Photocatalytic Degradation of 2,4,6-Trichlorophenol by MgO–MgFe2O4 Derived from Layered Double Hydroxide Structures

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. X-Ray Diffraction (XRD)
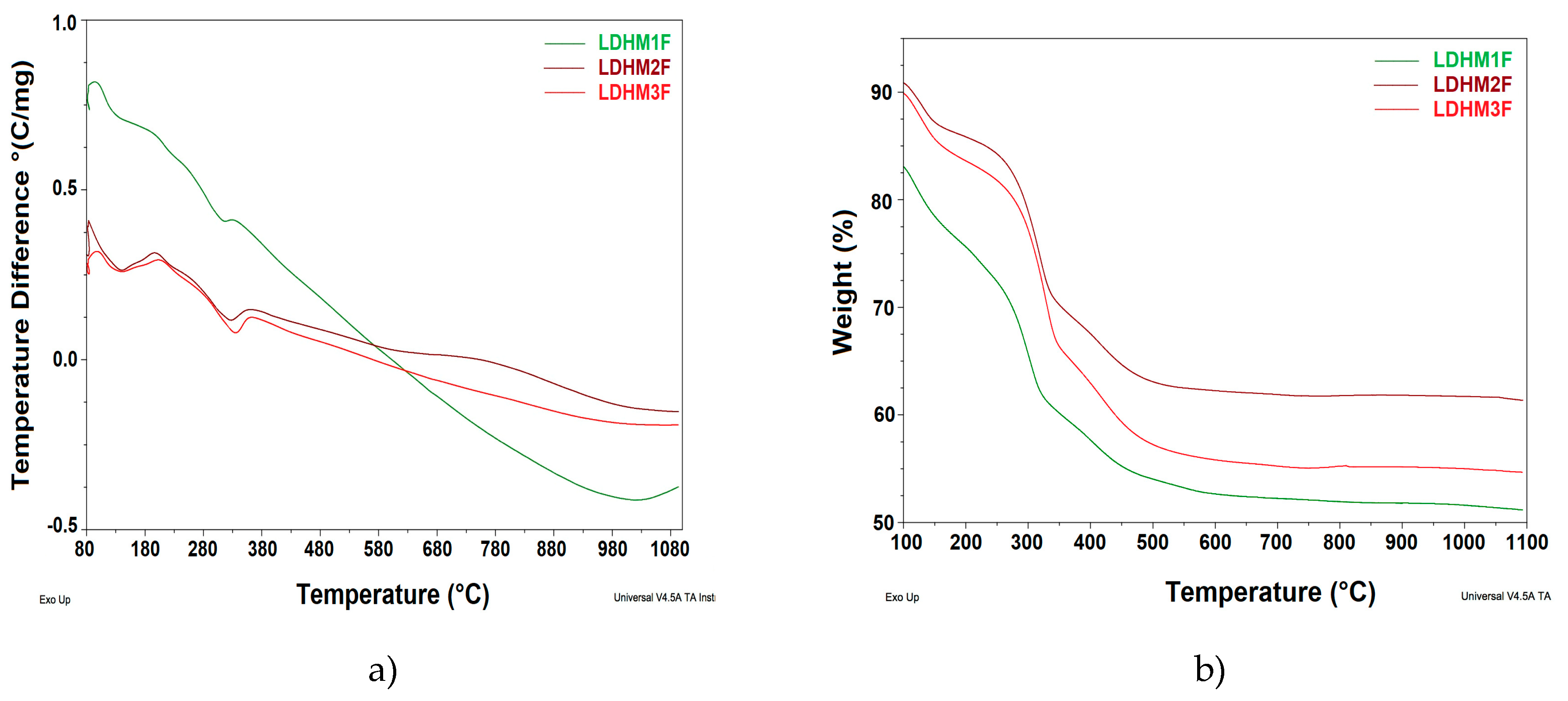
2.2. Thermal Analysis: DTA and TGA
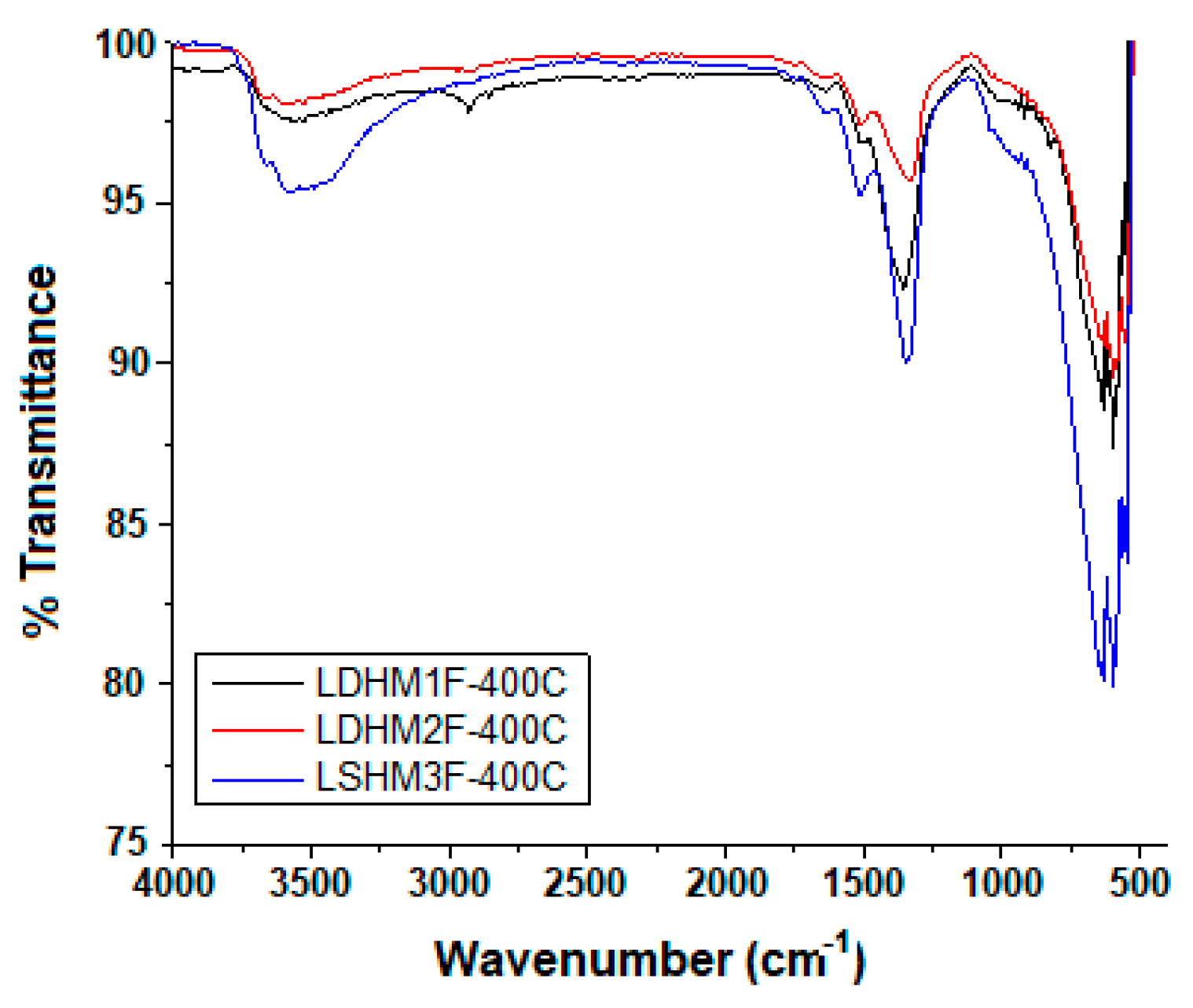
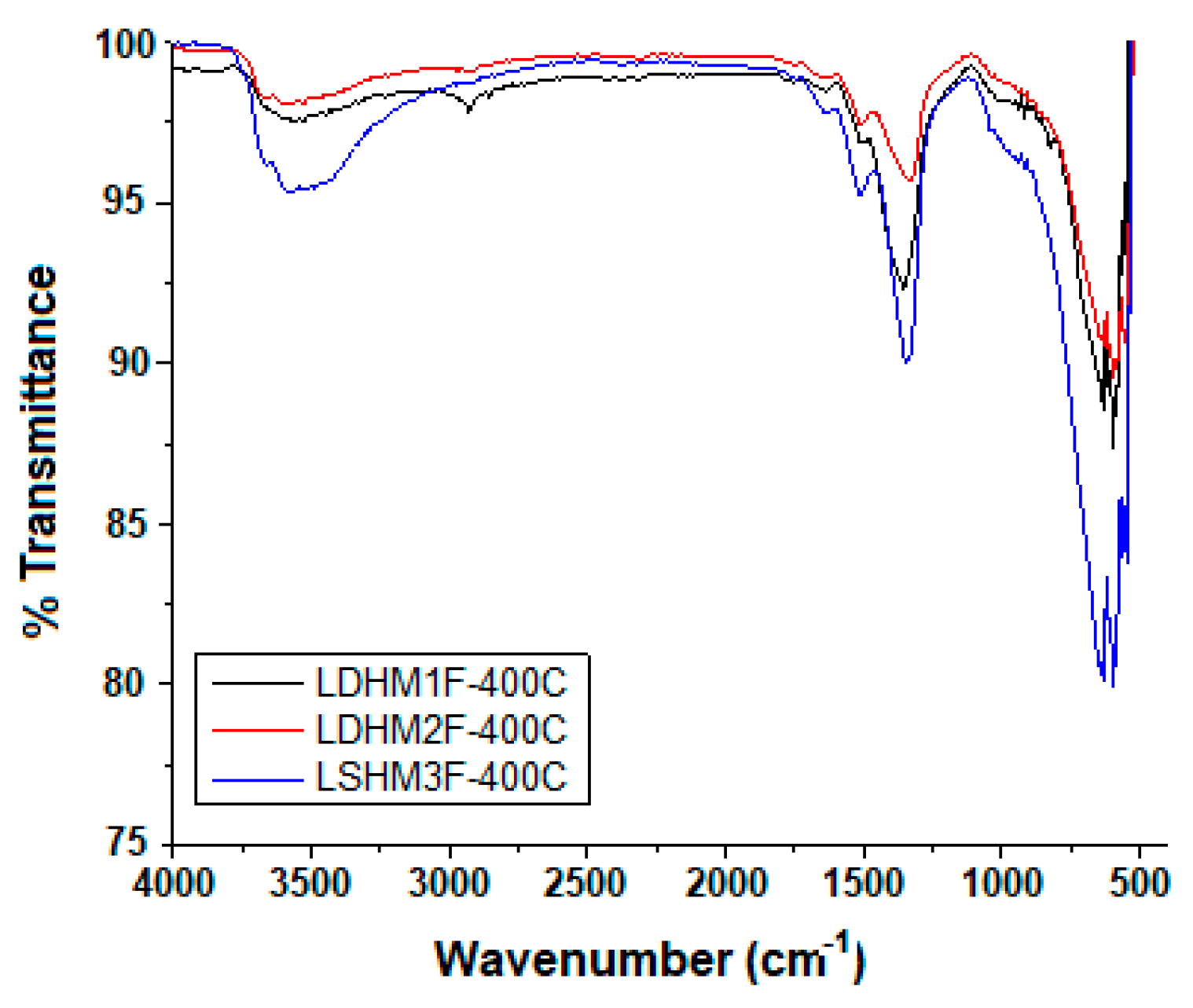
2.3. Fourier Transformed Infrared Spectrocopy (FTIR)
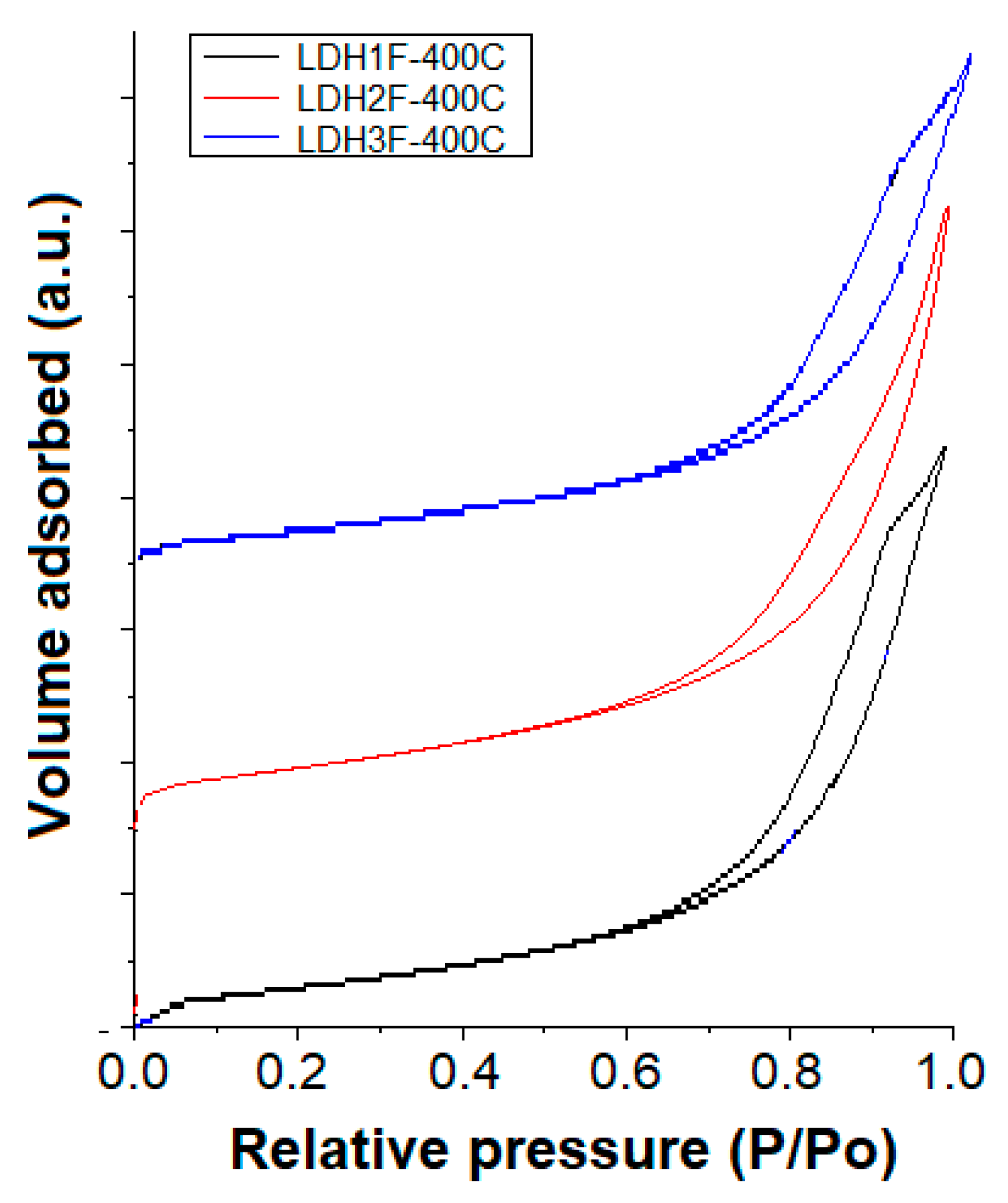
2.4. Textural Analysis
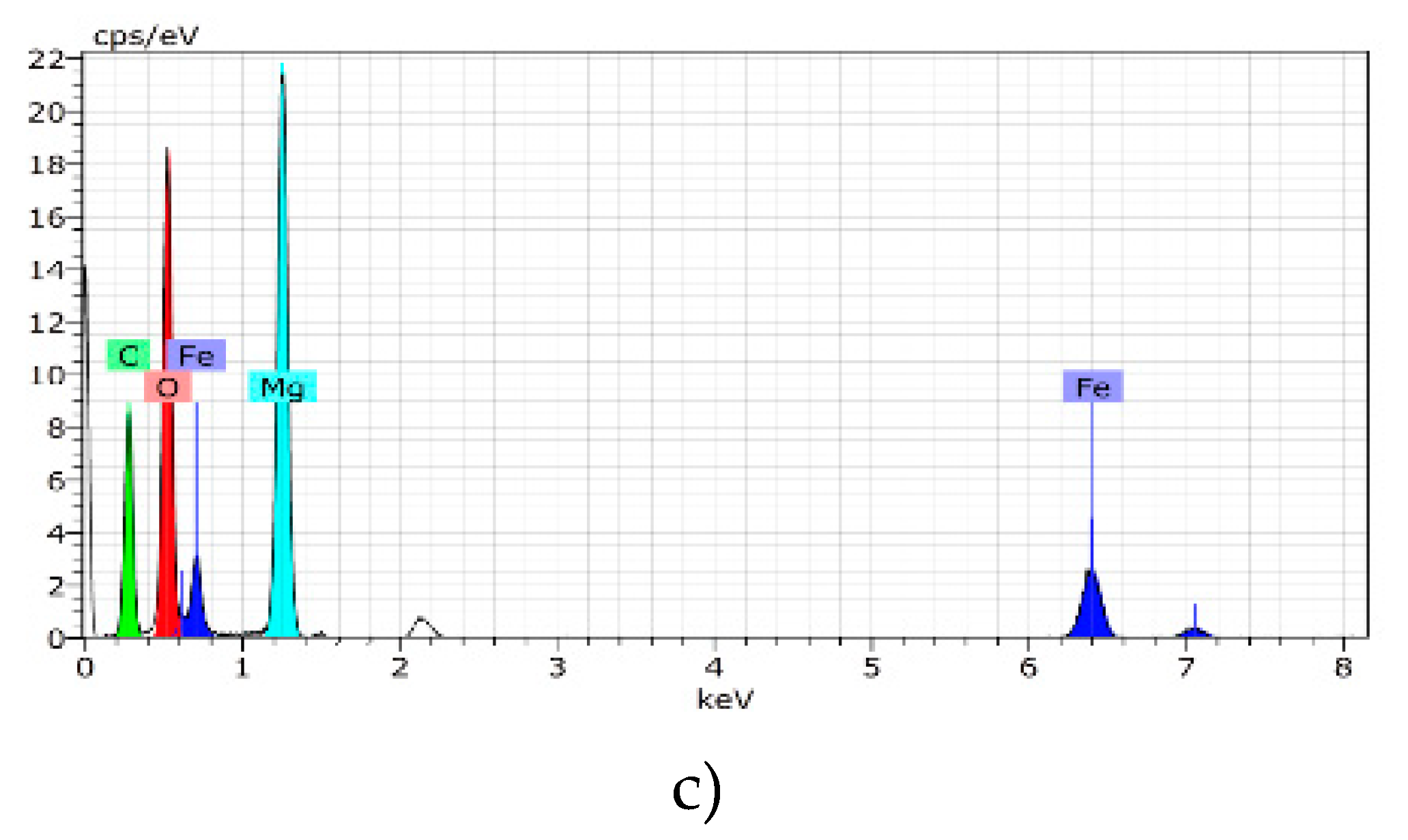
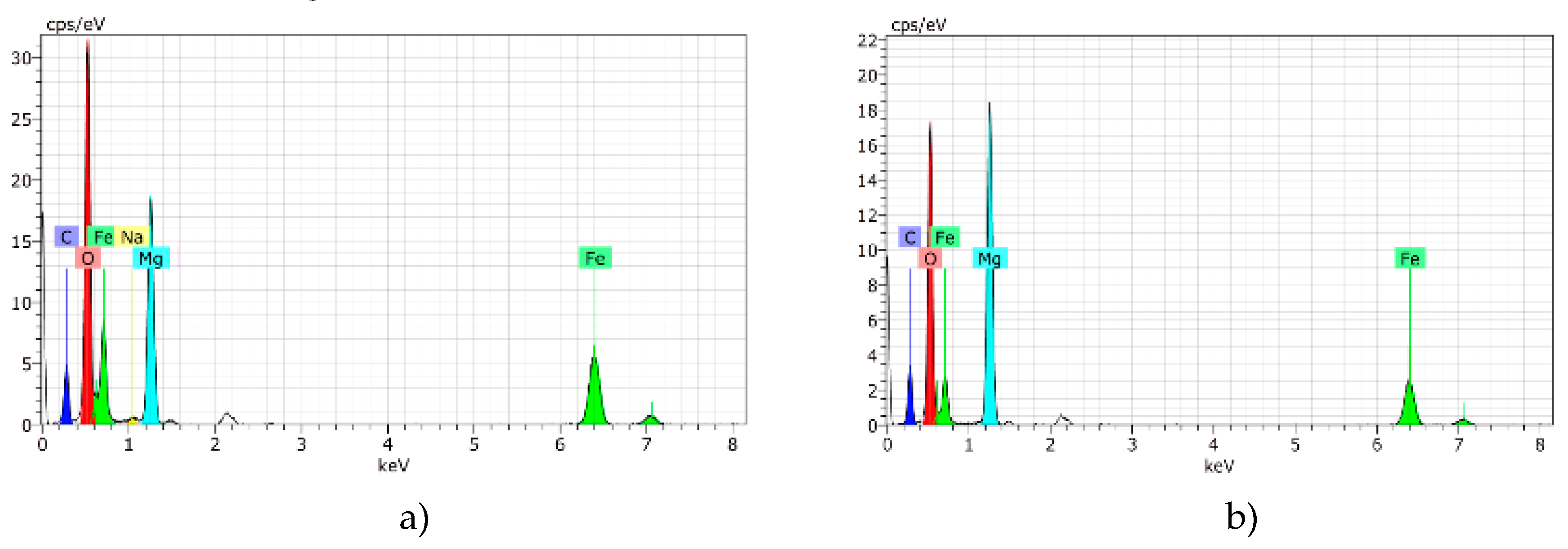
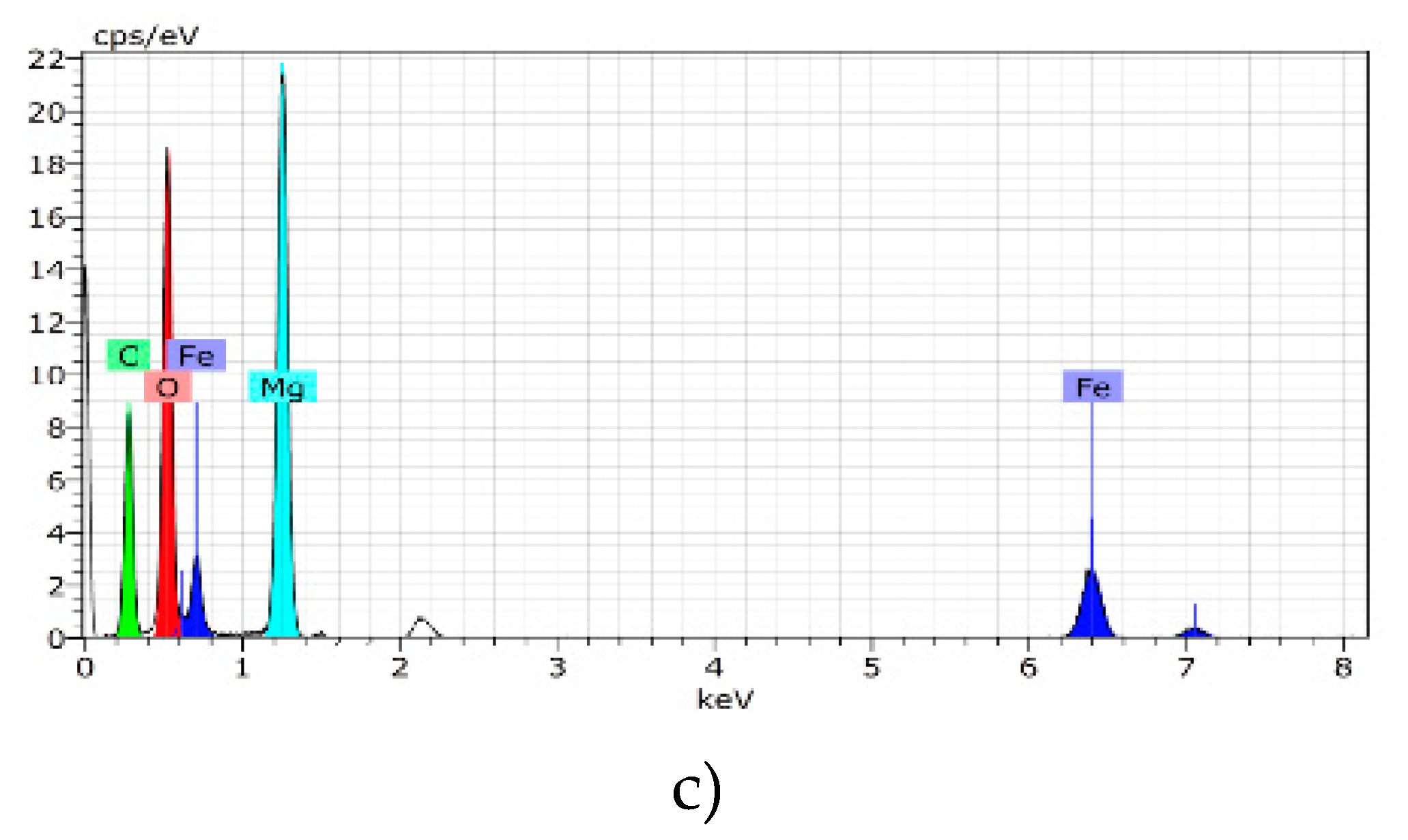
2.5. Scanning Electron Microscopy (SEM) with Energy-Dispersive X-Ray Spectroscopy (EDS)
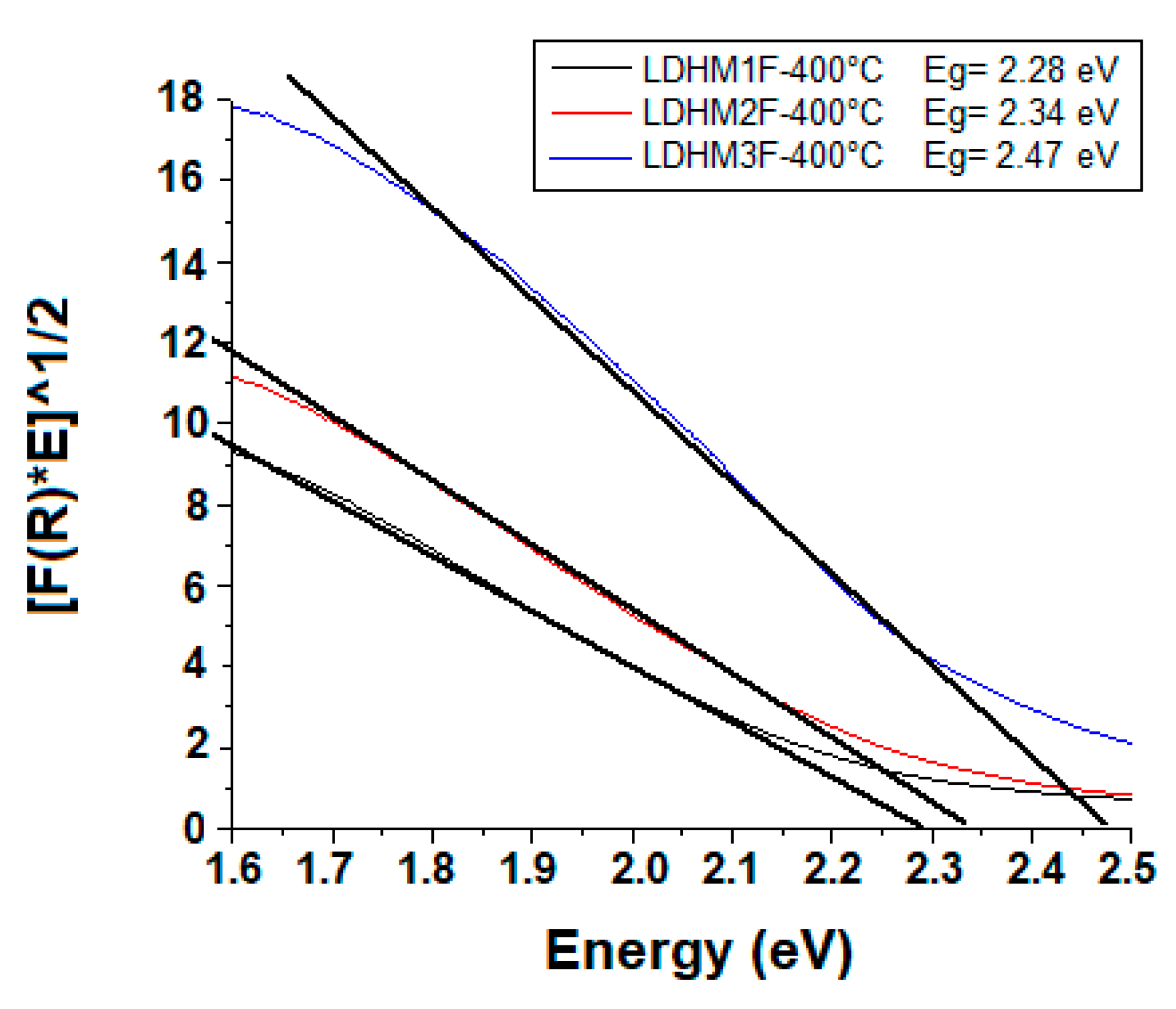
2.6. Diffuse Reflectance Spectroscopy (DRS)
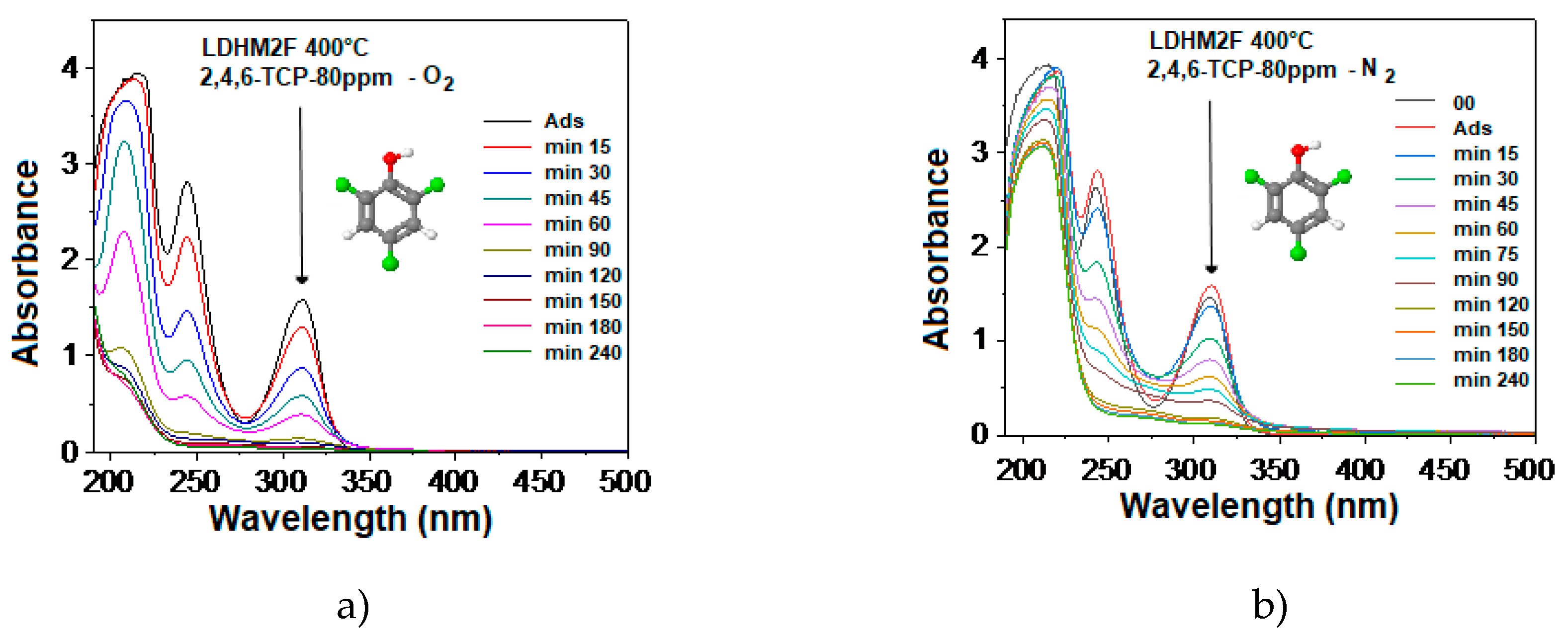
2.7. Photocatalytic Degradation of 2,4,6-Trichlorophenol
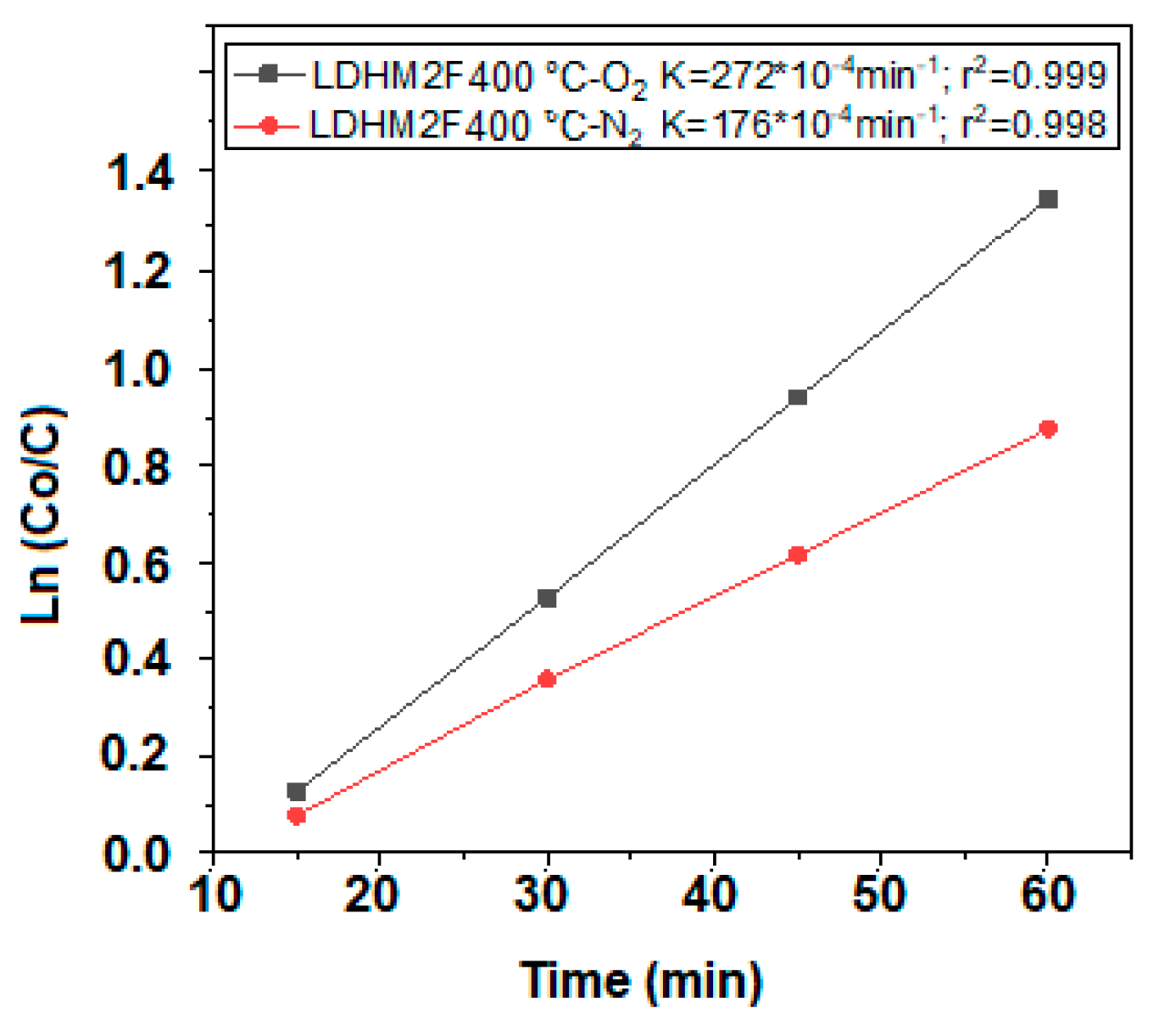
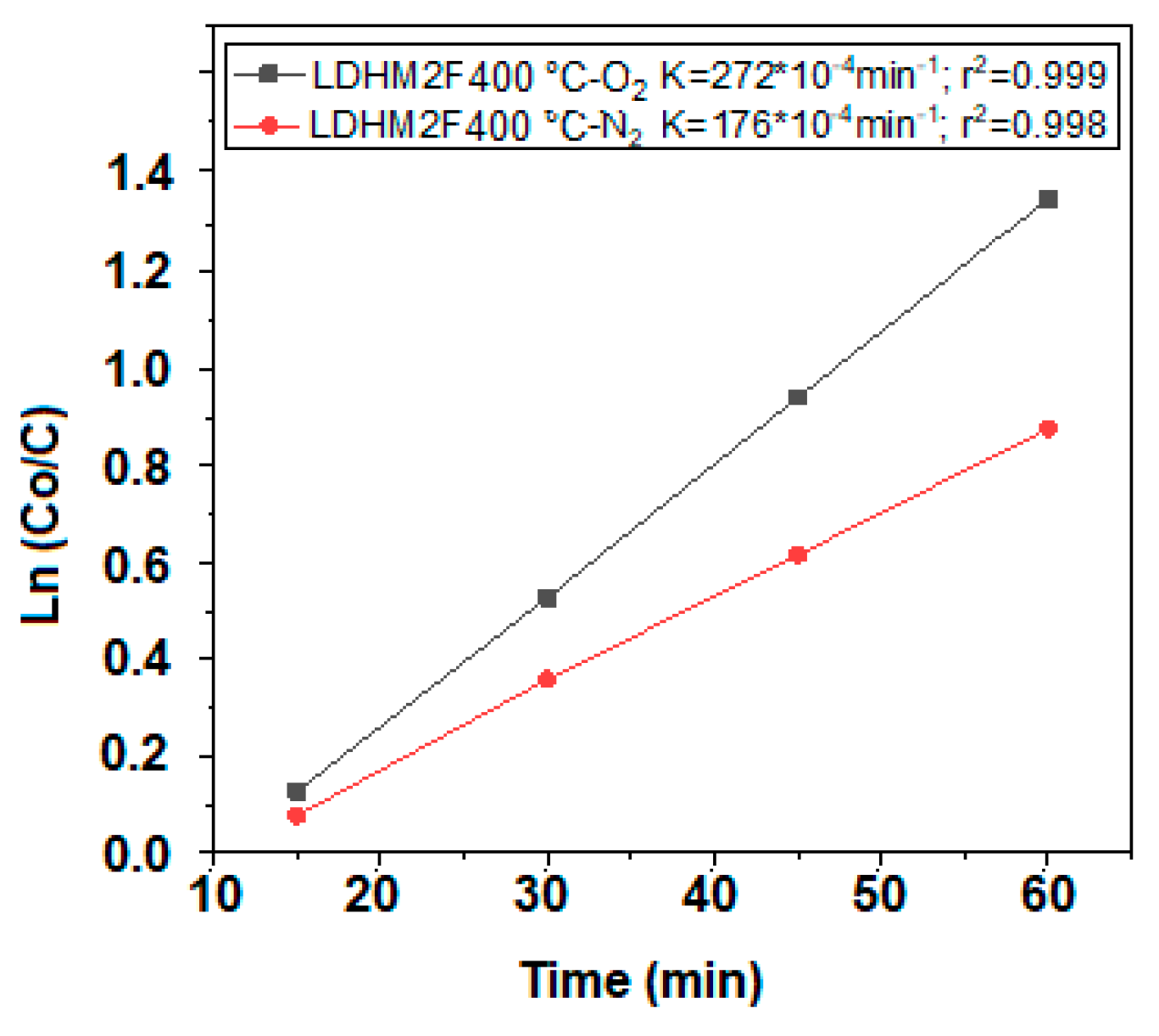
2.8. Kinetic Model Adjustment Study
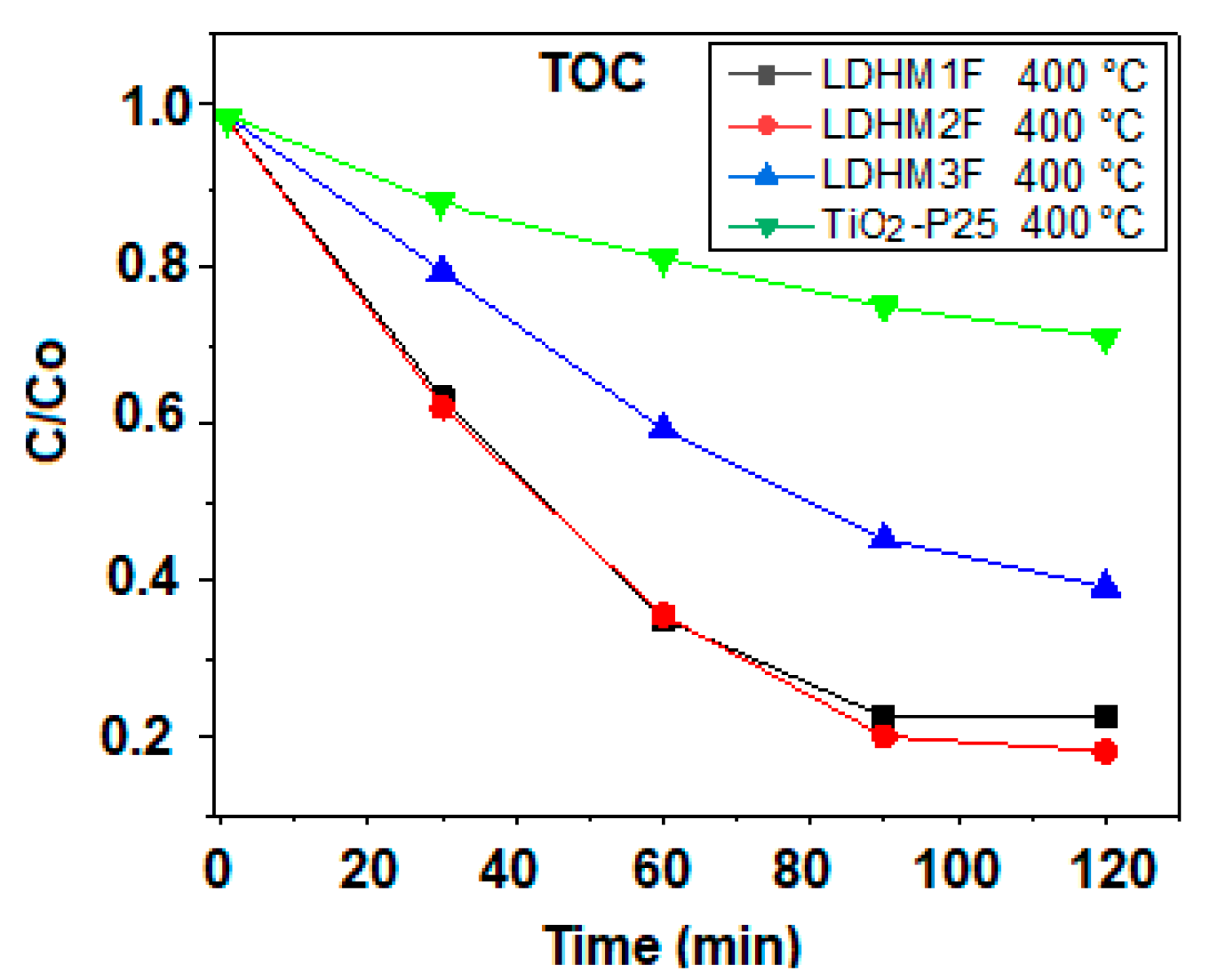
2.9. Mineralization Study
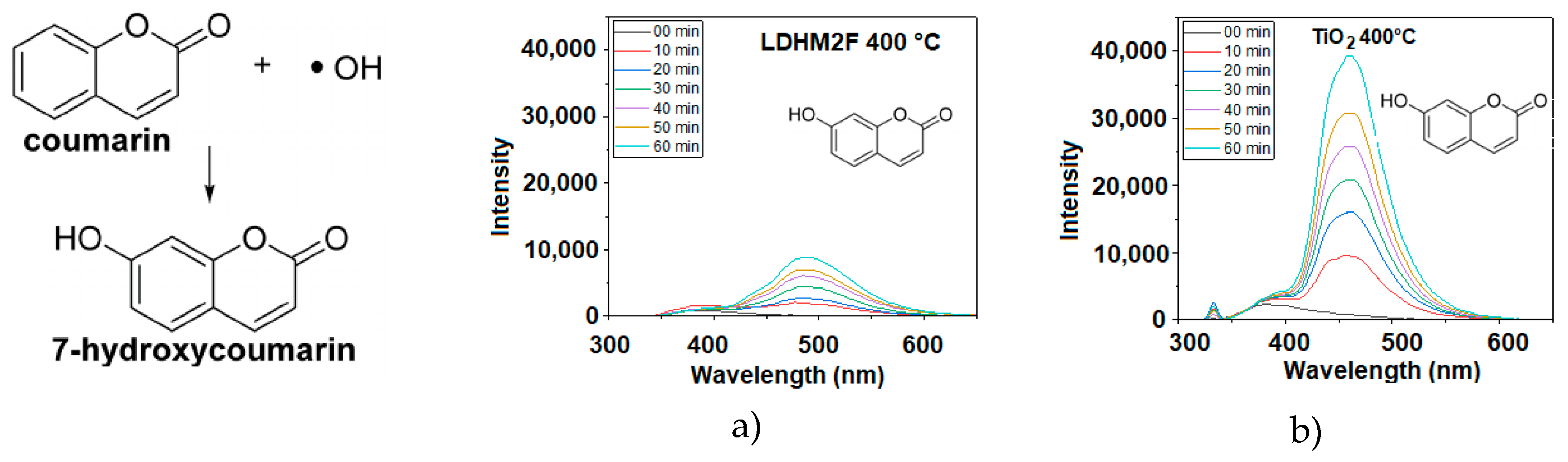
2.10. Detection of Hydroxyl Radicals (OH•)
2.11. Photocatalytic Evaluation of Superoxide Radicals (𝑂2−•)
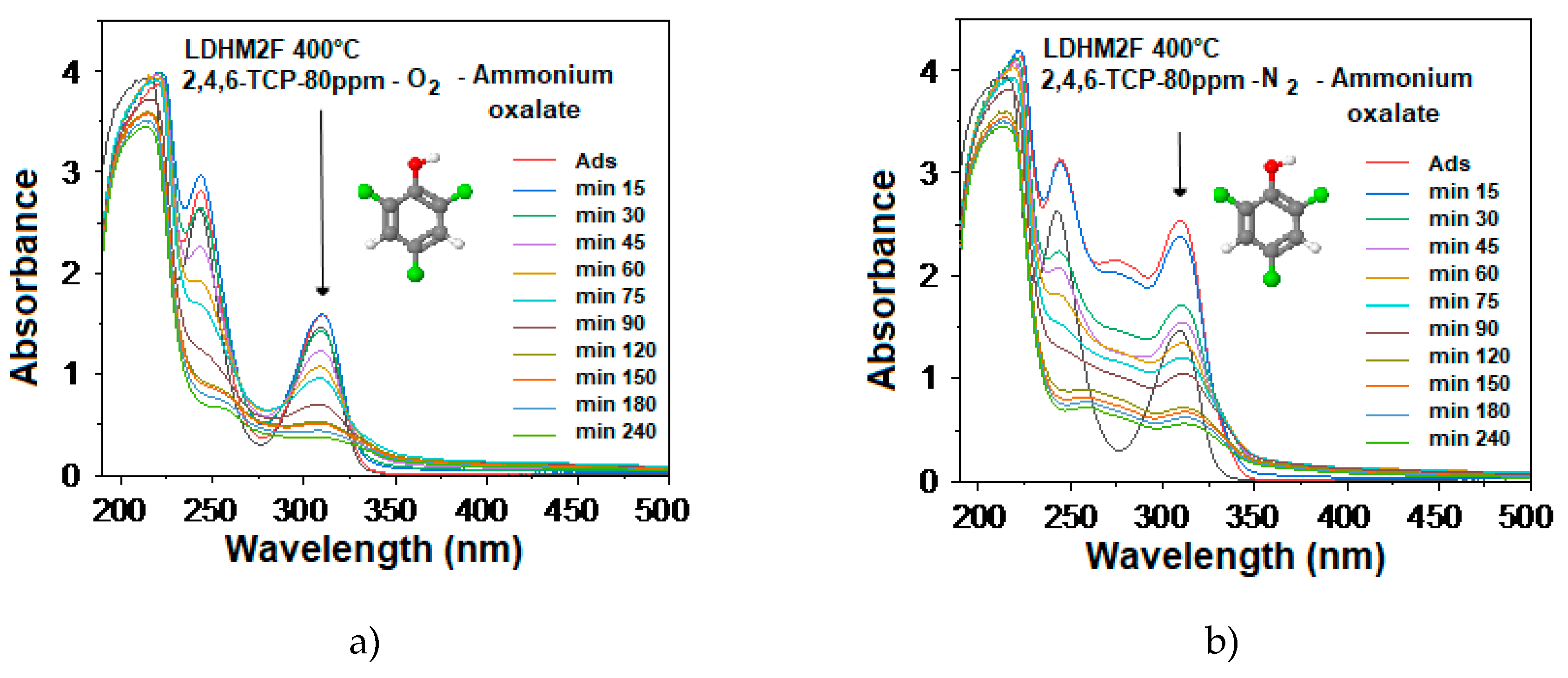
2.12. Photocatalytic Evaluation of the Hole Trap (h+)
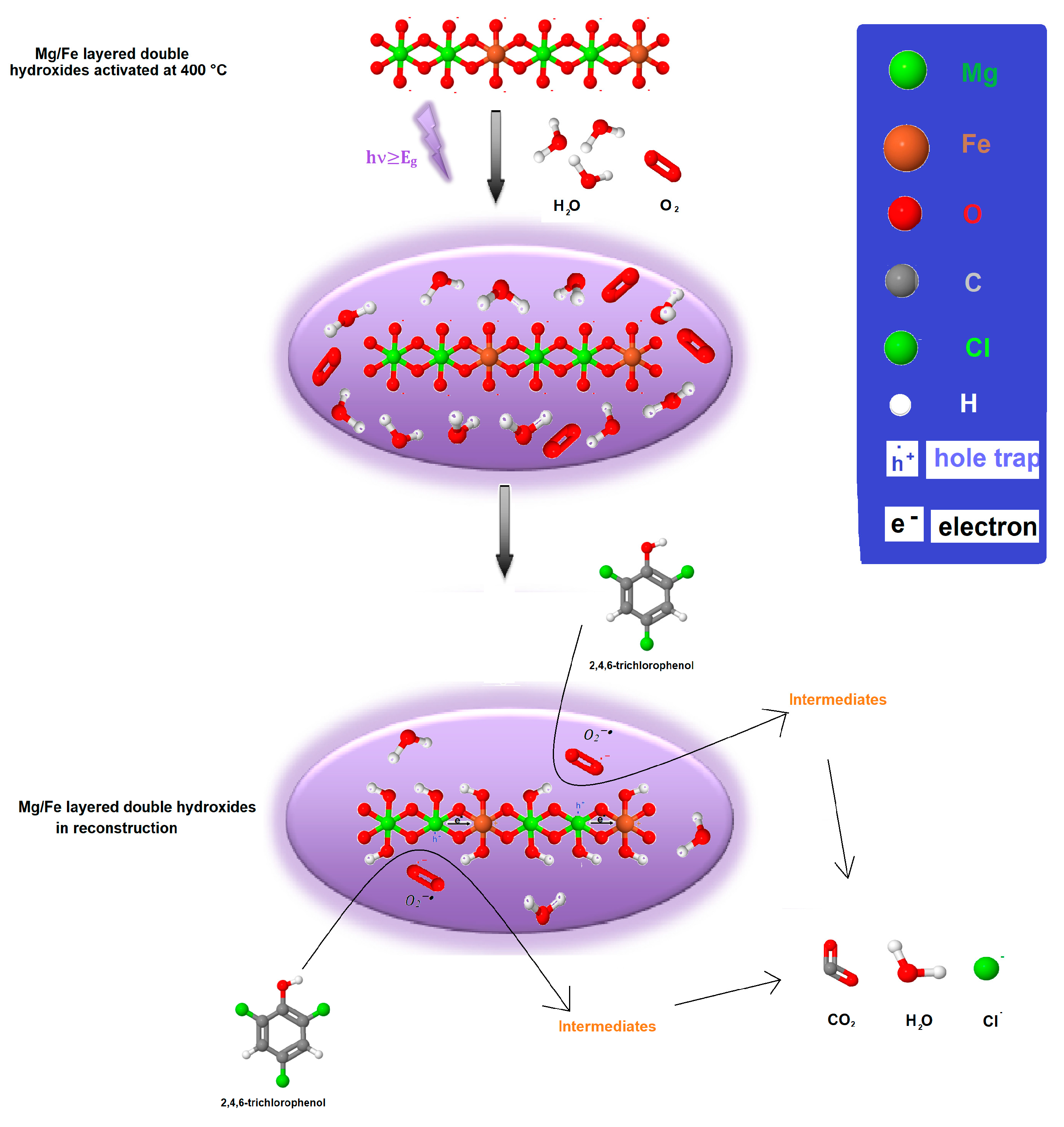
2.13. Possible 2,4,6-Trichlorophenol Degradation Mechanisms
3. Materials and Methods
3.1. Obtaining Photocatalysts
3.1.1. Synthesis of Mg/Fe-Layered Double Hydroxide Catalytic Pre-Cursors of Ratios 1, 2, and 3
3.1.2. Activation of Layered Double Hydroxides
3.2. Physicochemical Characterization of Catalytic Pre-Cursors and Catalysts
3.3. Evaluation of the Photocatalytic Activity in the Degradation of 2,4,6-Trichlorophenol
3.3.1. Degradation of 2,4,6-Trichlorophenol
3.3.2. Detection of Hydroxyl and Superoxide Radicals and Study of the Hole Trap
3.3.3. Photocatalytic Evaluation of Hole Capture with Ammonium Oxalate as a Sacrificial Agent
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miyata, S. Physico-chemical properties of synthetic hydrotalcites in relation to composition. Clays Clay Miner. 1980, 28, 50–56. [Google Scholar] [CrossRef]
- Mills, S.J.; Christy, A.G.; Génin, J.M.R.; Kameda, T.; Colombo, F. Nomenclature of the hydrotalcite supergroup: Natural layered double hydroxides. Mineral. Mag. 2012, 76, 1289–1336. [Google Scholar] [CrossRef]
- Roelofs, J.C.A.A.; Bokhoven, J.A.; Dillen, A.J.; Geus, J.W.; Jong, K.P. The Thermal Decomposition of Mg±Al Hydrotalcites: Effects of Interlayer Anions and Characteristics of the Final Structure. Chem. Eur. J. 2002, 8, 5571–5579. [Google Scholar] [CrossRef]
- Nguyen, H.K.D.; Nguyen, H.V.; Nguyen, V.A. Effect of synthetic conditions on the structure of mesoporous Mg-Al-Co hydrotalcite. J. Mol. Struct. 2018, 1171, 25–32. [Google Scholar] [CrossRef]
- Sikander, U.; Sufian, S.; Salam, M.A. A review of hydrotalcite based catalysts for hydrogen production systems. Int. J. Hydrogen Energy 2017, 42, 19851–19868. [Google Scholar] [CrossRef]
- Cavani, F.; Trifiro, F.; Vaccari, A. Hydrotalcite–type anionic clays: Preparation, properties and applications. Catal Today 1992, 11, 173–301. [Google Scholar] [CrossRef]
- Othman, M.R.; Helwani, Z.; Martunus, W.J.N. Synthetic hydrotalcites from different routes and their application as catalysts and gas adsorbents: A review. Appl. Organomet. Chem. 2009, 23, 335–346. [Google Scholar] [CrossRef]
- Yahyaoui, R.; Sanchez, P.E.; Pérez, L.A.; Nahdi, K.; Criado, J.M. Synthesis, characterization and combined kinetic analysis of thermal decomposition of hydrotalcite (Mg6Al2(OH)16CO3·4H2O). Thermochim. Acta 2018, 667, 177–184. [Google Scholar] [CrossRef]
- Vágvölgyi, V.; Palmer, S.J.; Kristóf, J.; Frost, R.L.; Horváth, E. Mechanism for hydrotalcite decomposition: A controlled rate thermal analysis study. J. Colloid Interface Sci. 2008, 318, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Mokhtar, M.; Inayat, A.; Ofili, J.; Schwieger, W. Thermal decomposition, gas phase hydration and liquid phase reconstruction in the system Mg/Al hydrotalcite/mixed oxide: A comparative study. Appl. Clay Sci. 2010, 50, 176–181. [Google Scholar] [CrossRef]
- Parida, K.M.; Sahoo, M.; Singha, S. Synthesis and characterization of a Fe(III)-Schiff base complex in a Zn-Al LDH host for cyclohexane oxidation. J. Mol. Catal A Chem. 2010, 329, 7–12. [Google Scholar] [CrossRef]
- Védrine, J.C. Heterogeneous Catalysis on Metal Oxides. Catalysts 2017, 7, 341. [Google Scholar] [CrossRef]
- Baloyi, J.; Ntho, T.; Mom, J. Synthesis and application of pillared clay heterogeneous catalysts for wastewater treatment: A review. RSC Adv. 2018, 8, 5197. [Google Scholar] [CrossRef]
- Prasad, C.; Tang, H.; Liu, W. Magnetic Fe3O4 based layered double hydroxides (LDHs) nanocomposites (Fe3O4/LDHs): Recent review of progress in synthesis, properties and applications. J. Nanostruct. Chem. 2018, 8, 393–412. [Google Scholar] [CrossRef]
- Moma, J.; Baloyi, J.; Ntho, T. Synthesis and characterization of an efficient and stable Al/Fe pillared clay catalyst for the catalytic wet air oxidation of phenol. RSC Adv. 2018, 8, 30115. [Google Scholar] [CrossRef]
- Valencia-Lopez, C.D.; Zafra-Calvo, M.; Martín de Vidales, M.J.; Blanco-Gutierrez, V.; Atanes-Sanchez, E.; Merayo, N.; Fernandez-Martinez, F.; Nieto-Marquez, A.; Dos santos-Garcia, J. Synthesis of NiFe2O4-LDH Composites with High Adsorption and Photocatalytic Activity for Methyl Orange Degradation. Inorganics 2018, 6, 98. [Google Scholar] [CrossRef]
- Li, Z.; Chen, H.; Liu, W. Full-Spectrum Photocatalytic Activity of ZnO/CuO/ZnFe2O4 Nanocomposite as a PhotoFenton-Like Catalyst. Catalysts 2018, 8, 557. [Google Scholar] [CrossRef]
- Descorme, C. Catalytic wastewater treatment: Oxidation and reduction processes. Recent studies on chlorophenols, Claude Descorme. Catal Today 2017, 297, 324–334. [Google Scholar] [CrossRef]
- Lazar, M.A.; Varghese, S.; Nair, S.S. Photocatalytic Water Treatment by Titanium Dioxide: Recent Updates. Catalysts 2012, 2, 572–601. [Google Scholar] [CrossRef] [Green Version]
- Pi, Y.; Wang, J. Pathway of the ozonation of 2,4,6-trichlorophenol in aqueous solution. Front. Environ. Sci. Eng. China 2007, 1, 179–183. [Google Scholar] [CrossRef]
- Yan, Y.; Wu, X.; Zhang, H. Catalytic wet peroxide oxidation of phenol over Fe2O3/MCM-41 in a fixed bed reactor. Sep. Purif. Technol. 2016, 171, 52–61. [Google Scholar] [CrossRef]
- Honda, M.; Kannan, K. Biomonitoring of chlorophenols in human urine from several Asian countries, Greece and the United States. Environ. Pollut. 2018, 232, 487–493. [Google Scholar] [CrossRef]
- Badanthadka, M.; Mehendale, H.M. Chlorophenols. In Encyclopedia of Toxicology, 3rd ed.; Elsiever: Amsterdam, The Netherlands, 2014; pp. 896–899. [Google Scholar] [CrossRef]
- Mostafalou, S.; Abdollahi, M. Pesticides: An update of human exposure and toxicity. Arch. Toxicol. 2017, 91, 549–599. [Google Scholar] [CrossRef]
- Michałowicz, J. Pentachlorophenol and its derivatives induce oxidative damage and morphological changes in human lymphocytes (in vitro). Arch. Toxicol. 2010, 84, 379–387. [Google Scholar] [CrossRef]
- Khorsandi, H.; Ghochlavi, N.; Aghapour, A.A. Biological Degradation of 2,4,6-Trichlorophenol by a Sequencing Batch Reactor. Environ. Process. 2018, 5, 907–917. [Google Scholar] [CrossRef]
- Yi, A.; Feng, Y.; Du, Z.; Li, H. Mechanism of 2, 4, 6-Trichlorophenol Degradation in Microbial Fuel Cells System with Microbe Isolated from Submarine Sediment. Int. J. Electrochem. Sci. 2015, 10, 1459–1468. [Google Scholar]
- Krishnaiah, D.; Anisuzzaman, S.M.; Bono, A.; Sarbatly, R. Adsorption of 2,4,6-trichlorophenol (TCP) onto activated carbon. J. King Saud Univ. Sci. 2013, 25, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Liu, X.; Tian, W.; Yan, D.; Sun, X.; Lei, X. Adsolubilization of 2,4,6-trichlorophenol from aqueous solution by surfactant intercalated ZnAl layered double hydroxides. Chem. Eng. J. 2015, 279, 597–604. [Google Scholar] [CrossRef]
- Saritha, P.; Suman, D.S.; Aparna, C.; Vijaya, P.N.; Himabindu, V.; Anjaneyulu, Y. Degradative Oxidation of 2,4,6 Trichlorophenol Using Advanced Oxidation Processes—A Comparative Study. Water Air Soil Pollut. 2009, 200, 169–179. [Google Scholar] [CrossRef]
- Kashyap, J.; Riaz, U. Facile synthesis of novel polypyrrole dispersed AgFeO2 nanohybrid with highly efficient photocatalytic activity towards 2,4,6-trichlorophenol degradation. RSC Adv. 2018, 8, 13218–13225. [Google Scholar] [CrossRef]
- Benbachir, H.; Gaffour, H.; Mokhtari, M. Photodegradation of 2,4,6-trichlorophenol using natural hematite modified with chloride of zirconium oxide. React. Kinet. Mech. Catal. 2017, 122, 635–653. [Google Scholar] [CrossRef]
- Pino-Chamorro, J.Á.; Ditrói, T.; Lente, G.; Fábián, I. A detailed kinetic study of the direct photooxidation of 2,4,6-trichlorophenol. J. Photochem. Photobiol. A 2016, 330, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Chaliha, S.; Bhattacharyya, K.G. Wet oxidative method for removal of 2,4,6-trichlorophenol in water using Fe(III), Co(II), Ni(II) supported MCM41 catalysts. J. Hazard. Mater. 2008, 150, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, H.; Gao, J.; Yan, T.; Zhou, F.; Cuin, S.; Bi, W. Synthesis of Fe3O4/g-C3N4 nanocomposites and their application in the photodegradation of 2,4,6-trichlorophenol under visible light. Mater. Lett. 2016, 164, 183–189. [Google Scholar] [CrossRef]
- Hee-Chan, K.; Sang-Hyup, L.; Dong-Ju, K.; Jae-Woo, C. Photocatalytic Activity of 2,4,6-Trichlorophenol by TiO2 Mesostructures: Effects of Surface Modification, Calcination Temperature and Initial pH. Water Air Soil Pollut. 2013, 224, 1459–1467. [Google Scholar] [CrossRef]
- Rozov, K.; Berner, U.R.; Kulik, D.A.; Diamond, L.W. Solubility and thermodynamic properties of carbonate-bearing hydrotalcite-pyroaurite solid solutions with a 3:1 Mg/(Al+Fe) mole ratio. Clays Clay Miner. 2011, 59, 215–232. [Google Scholar] [CrossRef]
- Lavand, A.B.; Malghe, Y.S. Nano sized C-doped TiO2 as a visible-light photocatalyst for the degradation of 2,4,6- trichlorophenol. Adv. Mater. Lett. 2015, 6, 695–700. [Google Scholar] [CrossRef]
- Skrabal, P.M. Spectroscopy: An Interdisciplinary Integral Description of Spectroscopy from UV to NMR, 1st ed.; vdf Hochschulverlag AG an der ETZ Zurich: Zurich, Switzerland, 2012; Volume 1, pp. 216–224. ISBN 978-3-7281-3385-4. [Google Scholar]
- Turchi, C.S.; Ollis, D.F. Mixed Reactant photocatalysis: Intermediates and Mutual Rate Inhibition. J. Catal. 1989, 119, 483–496. [Google Scholar] [CrossRef]
- Lin, Z.R.; Zhao, L.; Dong, Y.H. Quantitative characterization of hydroxyl radical generation in a goethite-catalyzed Fenton-like reaction Author links open overlay panel. Chemosphere 2015, 141, 7–12. [Google Scholar] [CrossRef]
- Fu, S.; Deng, B.; Ma, D.; Cheng, H.; Dong, S. Visible-Light-Driven Photocatalytic Fuel Cell with an Ag-TiO2 Carbon Foam Anode for Simultaneous 4-Chlorophenol Degradation and Energy Recovery. ChemEngineering 2018, 2, 20. [Google Scholar] [CrossRef]
- An, X.; Liu, H.; Qu, J.; Moniz, S.J.A.; Tang, J. Photocatalytic mineralisation of herbicide 2,4,5-trichlorophenoxyacetic acid: Enhanced performance by triple junction Cu–TiO2–Cu2O and the underlying reaction mechanism. New J. Chem. 2015, 39, 314–320. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Generation and Detection of Reactive Oxygen Species in Photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef]
- Chen, F.; Yang, Q.; Pehkonen, S.O.; Ray, M.B. Modeling of Gas-Phase Photodegradation of Chloroform and Carbon Tetrachloride. J. Air Waste Manag. Assoc. 2004, 54, 1281–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, E.M.; Márquez, G.; Tena, M.; Álvarez, P.M.; Beltrán, F.J. Determination of main species involved in the first steps of TiO2 photocatalytic degradation of organics with the use of scavengers: The case of ofloxacin. Appl. Catal. B 2015, 178, 44–53. [Google Scholar] [CrossRef]
- Wu, W.; Liang, S.; Chen, Y.; Shen, L.; Zheng, H.; Wu, L. High efficient photocatalytic reduction of 4-nitroaniline to p-phenylenediamine over microcrystalline SrBi2Nb2O9. Catal. Commun. 2012, 17, 39–42. [Google Scholar] [CrossRef]
- Xian, T.; Yang, H.; Di, L.; Ma, J.; Zhang, H.; Dai, J. Photocatalytic reduction synthesis of SrTiO3-graphene nanocomposites and their enhanced photocatalytic activity. Nanoscale Res. Lett. 2014, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wnag, M.; Xu, C.; Chen, S.; Fu, X. Significantly enhanced visible-light photocatalytic activity of g-C3N4 via ZnO modification and the mechanism study. J. Mol. Catal. A Chem. 2013, 368, 9–15. [Google Scholar] [CrossRef]
- Mascolo, G.; Mascolo, M.C. On the synthesis of layered double hydroxides (LDHs) by reconstruction method based on the “memory effect”. Microporous Mesoporous Mater. 2015, 214, 246–248. [Google Scholar] [CrossRef]
- Kim, B.K.; Gwak, G.H.; Okada, T.; Oh, J.M. Effect of particle size and local disorder on specific surface area of layered double hydroxides upon calcination-reconstruction. J. Solid State Chem. 2018, 263, 60–64. [Google Scholar] [CrossRef]
- Teodorescu, F.; Paladuta, A.M.; Pavel, O.D. Memory effect of hydrotalcites and its impact on cyanoethylation reaction. Mater. Res. Bull. 2013, 48, 2055–2059. [Google Scholar] [CrossRef]
- Czaplicka, M. Photo-degradation of chlorophenols in the aqueous solution. J. Hazard Mater. 2006, 134, 45–59. [Google Scholar] [CrossRef]
- Xu, L.; Wang, J. Degradation of 2,4,6-trichlorophenol using magnetic nanoscaled Fe3O4/CeO2 composite as a heterogeneous Fenton-like catalyst. Sep. Purif. Technol. 2015, 149, 255–264. [Google Scholar] [CrossRef]
- Odling, G.; Robertson, N. Bridging the gap between laboratory and application in photocatalytic water purification. Catal. Sci. Technol. 2019, 9, 533–545. [Google Scholar] [CrossRef]
- Guo, Q.; Zhou, C.; Ma, Z.; Ren, Z.; Fan, H.; Yang, X. Elementary Chemical Reactions in Surface Photocatalysis. Annu. Rev. Phys. Chem. 2018, 69, 451–472. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | BET Area (m2/g) | Pore Diameter (nm) | Pore Volume (cm3/g) |
|---|---|---|---|
| LDHMIF-400 °C | 282.2 | 8.72 | 0.0178 |
| LDHM2F-400 °C | 253.1 | 7.34 | 0.0391 |
| LDHM3F-400 °C | 248.9 | 6.30 | 0.0136 |
| Catalyst | Bandgap (eV) | Kapp*10−4 (min−1) | t1/2 (min) | %Degradation (UV-Vis) | %Mineralization (TOC) |
|---|---|---|---|---|---|
| LDHMIF-400 °C | 2.28 | 252 | 28 | 92 | 78 |
| LDHM2F-400 °C | 2.34 | 272 | 25 | 93 | 82 |
| LDHM3F-400 °C | 2.47 | 125 | 56 | 55 | 61 |
| TiO2-P25-400 °C | 3.20 | 51 | 136 | 18 | 29 |
| Catalyst | Kapp*10−4 (min−1) | %Degradation (UV-Vis) | %Mineralization (TOC) |
|---|---|---|---|
| LDHM2F-400 °C with O2 | 252 | 91 | 77 |
| LDHM2F-400 °C with N2 | 176 | 76 | 66 |
| LDHM2F-400 °C-O2 with AO | 87 | 47 | 20 |
| LDHM2F-400 °C with N2-AO | 83 | 44 | 18 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-Ramírez, E.; Tzompantzi-Morales, F.; Gutiérrez-Ortega, N.; Mojica-Calvillo, H.G.; Castillo-Rodríguez, J. Photocatalytic Degradation of 2,4,6-Trichlorophenol by MgO–MgFe2O4 Derived from Layered Double Hydroxide Structures. Catalysts 2019, 9, 454. https://doi.org/10.3390/catal9050454
Ramos-Ramírez E, Tzompantzi-Morales F, Gutiérrez-Ortega N, Mojica-Calvillo HG, Castillo-Rodríguez J. Photocatalytic Degradation of 2,4,6-Trichlorophenol by MgO–MgFe2O4 Derived from Layered Double Hydroxide Structures. Catalysts. 2019; 9(5):454. https://doi.org/10.3390/catal9050454
Chicago/Turabian StyleRamos-Ramírez, Esthela, Francisco Tzompantzi-Morales, Norma Gutiérrez-Ortega, Héctor G. Mojica-Calvillo, and Julio Castillo-Rodríguez. 2019. "Photocatalytic Degradation of 2,4,6-Trichlorophenol by MgO–MgFe2O4 Derived from Layered Double Hydroxide Structures" Catalysts 9, no. 5: 454. https://doi.org/10.3390/catal9050454