatment
3.8. GC Analysis
3.9. HPLC Analysis
3.10. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Priefert, H.; Rabenhorst, J.; Steinbuchel, A. Biotechnological production of vanillin. Appl. Microbiol. Biotechnol. 2001, 56, 296–314. [Google Scholar] [CrossRef] [PubMed]
- Walton, N.J.; Mayer, M.J.; Narbad, A. Vanillin. Phytochemistry 2003, 63, 505–515. [Google Scholar] [CrossRef]
- Kaur, B.; Chakraborty, D. Biotechnological and molecular approaches for vanillin production: A review. Appl. Biochem. Biotechnol. 2013, 169, 1353–1372. [Google Scholar] [CrossRef]
- Banerjee, G.; Chattopadhyay, P. Vanillin biotechnology: The perspectives and future. J. Sci. Food Agric. 2019, 99, 499–506. [Google Scholar] [CrossRef]
- Gallage, N.J.; Møller, B.L. Vanillin-bioconversion and bioengineering of the most popular plant flavor and its de novo biosynthesis in the vanilla orchid. Mol. Plant 2015, 8, 40–57. [Google Scholar] [CrossRef]
- Furuya, T.; Miura, M.; Kino, K. A coenzyme-independent decarboxylase/oxygenase cascade for the efficient synthesis of vanillin. ChemBioChem 2014, 15, 2248–2254. [Google Scholar] [CrossRef]
- Furuya, T.; Kuroiwa, M.; Kino, K. Biotechnological production of vanillin using immobilized enzymes. J. Biotechnol. 2017, 243, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Girvan, H.M.; Munro, A.W. Applications of microbial cytochrome P450 enzymes in biotechnology and synthetic biology. Curr. Opin. Chem. Biol. 2016, 31, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryle, M.J.; Stok, J.E.; De Voss, J.J. Reactions catalyzed by bacterial cytochromes P450. Aust. J. Chem. 2003, 56, 749–762. [Google Scholar] [CrossRef]
- Hammer, S.C.; Knight, A.M.; Arnold, F.H. Design and evolution of enzymes for non-natural chemistry. Curr. Opin. Green Sustain. Chem. 2017, 7, 23–30. [Google Scholar] [CrossRef]
- Arnold, F.H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed. 2018, 57, 4143–4148. [Google Scholar] [CrossRef]
- Brandenberg, O.F.; Fasan, R.; Arnold, F.H. Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol. 2017, 47, 102–111. [Google Scholar] [CrossRef]
- Narhi, L.O.; Fulco, A.J. Identification and characterization of two functional domains in cytochrome P450-BM3, a catalytically self-sufficient monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 1987, 262, 6683–6690. [Google Scholar]
- Miura, Y.; Fulco, A.J. (Omega-2) hydroxylation of fatty acids by a soluble system from Bacillus megaterium. J. Biol. Chem. 1974, 249, 1880–1888. [Google Scholar]
- Urlacher, V.B.; Lutz-Wahl, S.; Schmid, R.D. Microbial P450 enzymes in biotechnology. Appl. Microbiol. Biotechnol. 2004, 64, 317–325. [Google Scholar] [CrossRef]
- Fasan, R. Tuning P450 Enzymes as Oxidation Catalysts. ACS Catal. 2012, 2, 647–666. [Google Scholar] [CrossRef]
- O’Hanlon, J.A.; Ren, X.; Morris, M.; Wong, L.L.; Robertson, J. Hydroxylation of anilides by engineered cytochrome P450-BM3. Org. Biomol. Chem. 2017, 15, 8780–8787. [Google Scholar] [CrossRef]
- Denard, C.A.; Ren, H.; Zhao, H. Improving and repurposing biocatalysts via directed evolution. Curr. Opin. Chem. Biol. 2015, 25, 55–64. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, B.; Wang, F.; Yu, X.; Ma, L.; Li, A.; Reetz, M.T. Chemo- and Regioselective Dihydroxylation of Benzene to Hydroquinone Enabled by Engineered Cytochrome P450 Monooxygenase. Angew. Chem. Int. Ed. 2019, 58, 764–768. [Google Scholar] [CrossRef]
- Dennig, A.; Weingartner, A.M.; Kardashliev, T.; Müller, C.A.; Tassano, E.; Schürmann, M.; Ruff, A.J.; Schwaneberg, U. An Enzymatic Route to α-Tocopherol Synthons: Aromatic Hydroxylation of Pseudocumene and Mesitylene with P450 BM3. Chem. A Eur. J. 2017, 23, 17981–17991. [Google Scholar] [CrossRef]
- Dennig, A.; Lülsdorf, N.; Liu, H.; Schwaneberg, U. Regioselective o-hydroxylation of monosubstituted benzenes by P450 BM3. Angew. Chem. Int. Ed. 2013, 52, 8459–8462. [Google Scholar] [CrossRef]
- Munday, S.D.; Dezvarei, S.; Lau, I.C.-K.; Bell, S.G. Examination of Selectivity in the Oxidation of ortho- and meta-Disubstituted Benzenes by CYP102A1 (P450 BM3) Variants. ChemCatChem 2017, 9, 2512–2522. [Google Scholar] [CrossRef]
- Seifert, A.; Pleiss, J. Identification of selectivity-determining residues in cytochrome P450 monooxygenases: A systematic analysis of the substrate recognition site 5. Proteins 2009, 74, 1028–1035. [Google Scholar] [CrossRef]
- Seifert, A.; Vomund, S.; Grohmann, K.; Kriening, S.; Urlacher, V.B.; Laschat, S.; Pleiss, J. Rational design of a minimal and highly enriched CYP102A1 mutant library with improved regio-, stereo- and chemoselectivity. ChemBioChem 2009, 10, 853–861. [Google Scholar] [CrossRef]
- Whitehouse, C.J.C.; Bell, S.G.; Wong, L.-L. P450BM3 (CYP102A1): Connecting the dots. Chem. Soc. Rev. 2012, 41, 1218–1260. [Google Scholar] [CrossRef]
- Weber, E.; Seifert, A.; Antonovici, M.; Geinitz, C.; Pleiss, J.; Urlacher, V.B. Screening of a minimal enriched P450 BM3 mutant library for hydroxylation of cyclic and acyclic alkanes. Chem. Commun. 2011, 47, 944–946. [Google Scholar] [CrossRef]
- Whitehouse, C.J.; Rees, N.H.; Bell, S.G.; Wong, L.L. Dearomatisation of o-xylene by P450-BM3 (CYP102A1). Chemistry 2011, 17, 6862–6868. [Google Scholar] [CrossRef]
- Winter, R.T.; Van Beek, H.L.; Fraaije, M.W. The nose knows: Biotechnological production of vanillin. J. Chem. Educ. 2012, 89, 258–261. [Google Scholar] [CrossRef]
- de Jong, E.; van Berkel, W.J.; van der Zwan, R.P.; de Bont, J.A. Purification and characterization of vanillyl-alcohol oxidase from Penicillium simplicissimum. A novel aromatic alcohol oxidase containing covalently bound FAD. Eur. J. Biochem. 1992, 208, 651–657. [Google Scholar] [CrossRef]
- Fraaije, M.W.; Veeger, C.; van Berkel, W.J. Substrate specificity of flavin-dependent vanillyl-alcohol oxidase from Penicillium simplicissimum. Evidence for the production of 4-hydroxycinnamyl alcohols from 4-allylphenols. Eur. J. Biochem. 1995, 234, 271–277. [Google Scholar] [CrossRef]
- Fraaije, M.W.; van den Heuvel, R.H.; Roelofs, J.C.; van Berkel, W.J. Kinetic mechanism of vanillyl-alcohol oxidase with short-chain 4-alkylphenols. Eur. J. Biochem. 1998, 253, 712–719. [Google Scholar] [CrossRef] [Green Version]
- van den Heuvel, R.H.; Fraaije, M.W.; Laane, C.; van Berkel, W.J. Enzymatic synthesis of vanillin. J. Agric. Food Chem. 2001, 49, 2954–2958. [Google Scholar] [CrossRef]
- van den Heuvel, R.H.; van den Berg, W.A.; Rovida, S.; van Berkel, W.J. Laboratory-evolved vanillyl-alcohol oxidase produces natural vanillin. J. Biol. Chem. 2004, 279, 33492–33500. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Sattler, J.; Resch, V.; Mutti, F.G.; Kroutil, W. Recent biocatalytic oxidation-reduction cascades. Curr. Opin. Chem. Biol. 2011, 15, 249–256. [Google Scholar] [CrossRef]
- García-Junceda, E.; Lavandera, I.; Rother, D.; Schrittwieser, J.H. (Chemo)enzymatic cascades—Nature’s synthetic strategy transferred to the laboratory. J. Mol. Catal. B Enzym. 2015, 114, 1–6. [Google Scholar] [CrossRef]
- Muschiol, J.; Peters, C.; Oberleitner, N.; Mihovilovic, M.D.; Bornscheuer, U.T.; Rudroff, F. Cascade catalysis—Strategies and challenges en route to preparative synthetic biology. Chem. Commun. 2015, 51, 5798–5811. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef]
- Gruber, P.; Marques, M.P.C.; O’Sullivan, B.; Baganz, F.; Wohlgemuth, R.; Szita, N. Conscious coupling: The challenges and opportunities of cascading enzymatic microreactors. Biotechnol. J. 2017, 12, 1–13. [Google Scholar] [CrossRef]
- Otte, K.B.; Hauer, B. Enzyme engineering in the context of novel pathways and products. Curr. Opin. Biotechnol. 2015, 35, 16–22. [Google Scholar] [CrossRef]
- Hepworth, L.J.; France, S.P.; Hussain, S.; Both, P.; Turner, N.J.; Flitsch, S.L. Enzyme Cascades in Whole Cells for the Synthesis of Chiral Cyclic Amines. ACS Catal. 2017, 7, 2920–2925. [Google Scholar] [CrossRef] [Green Version]
- Carnell, A.J. Synthetic cascades by combining evolved biocatalysts and artificial enzymes. ChemCatChem 2014, 6, 958–960. [Google Scholar] [CrossRef]
- Oberleitner, N.; Peters, C.; Muschiol, J.; Kadow, M.; Saß, S.; Bayer, T.; Schaaf, P.; Iqbal, N.; Rudroff, F.; Mihovilovic, M.D.; et al. An enzymatic toolbox for cascade reactions: A showcase for an in vivo redox sequence in asymmetric synthesis. ChemCatChem 2013, 5, 3524–3528. [Google Scholar] [CrossRef]
- Wu, S.; Li, Z. Whole-Cell Cascade Biotransformations for One-Pot Multistep Organic Synthesis. ChemCatChem 2018, 2164–2178. [Google Scholar] [CrossRef]
- Sulistyaningdyah, W.T.; Ogawa, J.; Li, Q.S.; Maeda, C.; Yano, Y.; Schmid, R.D.; Shimizu, S. Hydroxylation activity of P450 BM3 mutant F87V towards aromatic compounds and its application to the synthesis of hydroquinone derivatives from phenolic compounds. Appl. Microbiol. Biotechnol. 2005, 67, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Shoji, O.; Kunimatsu, T.; Kawakami, N.; Watanabe, Y. Highly selective hydroxylation of benzene to phenol by wild-type cytochrome P450-BM3 assisted by decoy molecules. Angew. Chem. Int. Ed. 2013, 52, 6606–6610. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.; Antonovici, M.; Hauer, B.; Pleiss, J. An Efficient Route to Selective Bio-oxidation Catalysts: An Iterative Approach Comprising Modeling, Diversification, and Screening, Based on CYP102A1. ChemBioChem 2011, 12, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.R.; Ravishankar, G.A. Vanilla flavour: Production by conventional and biotechnological routes. J. Sci. Food Agric. 2000, 80, 289–304. [Google Scholar]
- Sambrook, J.; Russell, D. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; ISBN 978-087969577-4. [Google Scholar]
- Maurer, S.; Urlacher, V.; Schulze, H.; Schmid, R.D. Immobilisation of P450 BM3 and an NADP+ cofactor recycling system: Towards a technical application of heme-containing monooxygenases in fine chemical synthesis. Adv. Synth. Catal. 2003, 345, 802–810. [Google Scholar] [CrossRef]
- Klaus, T. Novel Route to Vanillin—An Enzyme-Catalyzed Multi-Step Cascade Synthesis. Ph.D. Thesis, Universitaet Stuttgart, Stuttgart, Germany, 10 May 2017. [Google Scholar] [CrossRef]
- Omura, T.; Sato, R. The Carbon Monoxide-Binding Pigment of Liver Microsomes. I. Evidence for Its Hemoprotein Nature. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [PubMed]
- Omura, T.; Sato, R. The Carbon Monoxide-Binding Pigment of Liver Microsomes. II. Solubilization, Purification, and Properties. J. Biol. Chem. 1964, 239, 2379–2385. [Google Scholar] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms For Highly Efficient, Load-Balanced, And Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Wensink, E.J.W.; Hoffmann, A.C.; van Maaren, P.J.; van der Spoel, D. Dynamic Properties Of Water/Alcohol Mixtures Studied By Computer Simulation. J. Chem. Phys. 2003, 119, 7308–7317. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Nose, S. A Unified Formulation of the Constant Temperature Molecular-Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical-Integration Of Cartesian Equations Of Motion Of A System With Constraints—Molecular-Dynamics Of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- York, D.M.; Darden, T.A.; Pedersen, L.G. The Effect Of Long-Range Electrostatic Interactions In Simulations Of Macromolecular Crystals—A Comparison Of The Ewald And Truncated List Methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System 2002; De Lano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.M.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [Green Version]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.ED. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The Missing Term In Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]






{kind=link}
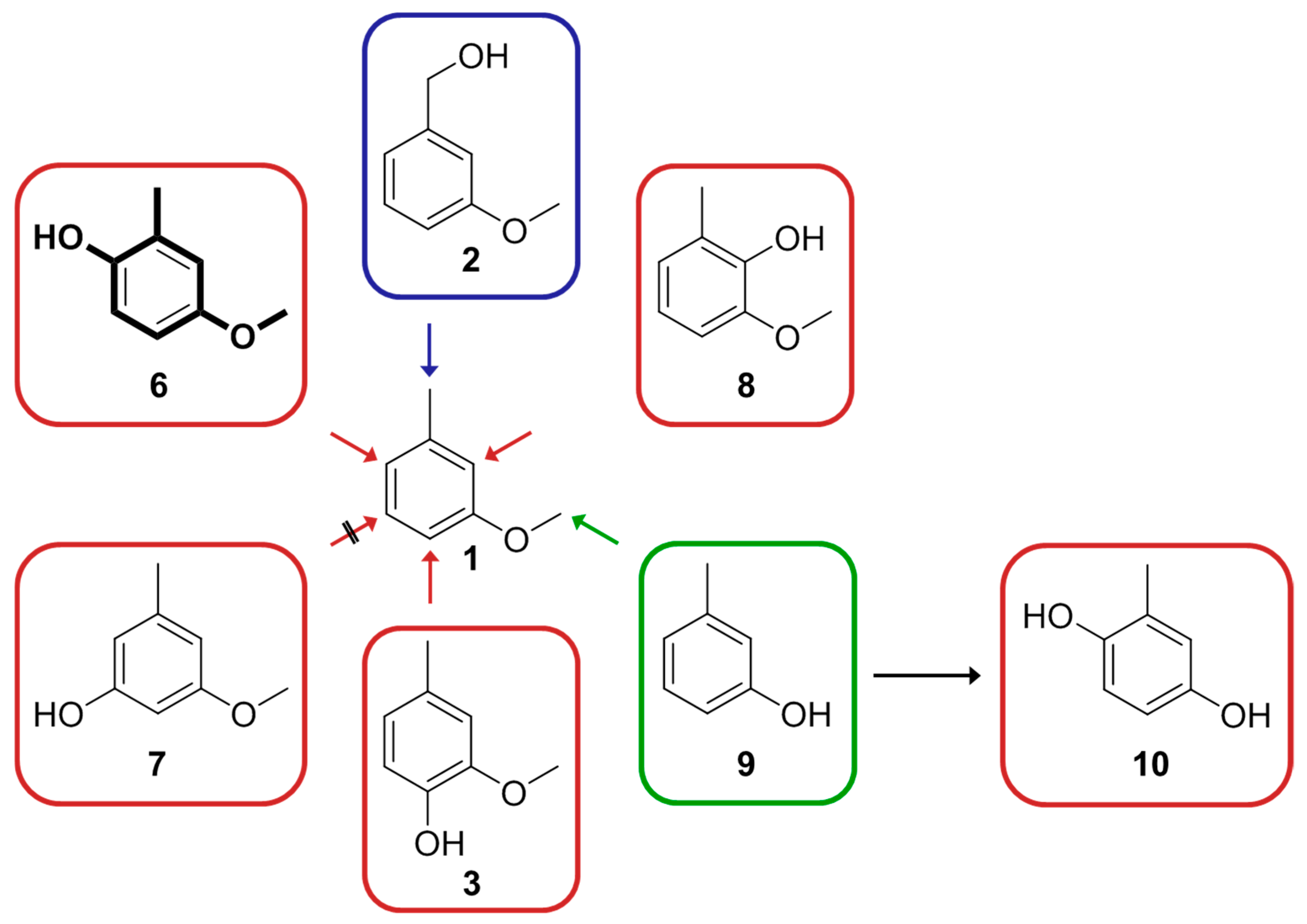{kind=link}
{kind=link}
{kind=link}
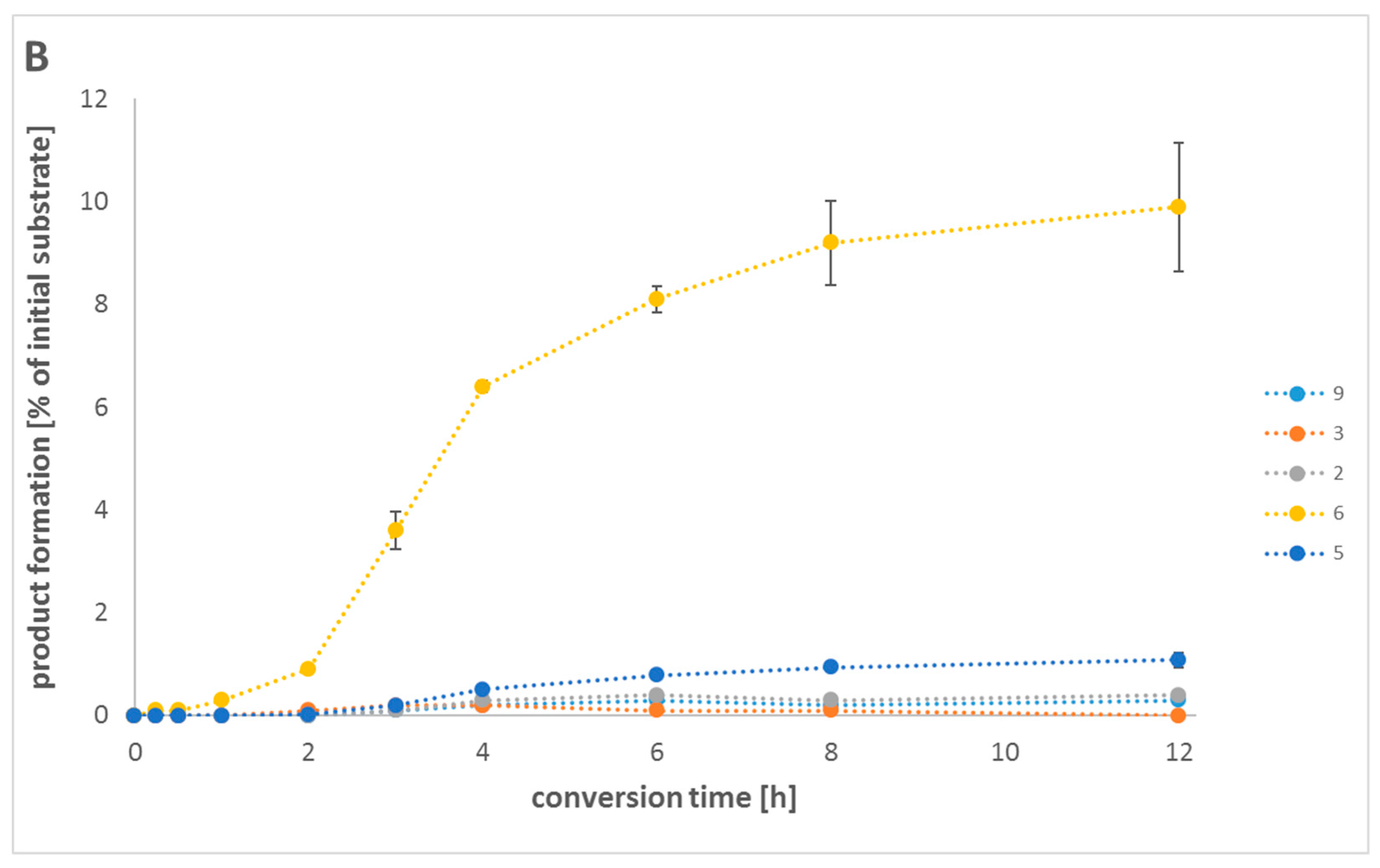{kind=link}
{kind=link}
| CYP102A1 Variant | Product Yield (% of Initial Substrate) | Total Conversion (%) | ||||
|---|---|---|---|---|---|---|
| 2 | 3 | 6 | 9 | 10 | ||
| wild-type | – | – | <1 | – | – | <1 |
| A328L | <1 | 2.7 ± 0.2 | 26.6 ± 2.5 | <1 | – | 31.0 ± 2.8 |
| F87V | <1 | <1 | 12.5 ± 0.2 | <1 | – | 14.9 ± 0.3 |
| F87V/A328L | 9.1 ± 0.2 | 1.6 ± 0.1 | 23.2 ± 0.4 | 12.0 ± 0.8 | 13.5 ± 0.8 | 59.3 ± 1.8 |
| F87V/A328L/L437I | 20.0 ± 0.9 | <1 | 14.0 ± 0.7 | 7.4 ± 1.3 | 19.3 ± 0.5 | 61.3 ± 2.9 |
| R47L/Y51F | <1 | – | 1.5 ± 0.1 | – | – | 1.6 ± 0.1 |
| R47L/Y51F/F87V/A328V | 5.0 ± 0.3 | <1 | 24.0 ± 0.8 | 3.0 ± 0.1 | 2.1 ± 0.2 | 34.9 ± 1.4 |
| R47L/Y51F/F87V/A328L/L437I | 13.5 ± 0.4 | <1 | 10.1 ± 0.5 | 7.7 ± 0.2 | 17.8 ± 0.8 | 50.4 ± 1.8 |
| Enzymes | Reaction Time (h) | Product Yield (% of Initial Substrate) | Total Conversion (%) | |||
|---|---|---|---|---|---|---|
| 2 | 3 | 5 | 6 | |||
| A328L R47L/Y51F/F87V/A328V | 2 | 2.2 ± 0.1 | 1.2 ± 0.1 | – | 19.3 ± 1.3 | 24.7 ± 1.6 |
| 1 + 1 | 1.2 ± 0.2 | 2.4 ± 0.5 | – | 25.6 ± 4.8 | 30.3 ± 5.7 | |
| A328L VAO_F454Y | 2 | <1 | <1 | <1 | 19.8 ± 2.7 | 22.9 ± 3.1 |
| 1 + 1 | 1.0 ± 0.1 | <1 | 1.2 ± 0.1 | 25.0 ± 2.2 | 29.0 ± 2.5 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klaus, T.; Seifert, A.; Häbe, T.; Nestl, B.M.; Hauer, B. An Enzyme Cascade Synthesis of Vanillin. Catalysts 2019, 9, 252. https://doi.org/10.3390/catal9030252
Klaus T, Seifert A, Häbe T, Nestl BM, Hauer B. An Enzyme Cascade Synthesis of Vanillin. Catalysts. 2019; 9(3):252. https://doi.org/10.3390/catal9030252
Chicago/Turabian StyleKlaus, Tobias, Alexander Seifert, Tim Häbe, Bettina M. Nestl, and Bernhard Hauer. 2019. "An Enzyme Cascade Synthesis of Vanillin" Catalysts 9, no. 3: 252. https://doi.org/10.3390/catal9030252
APA StyleKlaus, T., Seifert, A., Häbe, T., Nestl, B. M., & Hauer, B. (2019). An Enzyme Cascade Synthesis of Vanillin. Catalysts, 9(3), 252. https://doi.org/10.3390/catal9030252