Differences in the Catalytic Behavior of Au-Metalized TiO2 Systems During Phenol Photo-Degradation and CO Oxidation

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
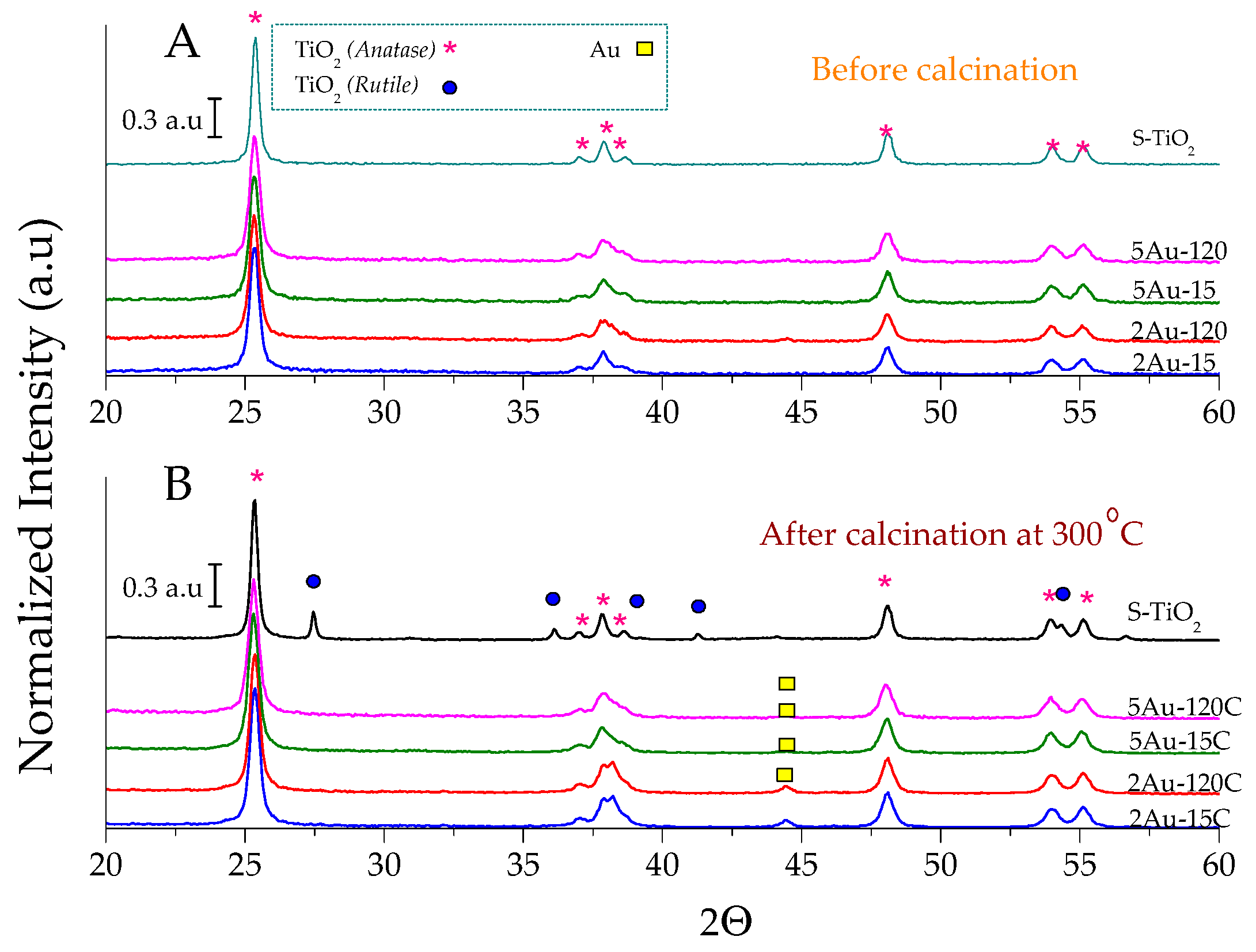
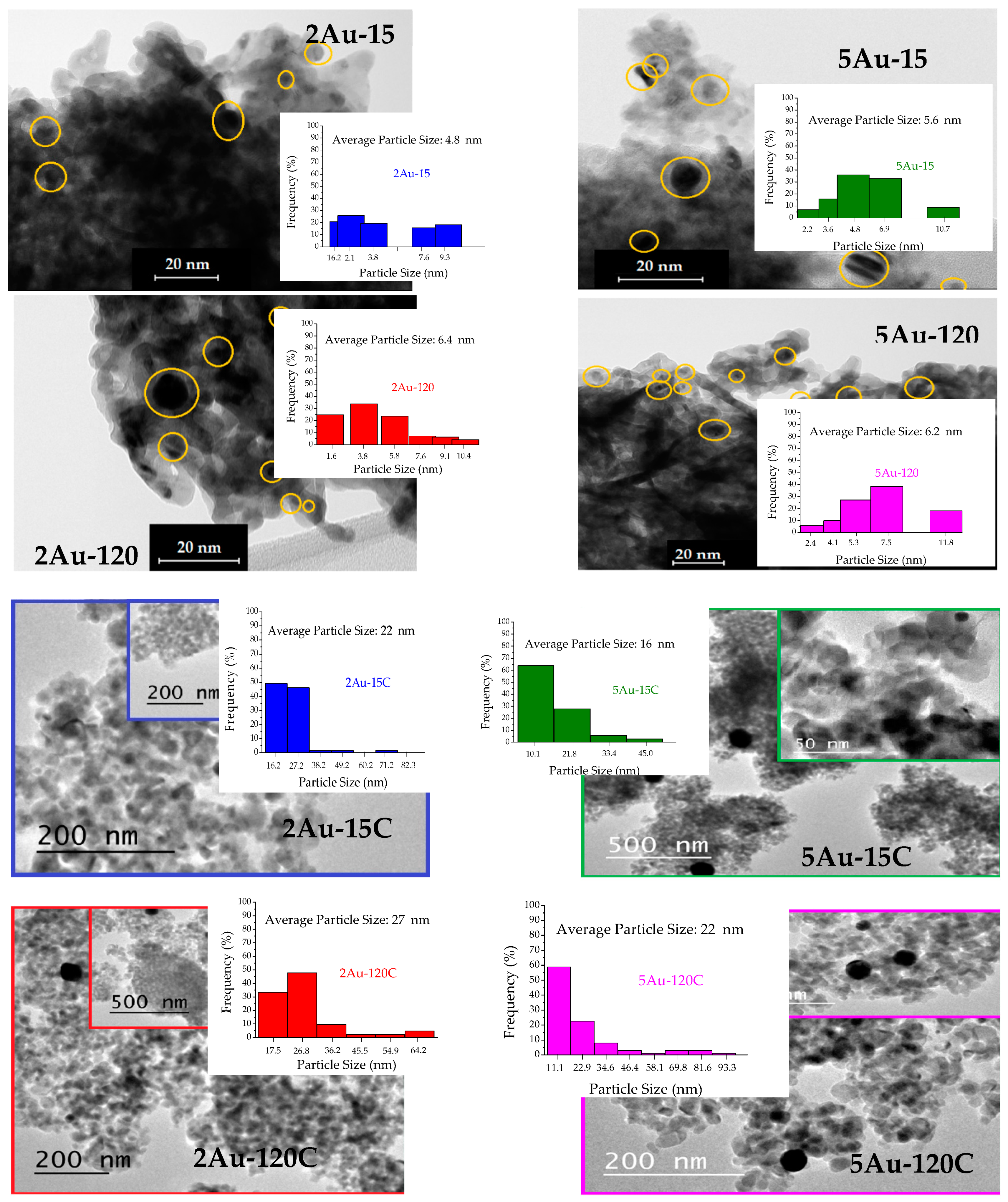
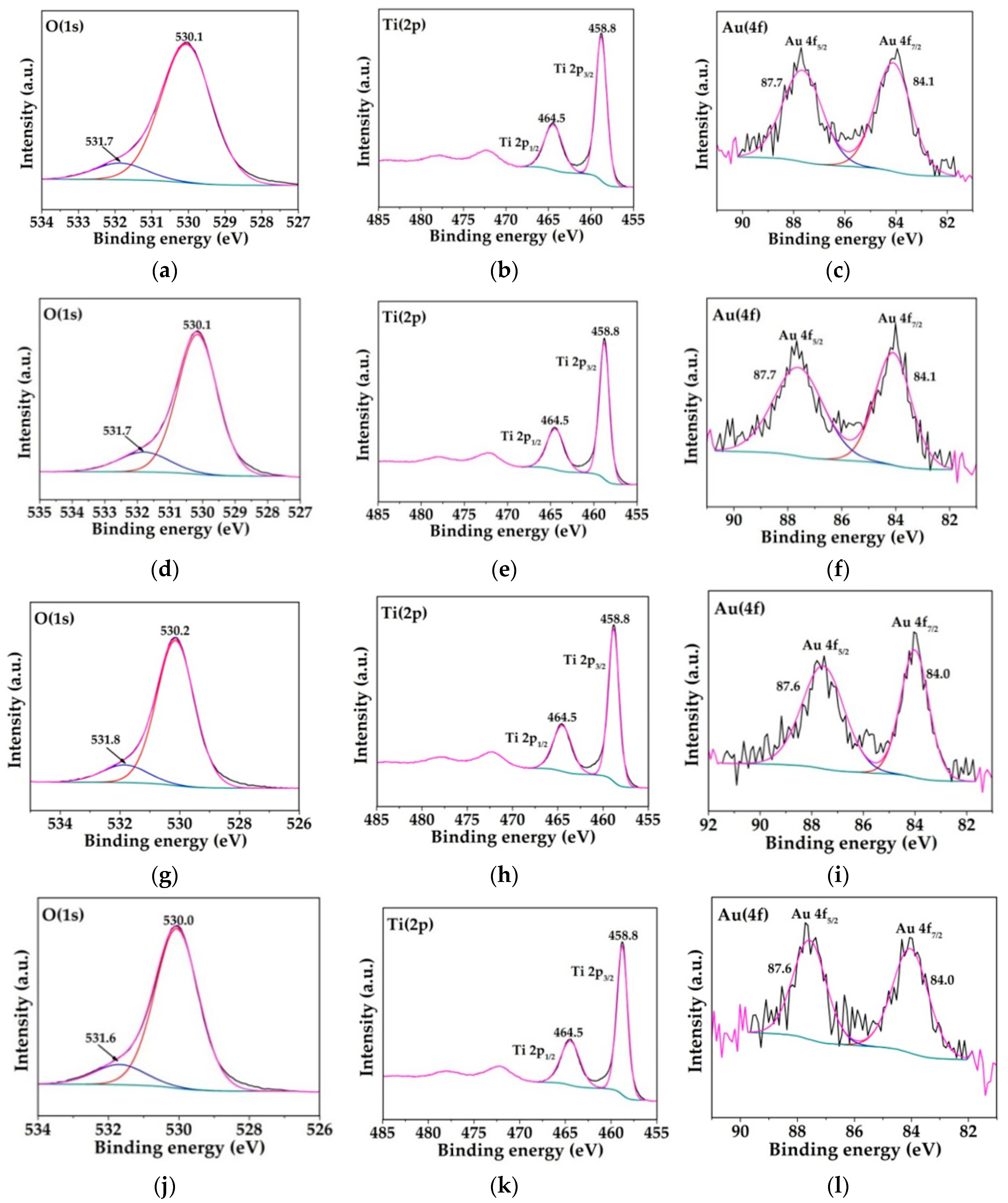
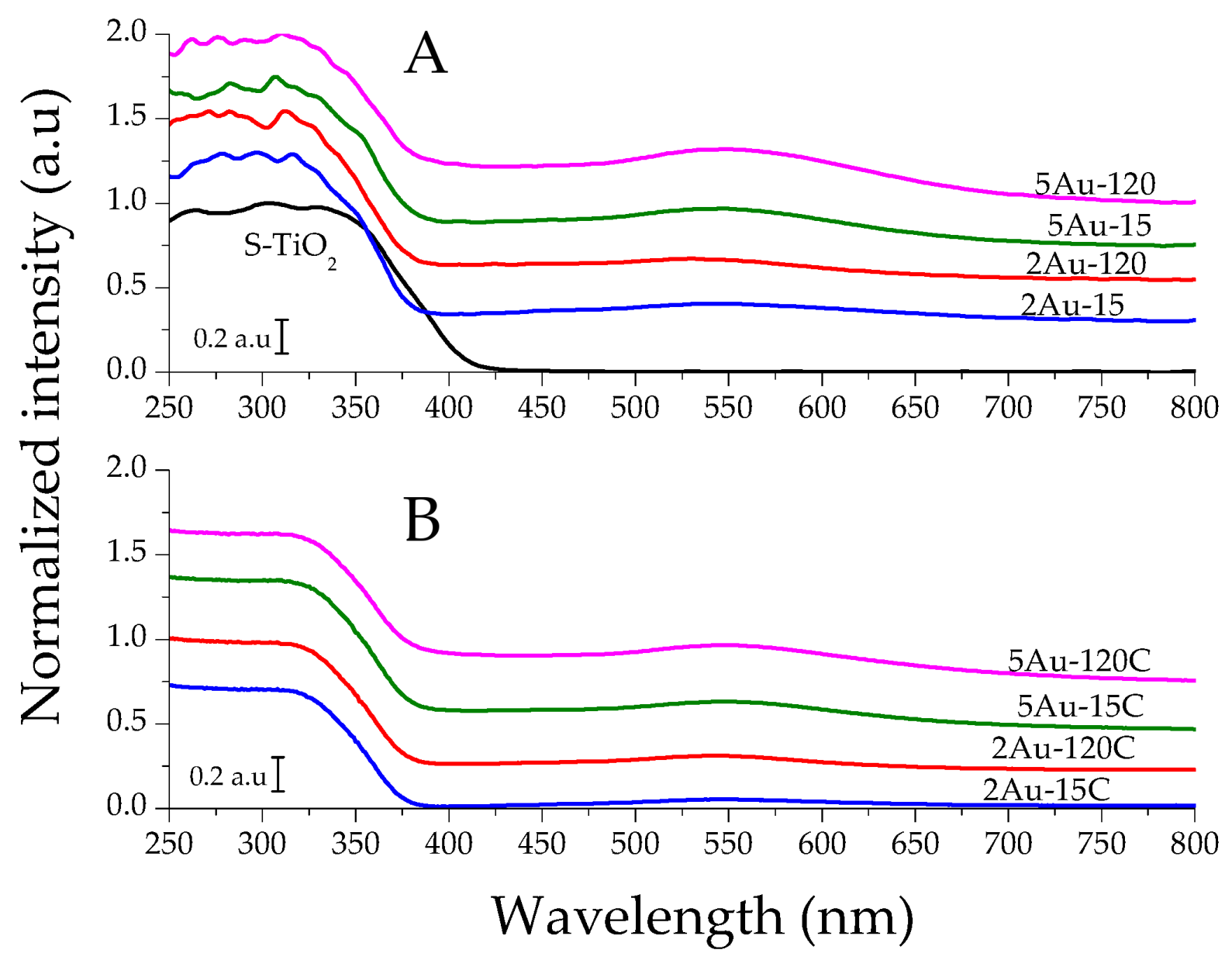
2.1. Physicochemical Characterization of the Obtained Materials
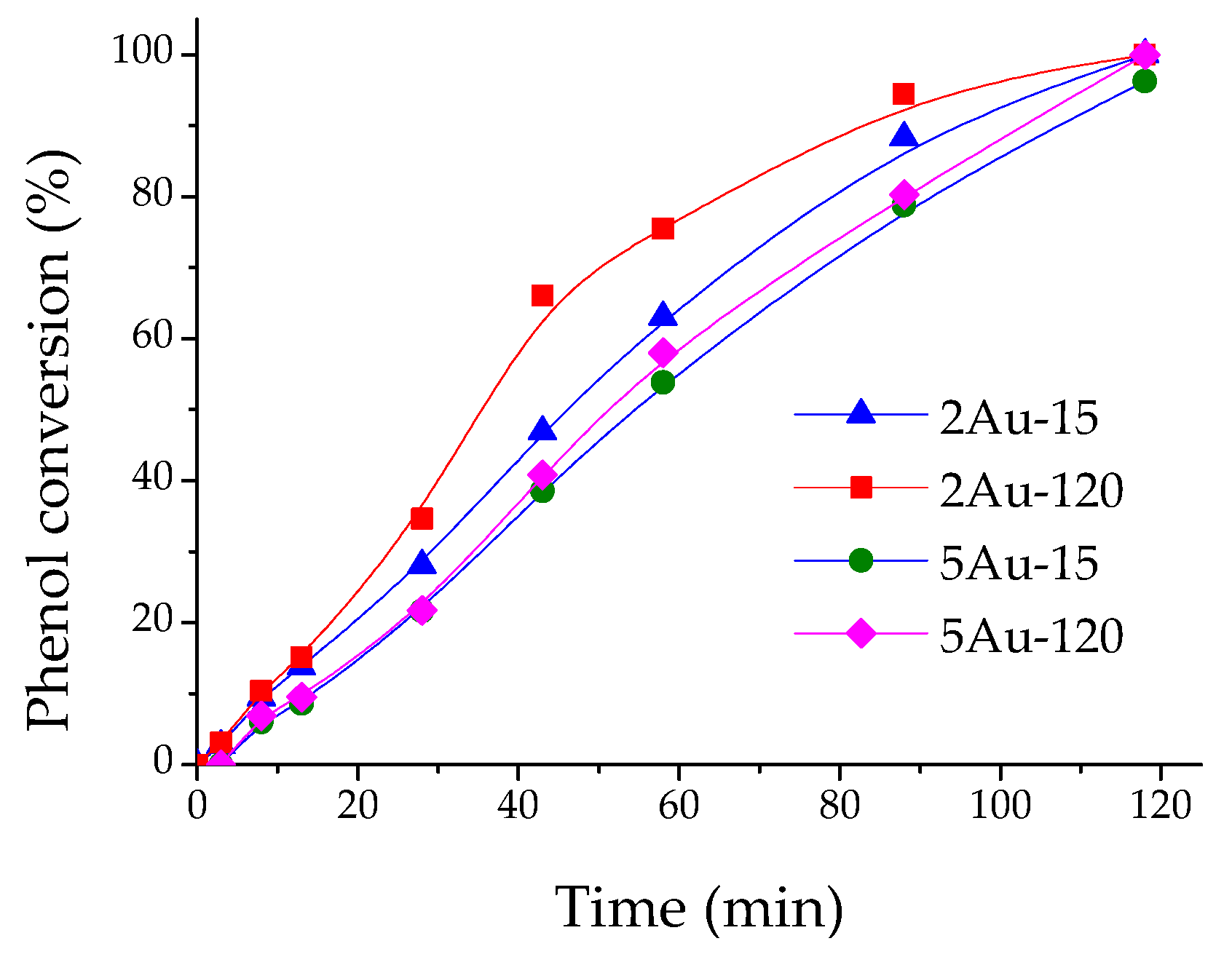
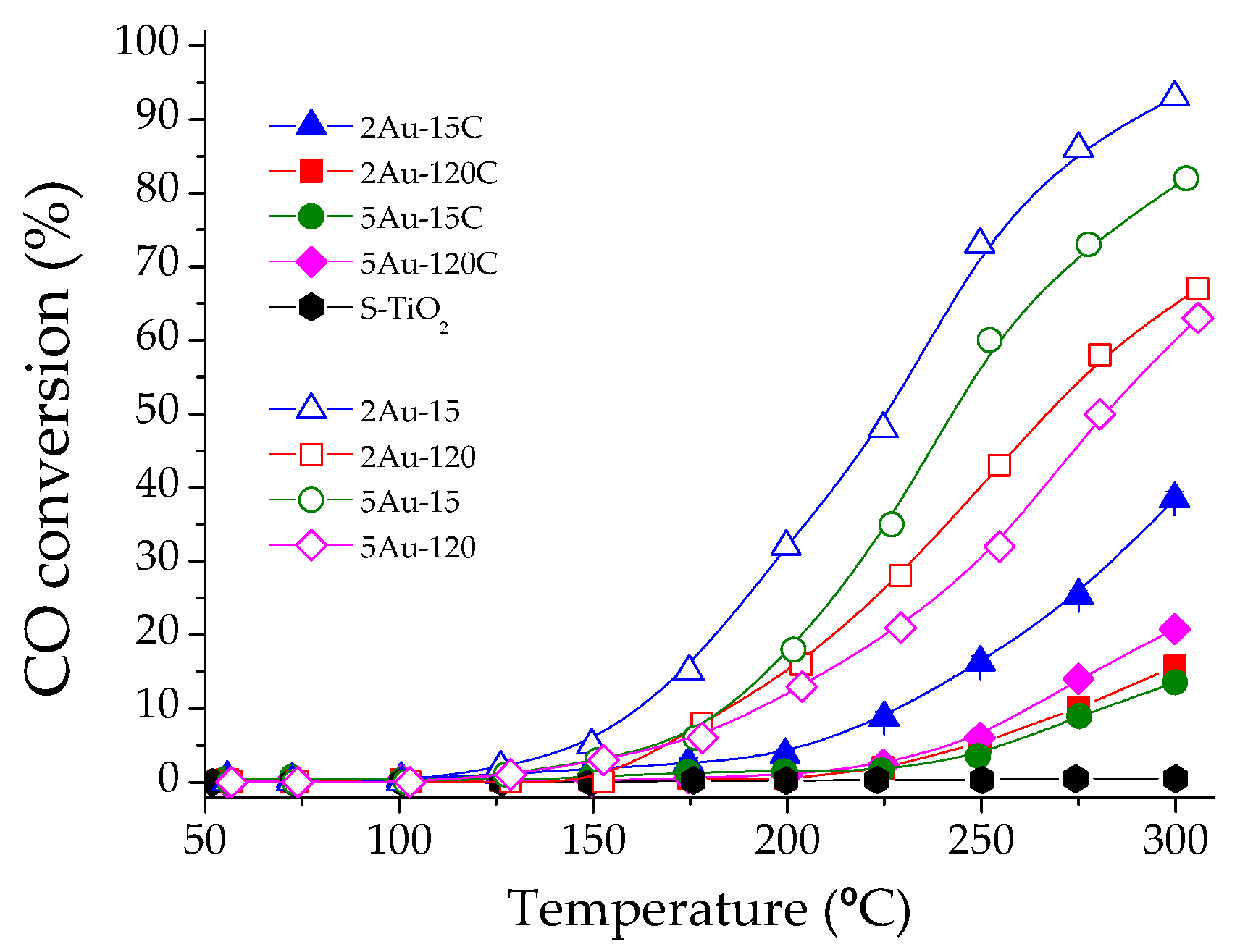
2.2. Catalytic Activity Measurements
3. Materials and methods
3.1. Synthesis of the Different Materials
3.2. Characterization of the Obtained Materials
3.3. Catalytic Activity Measurements
3.3.1. Phenol Photo-Catalytic Oxidation
3.3.2. CO Oxidation Reaction
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xie, X.; Li, Y.; Liu, Z.-Q.; Haruta, M.; Shen, W. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef]
- Sarria, F.R.; Plata, J.J.; Laguna, O.H.; Márquez, A.M.; Centeno, M.A.; Sanz, J.F.; Odriozola, J.A. Surface oxygen vacancies in gold based catalysts for CO oxidation. RSC Adv. 2014, 4, 13145–13152. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, T. CO-free fuel processing for fuel cell applications. Catal. Today 2002, 77, 65–78. [Google Scholar] [CrossRef]
- Costello, C.K.; Yang, J.H.; Law, H.Y.; Wang, Y.; Lin, J.N.; Marks, L.D.; Kung, M.C.; Kung, H.H. On the potential role of hydroxyl groups in CO oxidation over Au/Al2O3. Appl. Catal. A Gen. 2003, 243, 15–24. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T. Selective CO oxidation over CuO-CeO2 catalysts prepared via the urea–nitrate combustion method. Appl. Catal. A Gen. 2003, 244, 155–167. [Google Scholar] [CrossRef]
- Laguna, O.H.; Bobadilla, L.F.; Hernández, W.Y.; Centeno, M.A. Chapter 20: Low-Temperature CO oxidation. In Perovskites and Related Mixed Oxides: Concepts and Applications; Granger, P., Parvulescu, V.I., Kaliaguine, S., Prellier, W., Eds.; Wiley-VCH: Weinheim, Germany, 2016; pp. 453–475. [Google Scholar]
- Corma, A.; García, H. Supported Gold Nanoparticles as Oxidation Catalysts. In Nanoparticles and Catalysis; Astruc, D., Ed.; Willey-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 389–429. [Google Scholar]
- Santos, V.P.; Carabineiro, S.A.C.; Tavares, P.B.; Pereira, M.F.R.; Órfão, J.J.M.; Figueiredo, J.L. Oxidation of CO, ethanol and toluene over TiO2 supported noble metal catalysts. Appl. Catal. B Environ. 2010, 99, 198–205. [Google Scholar] [CrossRef]
- Bond, G.C.; Louis, C.; Thompson, D.T. Catalysis by Gold; Hutchings, G.J., Ed.; Catalytic Science Series; Imperial College Press: London, UK, 2006; pp. 180–182. [Google Scholar]
- Ide, Y.; Nakamura, N.; Hattori, H.; Ogino, R.; Ogawa, M.; Sadakane, M.; Sano, T. Sunlight-induced efficient and selective photocatalytic benzene oxidation on TiO2-supported gold nanoparticles under CO2 atmosphere. Chem. Commun. 2011, 47, 11531–11533. [Google Scholar] [CrossRef] [PubMed]
- Doustkhah, E.; Rostamnia, S.; Tsunoji, N.; Henzie, J.; Takei, T.; Yamauchi, Y.; Ide, Y. Templated synthesis of atomically-thin Ag nanocrystal catalysts in the interstitial space of a layered silicate. Chem. Commun. 2018, 54, 4402–4405. [Google Scholar] [CrossRef] [PubMed]
- Galusek, D.; Ghillányová, K. Ceramic Oxides. In Ceramics Science and Technology; Riedel, R., Chen, I.-W., Eds.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2010. [Google Scholar]
- Bagheri, S.; Julkapli, N.M.; Hamid, S.B.A. Titanium Dioxide as a Catalyst Support in Heterogeneous Catalysis. Sci. J. 2014, 2014, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Bamwenda, G.; Tsubota, S.; Nakamura, T.; Haruta, M. The influence of the preparation methods on the catalytic activity of platinum and gold supported on TiO2 for CO oxidation. Catal. Lett. 1997, 44, 83–87. [Google Scholar] [CrossRef]
- Sui, X.-L.; Wang, Z.-B.; Yang, M.; Huo, L.; Gu, D.-M.; Yin, G.-P. Investigation on C–TiO2 nanotubes composite as Pt catalyst support for methanol electrooxidation. J. Sources 2014, 255, 43–51. [Google Scholar] [CrossRef]
- Plata, J.J.; Romero-Sarria, F.; Amaya-Suarez, J.; Márquez, A.M.; Laguna, O.H.; Odriozola, J.A.; Sanz, J.F.; Ramos, J.J.P.; Sanz, J.F. Improving the activity of gold nanoparticles for the water-gas shift reaction using TiO2–Y2O3: An example of catalyst design. Phys. Chem. Chem. Phys. 2018, 20, 22076–22083. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.C.; Murcia, J.; Navío, J.A.; Colón, G.; Mesa, J.J.M. Photodeposition of gold on titanium dioxide for photocatalytic phenol oxidation. Appl. Catal. A Gen. 2011, 397, 112–120. [Google Scholar] [CrossRef]
- Colón, G.; Hidalgo, M.C.; Navío, J.A. Photocatalytic behaviour of sulphated TiO2 for phenol degradation. Appl. Catal. B Environ. 2003, 45, 39–50. [Google Scholar] [CrossRef]
- Chen, X.; Sun, H.; Zhang, J.; Guo, Y.; Kuo, D.-H. Cationic S-doped TiO2/SiO2 visible-light photocatalyst synthesized by co-hydrolysis method and its application for organic degradation. J. Mol. Liq. 2019, 273, 50–57. [Google Scholar] [CrossRef]
- Murcia, J.J.; Navio, J.A.; Hidalgo, M.C. Insights towards the influence of Pt features on the photocatalytic activity improvement of TiO2 by platinisation. Appl. Catal. B Environ. 2012, 126, 76–85. [Google Scholar] [CrossRef]
- Chenakin, S.; Kruse, N. Combining XPS and ToF-SIMS for assessing the CO oxidation activity of Au/TiO2 catalysts. J. Catal. 2018, 358, 224–236. [Google Scholar] [CrossRef]
- Chenakin, S.P.; Kruse, N. Au 4f spin–orbit coupling effects in supported gold nanoparticles. Phys. Chem. Chem. Phys. 2016, 18, 22778–22782. [Google Scholar] [CrossRef]
- Okazaki, K.; Morikawa, Y.; Tanaka, S.; Tanaka, K.; Kohyama, M. Electronic structures of Au onTiO2 (110) by first-principles calculations. Phys. Rev. B 2004, 69, 235404. [Google Scholar] [CrossRef]
- Vittadini, A.; Selloni, A. Small gold clusters on stoichiometric and defected TiO2 anatase (101) and their interaction with CO: A density functional study. J. Chem. Phys. 2002, 117, 353–361. [Google Scholar] [CrossRef]
- Li, B.; Hao, Y.; Shao, X.; Tang, H.; Wang, T.; Zhu, J.; Yan, S. Synthesis of hierarchically porous metal oxides and Au/TiO2 nanohybrids for photodegradation of organic dye and catalytic reduction of 4-nitrophenol. J. Catal. 2015, 329, 368–378. [Google Scholar] [CrossRef]
- Sanchez, V.M.; Martínez, E.D.; Ricci, M.L.M.; Troiani, H.; Soler-Illia, G.J.A.A. Optical Properties of Au Nanoparticles Included in Mesoporous TiO2 Thin Films: A Dual Experimental and Modeling Study. J. Phys. Chem. C 2013, 117, 7246–7259. [Google Scholar] [CrossRef]
- Chen, H.; Shao, L.; Li, Q.; Wang, J. Gold nanorods and their plasmonic properties. Chem. Soc. Rev. 2013, 42, 2679–2724. [Google Scholar] [CrossRef] [PubMed]
- Link, S.; El-Sayed, M.A. Size and Temperature Dependence of the Plasmon Absorption of Colloidal Gold Nanoparticles. J. Phys. Chem. B 1999, 103, 4212–4217. [Google Scholar] [CrossRef] [Green Version]
- Ayati, A.; Ahmadpour, A.; Bamoharram, F.F.; Tanhaei, B.; Mänttäri, M.; Sillanpää, M. A review on catalytic applications of Au/TiO2 nanoparticles in the removal of water pollutant. Chemosphere 2014, 107, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Maicu, M.; Hidalgo, M.C.; Colón, G.; Navío, J.A. Comparative study of the photodeposition of Pt, Au and Pd on pre-sulphated TiO2 for the photocatalytic decomposition of phenol. J. Photochem. Photobiol. A Chem. 2011, 217, 275–283. [Google Scholar] [CrossRef]
- Gołąbiewska, A.; Malankowska, A.; Jarek, M.; Lisowski, W.; Nowaczyk, G.; Jurga, S.; Zaleska-Medynska, A. The effect of gold shape and size on the properties and visible light-induced photoactivity of Au-TiO2. Appl. Catal. B Environ. 2016, 196, 27–40. [Google Scholar] [CrossRef]
- Sornalingam, K.; McDonagh, A.; Zhou, J.L.; Johir, M.A.H.; Ahmed, M.B. Photocatalysis of estrone in water and wastewater: Comparison between Au-TiO2 nanocomposite and TiO2, and degradation by-products. Sci. Total Environ. 2018, 610, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Pal, B. Size and shape dependent attachments of Au nanostructures to TiO2 for optimum reactivity of Au–TiO2 photocatalysis. J. Mol. Catal. A Chem. 2012, 355, 39–43. [Google Scholar] [CrossRef]
- Panayotov, D.A.; Morris, J.R. Surface chemistry of Au/TiO2: Thermally and photolytically activated reactions. Surf. Sci. Rep. 2016, 71, 77–271. [Google Scholar] [CrossRef]
- Maciejewski, M.; Fabrizioli, P.; Grunwaldt, J.-D.; Becker, O.S.; Baiker, A. Supported gold catalysts for CO oxidation: Effect of calcination on structure, adsorption and catalytic behaviour. Phys. Chem. Chem. Phys. 2001, 3, 3846–3855. [Google Scholar] [CrossRef]
- Yang, J.H.; Henao, J.D.; Raphulu, M.C.; Wang, Y.; Caputo, T.; Groszek, A.J.; Kung, M.C.; Scurrell, M.S.; Miller, J.T.; Kung, H.H. Activation of Au/TiO2 Catalyst for CO Oxidation. J. Phys. Chem. B 2005, 109, 10319–10326. [Google Scholar] [CrossRef]
- Weiher, N.; Beesley, A.M.; Tsapatsaris, N.; Delannoy, L.; Louis, C.; Van Bokhoven, J.A.; Schroeder, S.L.M. Activation of Oxygen by Metallic Gold in Au/TiO2 Catalysts. J. Am. Chem. Soc. 2007, 129, 2240–2241. [Google Scholar] [CrossRef]
- Wei, S.; Fu, X.-P.; Wang, W.-W.; Jin, Z.; Song, Q.-S.; Jia, C. Au/TiO2 Catalysts for CO Oxidation: Effect of Gold State to Reactivity. J. Phys. Chem. C 2018, 122, 4928–4936. [Google Scholar] [CrossRef]
- Laguna, O.H.; Domínguez, M.I.; Romero-Sarria, F.; Odriozola, J.A.; Centeno, M.A. Role of Oxygen Vacancies in Gold Oxidation Catalysis; Royal Society of Chemistry (RSC): London, UK, 2014; Chapter 13; pp. 489–511. [Google Scholar]
- Sarria, F.R.; Martínez T, L.M.; Centeno, M.A.; Odriozola, J.A. Surface Dynamics of Au/CeO2 Catalysts during CO Oxidation. J. Phys. Chem. C 2007, 111, 14469–14475. [Google Scholar] [CrossRef]
- Kamiuchi, N.; Sun, K.; Aso, R.; Tane, M.; Tamaoka, T.; Yoshida, H.; Takeda, S. Self-activated surface dynamics in gold catalysts under reaction environments. Nat. Commun. 2018, 9, 2060. [Google Scholar] [CrossRef] [PubMed]
- An, T.; Selloni, A.; Wang, H. Effect of reducible oxide–metal cluster charge transfer on the structure and reactivity of adsorbed Au and Pt atoms and clusters on anatase TiO2. J. Chem. Phys. 2017, 146, 184703. [Google Scholar]
- Kandoi, S.; Gokhale, A.; Grabow, L.; Dumesic, J.; Mavrikakis, M. Why Au and Cu Are More Selective Than Pt for Preferential Oxidation of CO at Low Temperature. Catal. Lett. 2004, 93, 93–100. [Google Scholar] [CrossRef]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.; Delmon, B. Low-Temperature Oxidation of CO over Gold Supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Cunningham, D.; Tsubota, S.; Kamijo, N.; Haruta, M. Preparation and catalytic behaviour of subnanometer gold deposited on TiO2 by vaccum calcination. Res. Chem. Intermed. 1993, 19, 1–13. [Google Scholar] [CrossRef]
- Murcia, J.J.; Ávila-Martínez, E.G.; Rojas, H.; Navío, J.A.; Hidalgo, M.C. Study of the E. coli elimination from urban wastewater over photocatalysts based on metallized TiO2. Appl. Catal. B Environ. 2017, 200, 469–476. [Google Scholar] [CrossRef]
- Sturges, H.A. The choice of a class interval Case I Computations involving a single. J. Am. Stat. Assoc. 1926, 21, 65–66. [Google Scholar] [CrossRef]
- Channei, D.; Inceesungvorn, B.; Wetchakun, N.; Ukritnukun, S.; Nattestad, A.; Chen, J.; Phanichphant, S. Photocatalytic Degradation of Methyl Orange by CeO2 and Fe–doped CeO2 Films under Visible Light Irradiation. Sci. Rep. 2014, 4, 5757. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Noble Metal Content (Au wt.%) | Area BET (m2/g) | Direct Band Gap (eV) | * Crystallite Size of the Metallic Species (nm) | Δ Average Particle Size of the Metallic Species (nm) |
|---|---|---|---|---|---|
| S–TiO2 | - | 58 | 3.20 | - | - |
| 2Au-15 | 0.28 | 53 | 3.56 | not detected | 4.8 |
| 2Au-120 | 0.77 | 53 | 3.53 | 6.4 | |
| 5Au-15 | 0.43 | 50 | 3.51 | 5.6 | |
| 5Au-120 | 0.73 | 48 | 3.51 | 6.2 | |
| 2Au-15C | 0.28 | 32 | 3.16 | 29 | 22 |
| 2Au-120C | 0.77 | 35 | 3.02 | 25 | 27 |
| 5Au-15C | 0.43 | 33 | 3.03 | 17 | 16 |
| 5Au-120C | 0.73 | 34 | 3.04 | 30 | 22 |
| Catalyst | Binding Energy (eV) | O/Ti Atomic Ratio | |
|---|---|---|---|
| Ti (2p3/2) | O (1s) | ||
| S–TiO2 | 458.5 | 529.8 | 1.70 |
| 2Au-15 | 458.6 | 530.0 | 1.83 |
| 2Au-120 | 458.5 | 529.8 | 1.85 |
| 5Au-15 | 458.9 | 530.2 | 1.86 |
| 5Au-120 | 458.5 | 529.9 | 1.95 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laguna, O.H.; Murcia, J.J.; Rojas, H.; Jaramillo-Paez, C.; Navío, J.A.; Hidalgo, M.C. Differences in the Catalytic Behavior of Au-Metalized TiO2 Systems During Phenol Photo-Degradation and CO Oxidation. Catalysts 2019, 9, 331. https://doi.org/10.3390/catal9040331
Laguna OH, Murcia JJ, Rojas H, Jaramillo-Paez C, Navío JA, Hidalgo MC. Differences in the Catalytic Behavior of Au-Metalized TiO2 Systems During Phenol Photo-Degradation and CO Oxidation. Catalysts. 2019; 9(4):331. https://doi.org/10.3390/catal9040331
Chicago/Turabian StyleLaguna, Oscar H., Julie J. Murcia, Hugo Rojas, Cesar Jaramillo-Paez, Jose A. Navío, and Maria C. Hidalgo. 2019. "Differences in the Catalytic Behavior of Au-Metalized TiO2 Systems During Phenol Photo-Degradation and CO Oxidation" Catalysts 9, no. 4: 331. https://doi.org/10.3390/catal9040331