Effect of Metal Loading in Unpromoted and Promoted CoMo/Al2O3–TiO2 Catalysts for the Hydrodeoxygenation of Phenol


 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Effect of Mo Loading
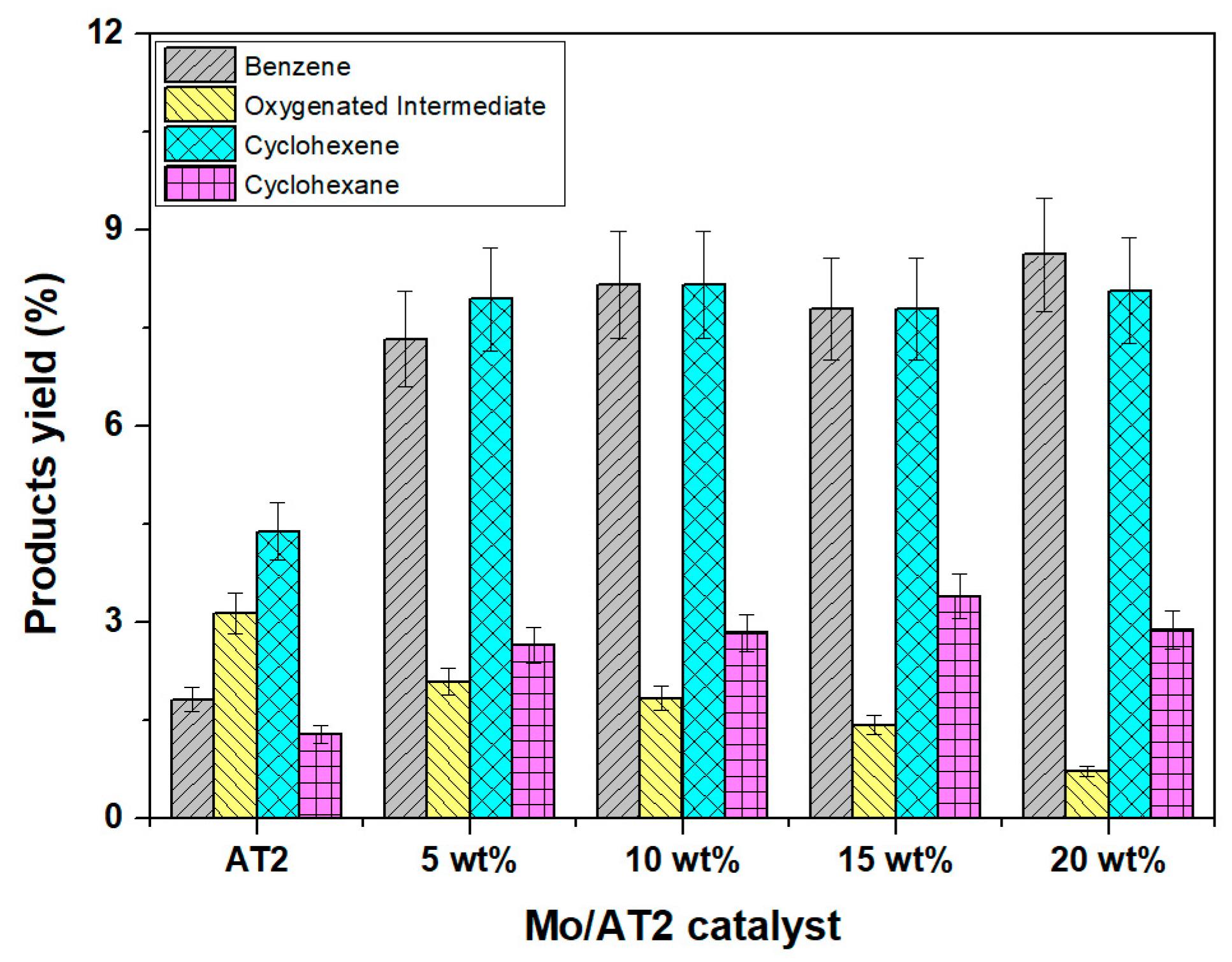
2.1.1. Catalytic Activity
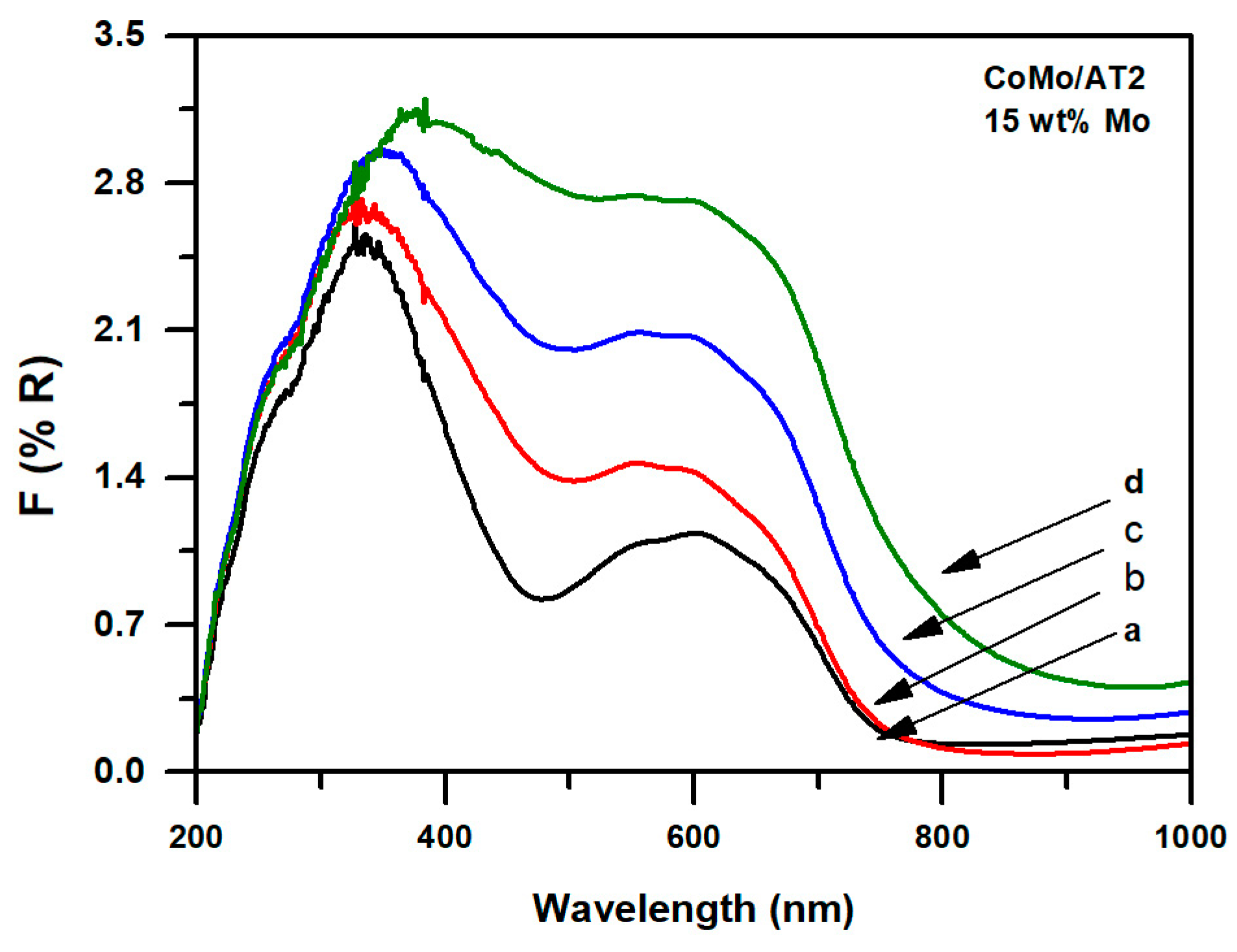
2.1.2. Diffuse Reflectance Spectroscopy UV-Vis
2.1.3. Laser Raman Spectroscopy
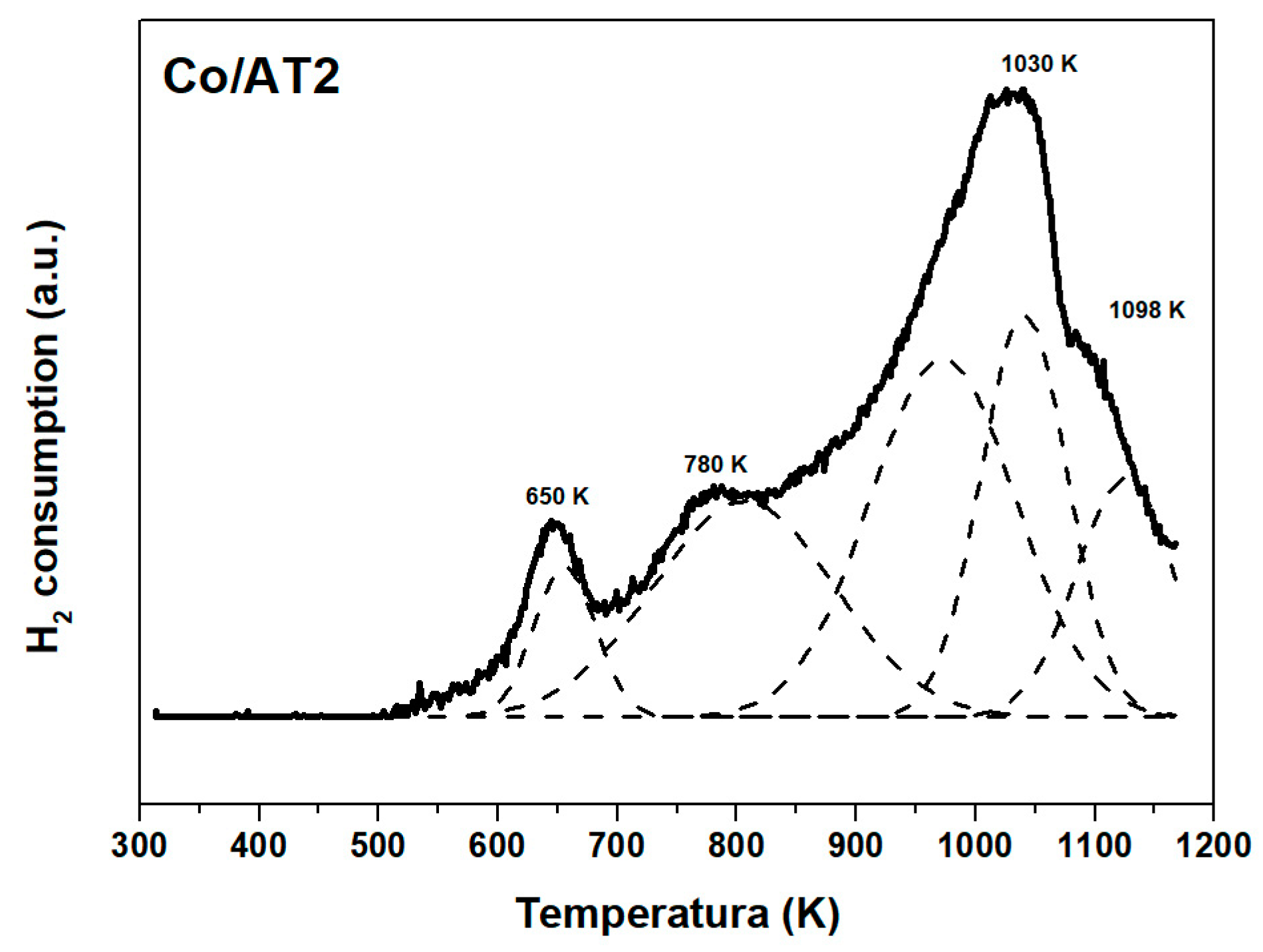
2.1.4. Temperature Programed Reduction
2.2. Effect of Co Loading
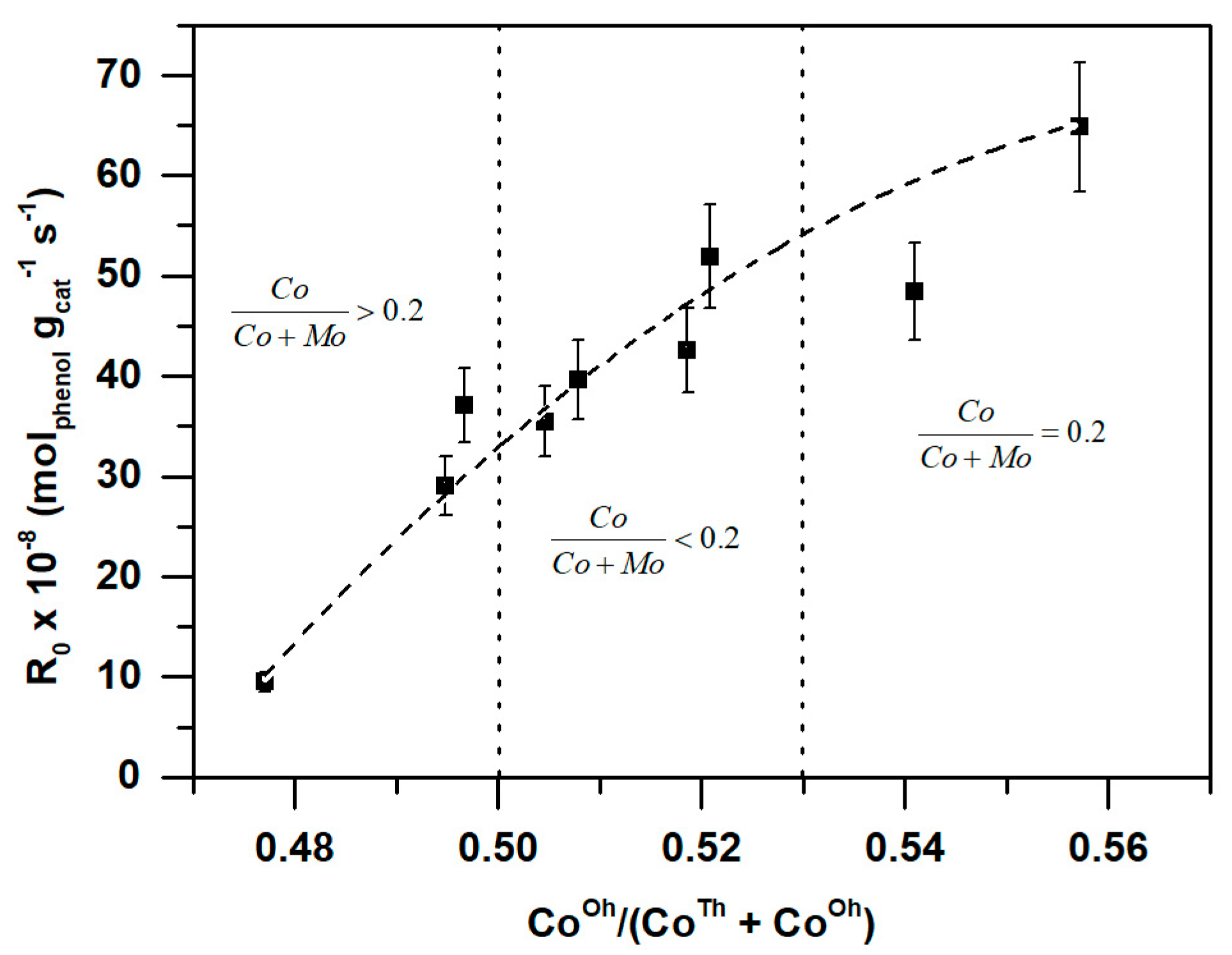
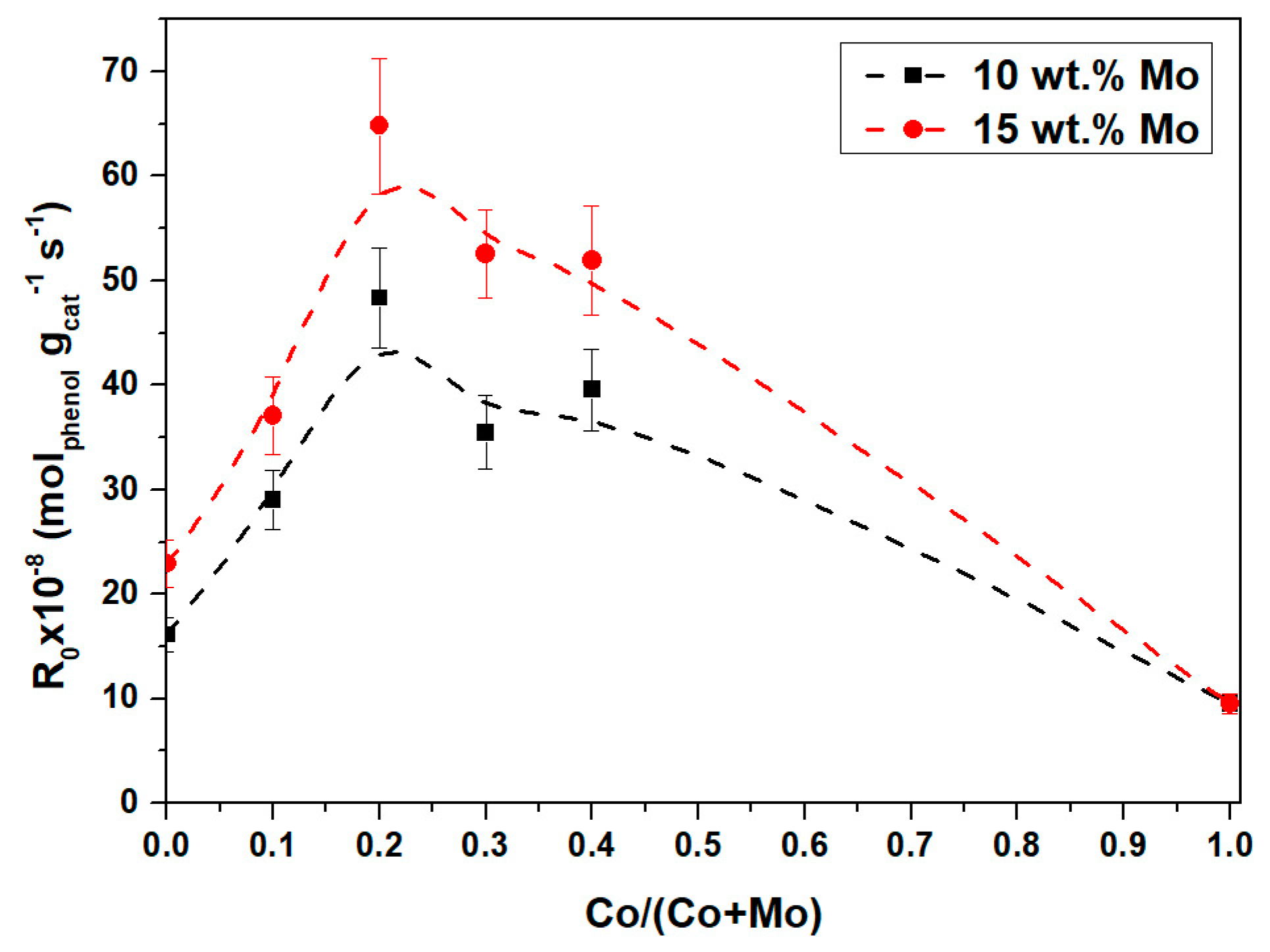
2.2.1. Catalytic Activity
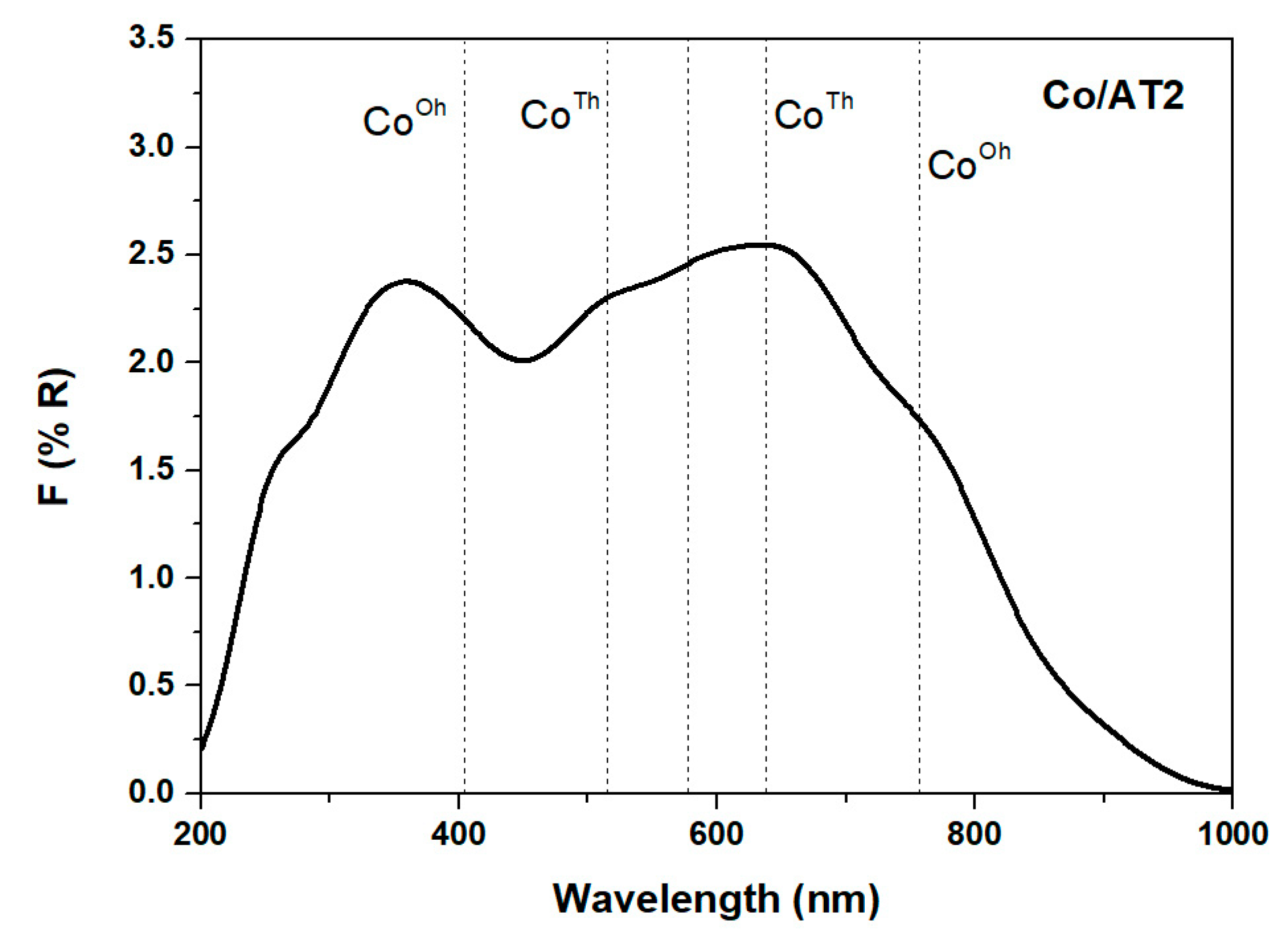
2.2.2. Diffuse Reflectance Spectroscopy UV-Vis
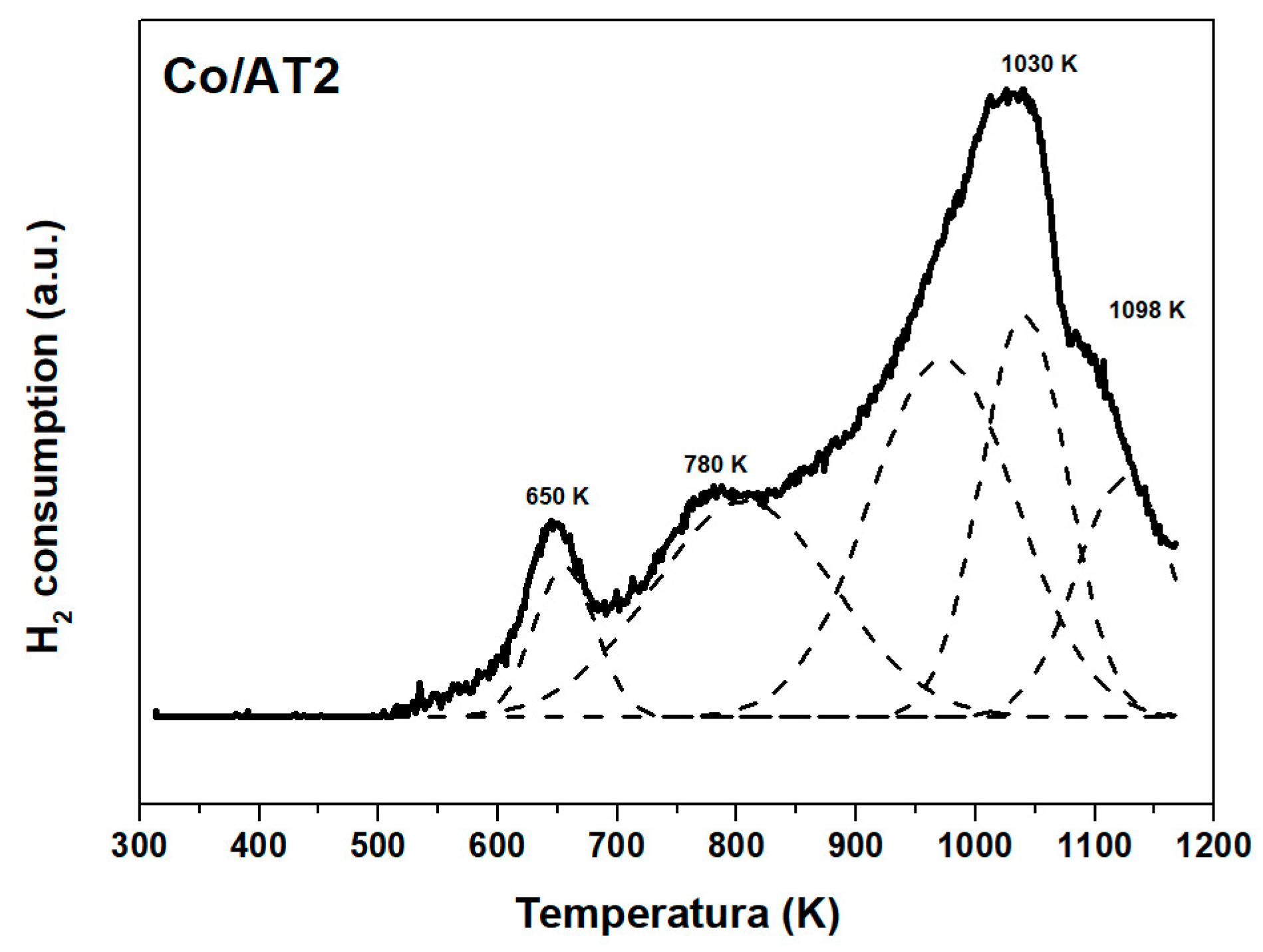
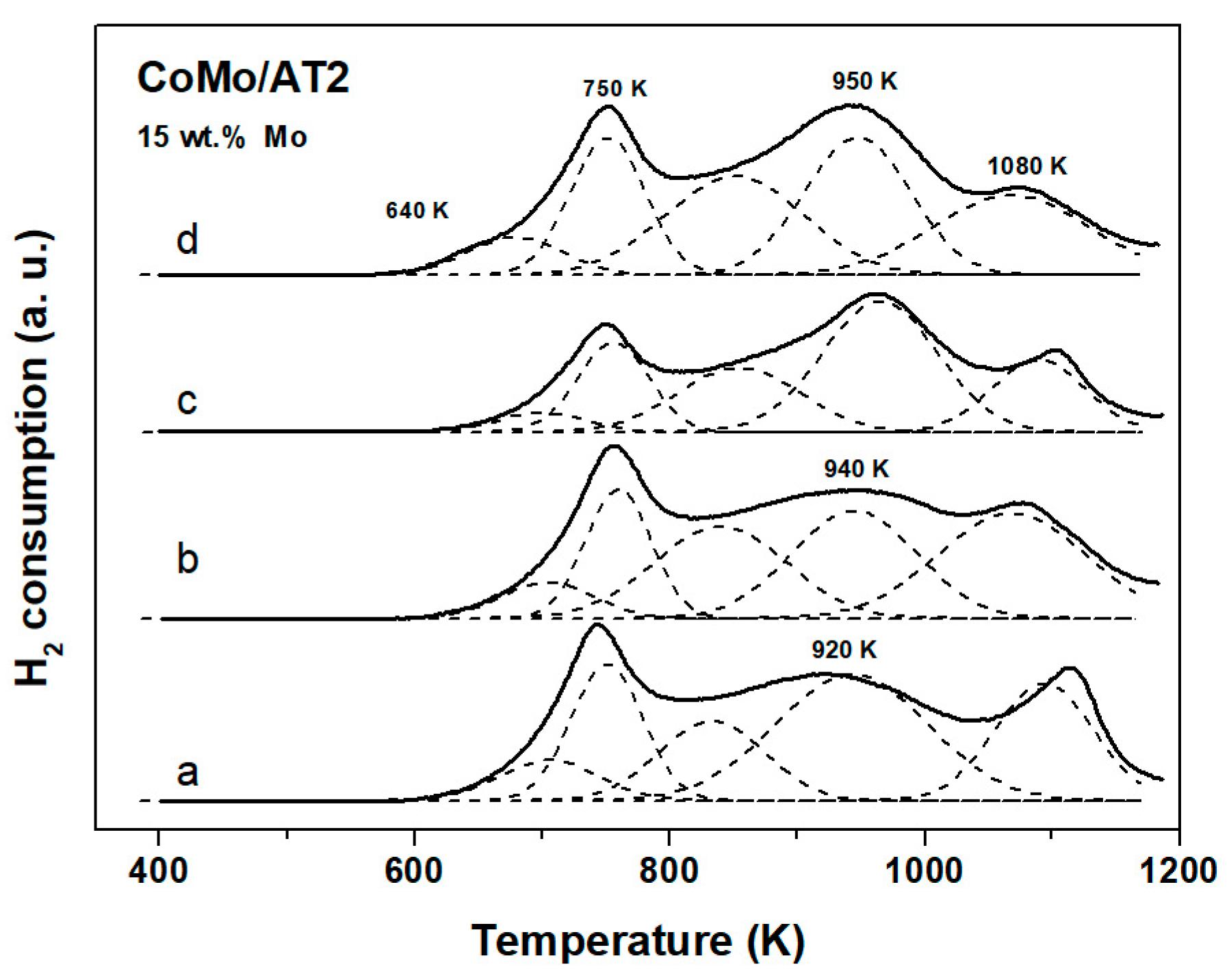
2.2.3. Temperature Programmed Reduction
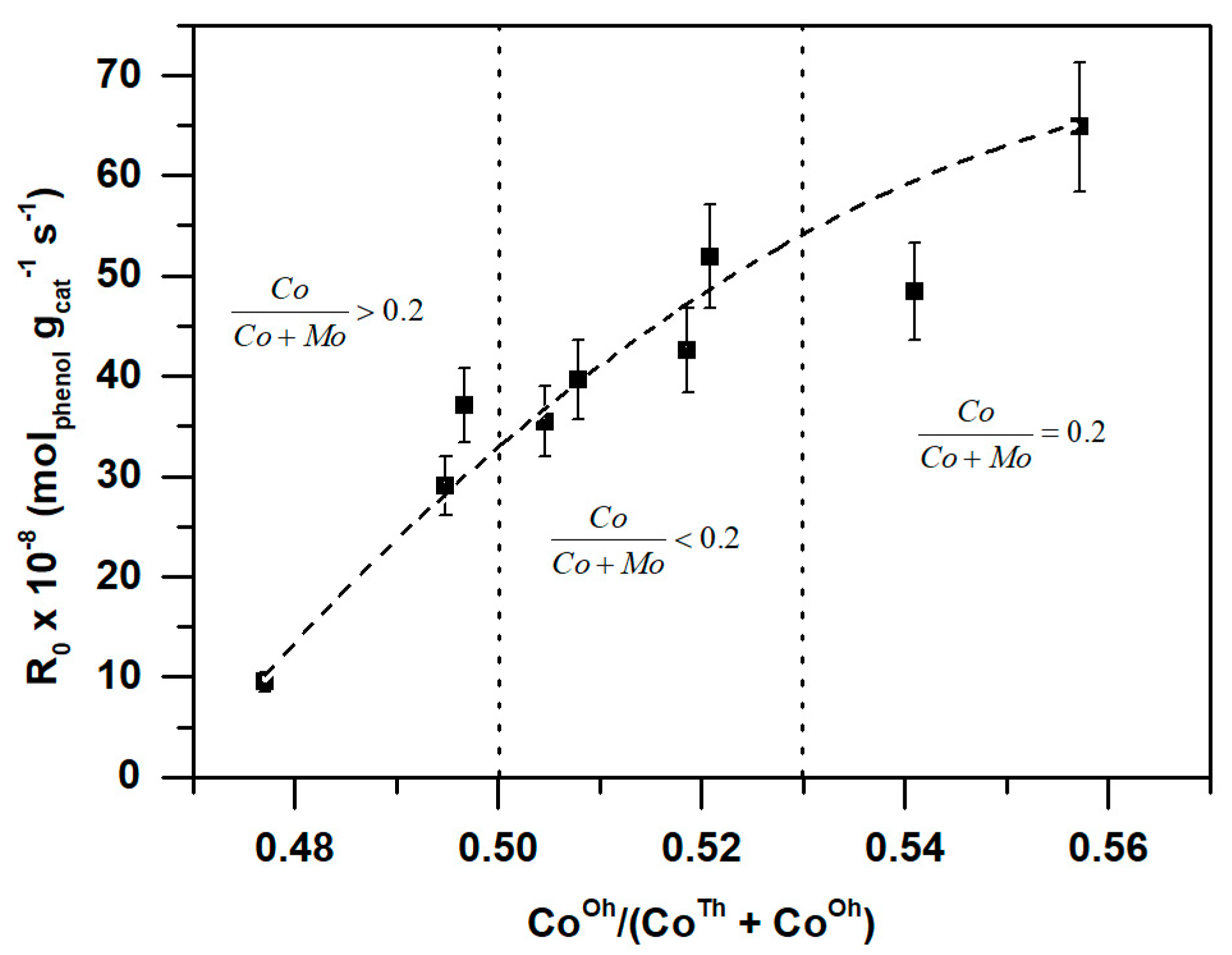
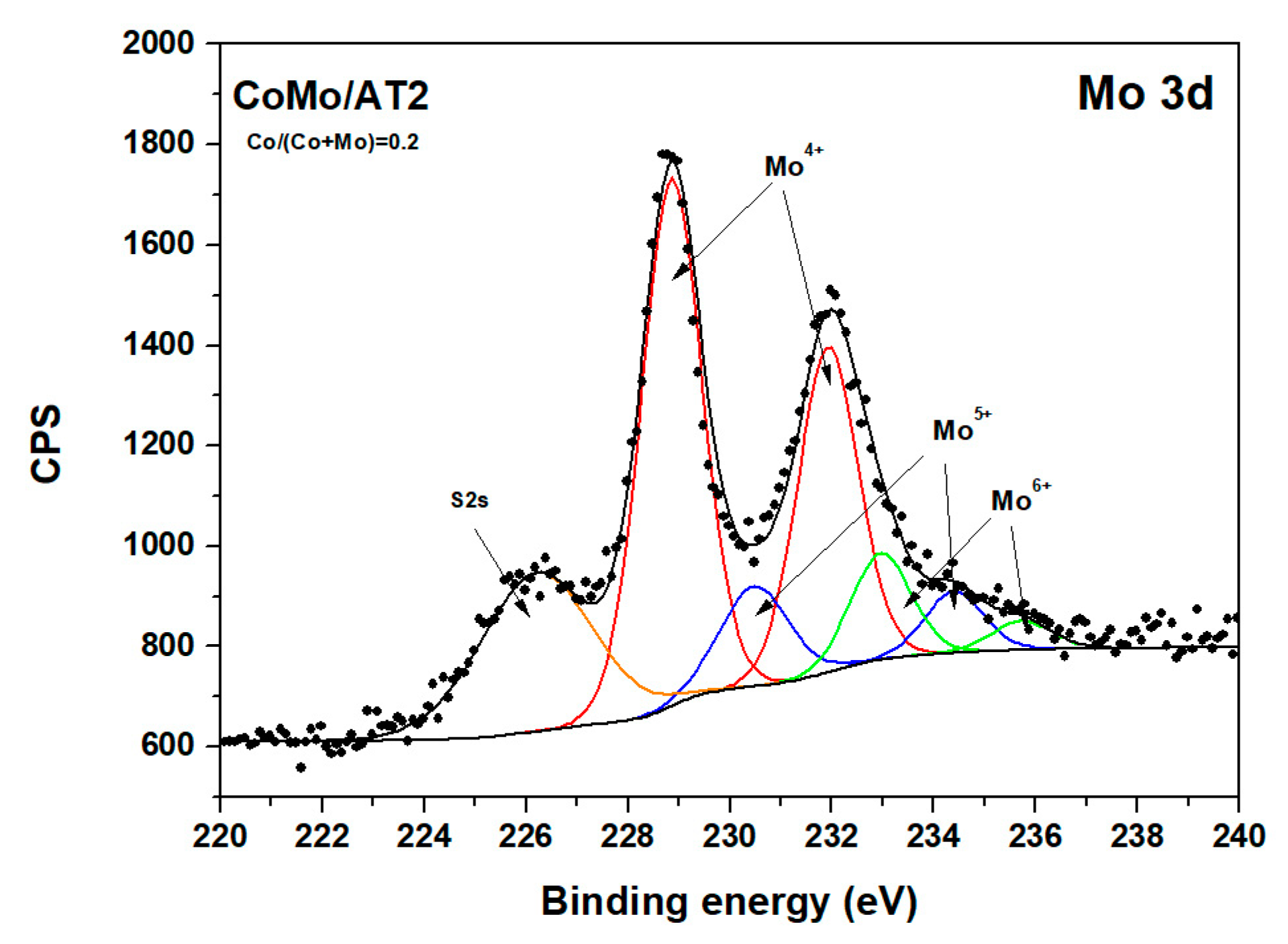
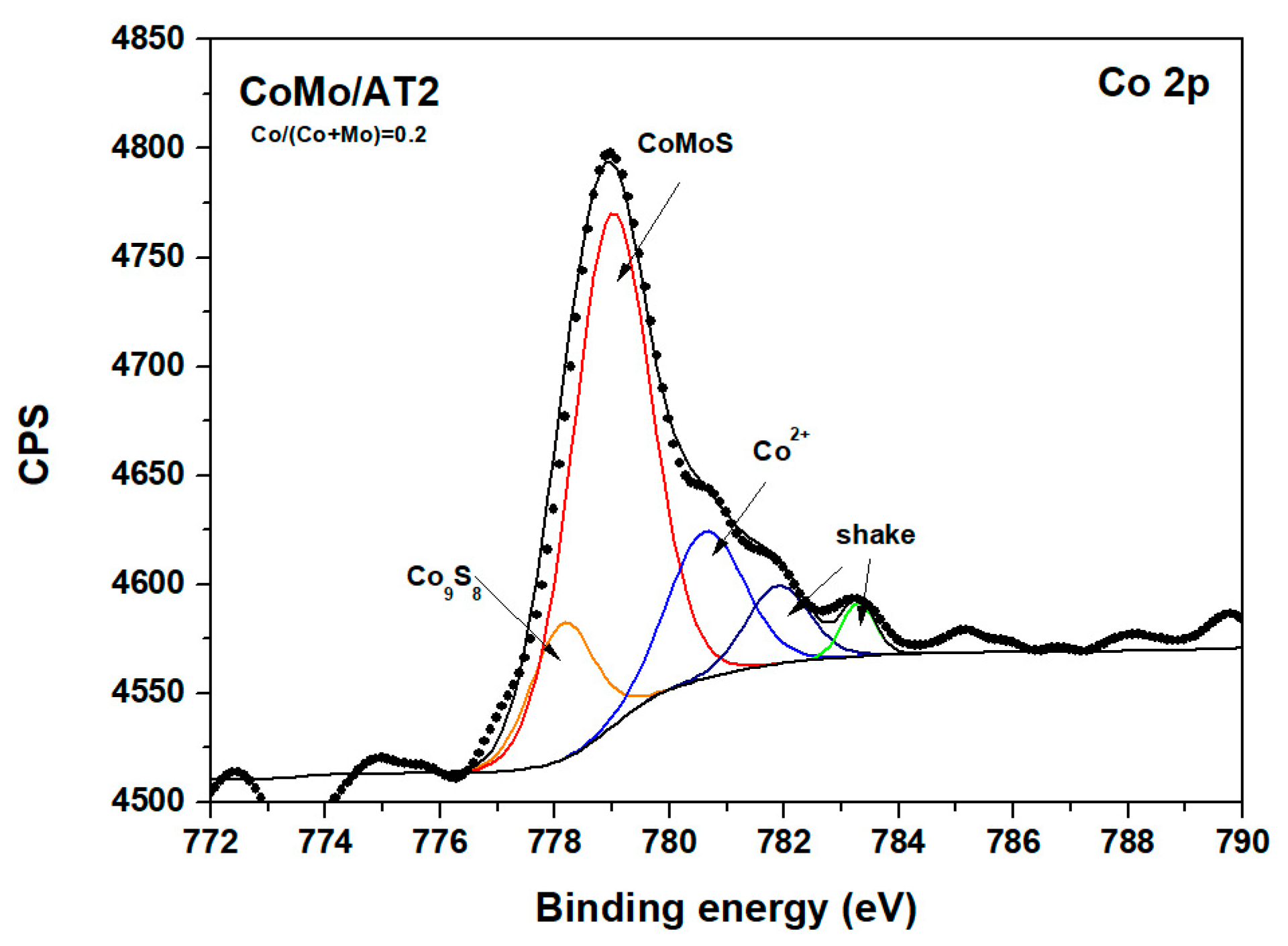
2.2.4. X-Ray Photoelectron Spectroscopy
3. Materials and Methods
3.1. Support and Catalysts Synthesis
3.1.1. Support Synthesis
3.1.2. Catalyst Synthesis
3.2. Materials Characterization
3.2.1. Diffuse Reflectance Ultraviolet-Visible Spectroscopy
3.2.2. Laser Raman Spectroscopy
3.2.3. Temperature Programmed Reduction
3.2.4. X-Ray Photoelectron Spectroscopy (XPS)
3.2.5. Catalytic Performance
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pourzolfaghar, H.; Abnisa, F.; Wan Daud, W.M.A.; Aroua, M.K. Atmospheric Hydrodeoxygenation of Bio-Oil Oxygenated Model Compounds: A Review. J. Anal. Appl. Pyrolysis 2018, 133, 117–127. [Google Scholar] [CrossRef]
- Li, X.; Chen, G.; Liu, C.; Ma, W.; Yan, B.; Zhang, J. Hydrodeoxygenation of Lignin-Derived Bio-Oil Using Molecular Sieves Supported Metal Catalysts: A Critical Review. Renew. Sustain. Energy Rev. 2017, 71, 296–308. [Google Scholar] [CrossRef]
- Dabros, T.M.H.; Stummann, M.Z.; Høj, M.; Jensen, P.A.; Grunwaldt, J.-D.; Gabrielsen, J.; Mortensen, P.M.; Jensen, A.D. Transportation Fuels from Biomass Fast Pyrolysis, Catalytic Hydrodeoxygenation, and Catalytic Fast Hydropyrolysis. Prog. Energy Combust. Sci. 2018, 68, 268–309. [Google Scholar] [CrossRef]
- Linck, M.; Felix, L.; Marker, T.; Roberts, M. Integrated Biomass Hydropyrolysis and Hydrotreating: A Brief Review. Wiley Interdiscip. Rev. Energy Environ. 2014, 3, 575–581. [Google Scholar] [CrossRef]
- Furimsky, E. Hydroprocessing Challenges in Biofuels Production. Catal. Today 2013, 217, 13–56. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass Chemistry Catalysts and Enginneering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, J.; Liu, B.; Liu, J.; Wang, C.; Chen, G. Hydrodeoxygenation of Lignin-Derived Phenols to Produce Hydrocarbons over Ni/Al-SBA-15 Prepared with Different Impregnants. Fuel 2019, 243, 314–321. [Google Scholar] [CrossRef]
- Jahromi, H.; Agblevor, F. Hydrogenation of Aqueous-Phase Catalytic Pyrolysis Oil to Liquid Hydrocarbons Using Multifunctional Nickel Catalyst. Ind. Eng. Chem. Res. 2018, 57, 13257–13268. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Delichère, P.; Geantet, C. Hydrodeoxygenation of Guaiacol: Part II: Support Effect for CoMoS Catalysts on HDO Activity and Selectivity. Appl. Catal. B Environ. 2011, 101, 246–255. [Google Scholar] [CrossRef]
- Şenol, O.İ.; Ryymin, E.-M.; Viljava, T.-R.; Krause, A.O.I. Effect of Hydrogen Sulphide on the Hydrodeoxygenation of Aromatic and Aliphatic Oxygenates on Sulphided Catalysts. J. Mol. Catal. A Chem. 2007, 277, 107–112. [Google Scholar] [CrossRef]
- Bu, Q.; Lei, H.; Zacher, A.H.; Wang, L.; Ren, S.; Liang, J.; Wei, Y.; Liu, Y.; Tang, J.; Zhang, Q.; et al. A Review of Catalytic Hydrodeoxygenation of Lignin-Derived Phenols from Biomass Pyrolysis. Bioresour. Technol. 2012, 124, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Echeandia, S.; Arias, P.L.; Barrio, V.L.; Pawelec, B.; Fierro, J.L.G. Synergy Effect in the HDO of Phenol over Ni–W Catalysts Supported on Active Carbon: Effect of Tungsten Precursors. Appl. Catal. B Environ. 2010, 101, 1–12. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Afanasiev, P.; Geantet, C. Hydrodeoxygenation of Guaiacol with CoMo Catalysts. Part I: Promoting Effect of Cobalt on HDO Selectivity and Activity. Appl. Catal. B Environ. 2011, 101, 239–245. [Google Scholar] [CrossRef]
- Honkela, M.L.; Viljava, T.-R.; Gutierrez, A.; Krause, A.O.I. Chapter 11 Hydrotreating for Bio-Oil Upgrading. In Thermochemical Conversion of Biomass to Liquid Fuels and Chemicals; Crocker, M., Ed.; The Royal Society of Chemistry: London, UK, 2010; pp. 288–306. [Google Scholar]
- Zhang, J.; Dong, K.; Luo, W. PdCl2-Catalyzed Hydrodeoxygenation of 5-Hydroxymethylfurfural into 2,5-Dimethylfuran at Room-Temperature using Polymethylhydrosiloxane as the Hydrogen Donor. Chem. Eng. Sci. 2019, 201, 467–474. [Google Scholar] [CrossRef]
- Viljava, T.-R.; Komulainen, S.; Selvam, T.; Krause, A.O.I. Stability of CoMo/Al2O3 Catalysts: Effect of HDO Cycles on HDS. In Studies in Surface Science and Catalysis: Hydrotreatment and Hydrocracking of Oil Fractions Proceedings of the 2nd International Symposium/7th European Workshop; Delmon, B., Froment, G.F., Grange, P., Eds.; Elsevier: Antwerpen, Belgium, 1999; Volume 127, pp. 145–152. [Google Scholar] [CrossRef]
- Massoth, F.E.; Politzer, P.; Concha, M.C.; Murray, J.S.; Jakowski, J.; Simons, J. Catalytic Hydrodeoxygenation of Methyl-Substituted Phenols: Correlations of Kinetic Parameters with Molecular Properties. J. Phys. Chem. B 2006, 110, 14283–14291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawi, M.; Paul, J.F.; Cristol, S.; Payen, E.; Romero, Y.; Richard, F.; Brunet, S.; Lambert, D.; Portier, X.; Popov, A.; et al. Effect of Water on the Stability of Mo and CoMo Hydrodeoxygenation Catalysts: A Combined Experimental and DFT Study. J. Catal. 2011, 282, 155–164. [Google Scholar] [CrossRef]
- Wildschut, J.; Mahfud, F.H.; Venderbosch, R.H.; Heeres, H.J. Hydrotreatment of Fast Pyrolysis Oil Using Heterogeneous Noble-Metal Catalysts. Ind. Eng. Chem. Res. 2009, 48, 10324–10334. [Google Scholar] [CrossRef]
- Lauritsen, J.V.; Besenbacher, F. Atom-Resolved Scanning Tunneling Microscopy Investigations of Molecular Adsorption on MoS2 and CoMoS Hydrodesulfurization Catalysts. J. Catal. 2015, 328, 49–58. [Google Scholar] [CrossRef]
- Topsøe, H.; Clausen, B.S.; Massoth, F.E. Hydrotreating Catalysis BT—Catalysis: Science and Technology; Anderson, J.R., Boudart, M., Eds.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 1996; 269p. [Google Scholar]
- Lauritsen, J.V.; Nyberg, M.; Nørskov, J.K.; Clausen, B.S.; Topsøe, H.; Lægsgaard, E.; Besenbacher, F. Hydrodesulfurization Reaction Pathways on MoS2 Nanoclusters Revealed by Scanning Tunneling Microscopy. J. Catal. 2004, 224, 94–106. [Google Scholar] [CrossRef]
- Zhu, Y.; Ramasse, Q.M.; Brorson, M.; Moses, P.G.; Hansen, L.P.; Topsøe, H.; Kisielowski, C.F.; Helveg, S. Location of Co and Ni Promoter Atoms in Multi-Layer MoS2 Nanocrystals for Hydrotreating Catalysis. Catal. Today 2016, 261, 75–81. [Google Scholar] [CrossRef]
- Liu, B.; Liu, L.; Chai, Y.; Zhao, J.; Liu, C. Essential Role of Promoter Co on the MoS2 Catalyst in Selective Hydrodesulfurization of FCC Gasoline. J. Fuel Chem. Technol. 2018, 46, 441–450. [Google Scholar] [CrossRef]
- Kim, D.S.; Segawa, K.; Soeya, T.; Wachs, I.E. Surface Structures of Supported Molybdenum Oxide Catalysts under Ambient Conditions. J. Catal. 1992, 136, 539–553. [Google Scholar] [CrossRef]
- Ng, K.Y.S.; Gulari, E. Molybdena on Titania: I. Preparation and Characterization by Raman and Fourier Transform Infrared Spectroscopy. J. Catal. 1985, 92, 340–354. [Google Scholar] [CrossRef]
- Cáceres, C.V.; Fierro, J.L.G.; Lázaro, J.; López Agudo, A.; Soria, J. Effect of Support on the Surface Characteristics of Supported Molybdena Catalysts. J. Catal. 1990, 122, 113–125. [Google Scholar] [CrossRef]
- Breysse, M.; Afanasiev, P.; Geantet, C.; Vrinat, M. Overview of Support Effects in Hydrotreating Catalysts. Catal. Today 2003, 86, 5–16. [Google Scholar] [CrossRef]
- Gutiérrez-Alejandre, A.; Ramírez, J.; Val, I.J.; Peñuelas-Galaz, M.; Sánchez-Neri, P.; Torres-Mancera, P. Activity of NiW Catalysts Supported on TiO2-Al2O3 Mixed Oxides: Effect of Ti Incorporation Method on the HDS of 4,6-DMDBT. Catal. Today 2005, 107–108, 879–884. [Google Scholar] [CrossRef]
- Escobar, J.; Antonio De Los Reyes, J.; Viveros, T. Nickel on TiO2-Modified Al2O3 Sol–Gel Oxides: Effect of Synthesis Parameters on the Supported Phase Properties. Appl. Catal. A Gen. 2003, 253, 151–163. [Google Scholar] [CrossRef]
- Núñez, S.; Escobar, J.; Vázquez, A.; de los Reyes, J.A.; Hernández-Barrera, M. 4,6-Dimethyl-Dibenzothiophene Conversion over Al2O3–TiO2-Supported Noble Metal Catalysts. Mater. Chem. Phys. 2011, 126, 237–247. [Google Scholar] [CrossRef]
- Tavizón-Pozos, J.A.; Suárez-Toriello, V.A.; de los Reyes, J.A.; Guevara-Lara, A.; Pawelec, B.; Fierro, J.L.G.; Vrinat, M.; Geantet, C. Deep Hydrodesulfurization of Dibenzothiophenes Over NiW Sulfide Catalysts Supported on Sol–Gel Titania–Alumina. Top. Catal. 2016, 59, 241–251. [Google Scholar] [CrossRef]
- Suárez-Toriello, V.A.; Santolalla-Vargas, C.E.; de los Reyes, J.A.; Vázquez-Zavala, A.; Vrinat, M.; Geantet, C. Influence of the Solution PH in Impregnation with Citric Acid and Activity of Ni/W/Al2O3 Catalysts. J. Mol. Catal. A Chem. 2015, 404–405, 36–46. [Google Scholar] [CrossRef]
- Tavizón-Pozos, J.A.; Suárez-Toriello, V.A.; del Ángel, P.; de los Reyes, J.A. Hydrodeoxygenation of Phenol Over Sulfided CoMo Catalysts Supported on a Mixed Al2O3-TiO2 Oxide. Int. J. Chem. React. Eng. 2016, 14, 1211–1223. [Google Scholar] [CrossRef]
- Escobar, J.; Núñez, S.; Montesinos-Castellanos, A.; de los Reyes, J.A.; Rodríguez, Y.; González, O.A. Dibenzothiophene Hydrodesulfurization over PdPt/Al2O3–TiO2. Influence of Ti-Addition on Hydrogenating Properties. Mater. Chem. Phys. 2016, 171, 185–194. [Google Scholar] [CrossRef]
- Olguin, E.; Vrinat, M.; Cedeño, L.; Ramirez, J.; Borque, M.; López-Agudo, A. The Use of TiO2—Al2O3 Binary Oxides as Supports for Mo-Based Catalysts in Hydrodesulfurization of Thiophene and Dibenzothiophene. Appl. Catal. A Gen. 1997, 165, 1–13. [Google Scholar] [CrossRef]
- Ramírez, J.; Macías, G.; Cedeño, L.; Gutiérrez-Alejandre, A.; Cuevas, R.; Castillo, P. The Role of Titania in Supported Mo, CoMo, NiMo, and NiW Hydrodesulfurization Catalysts: Analysis of Past and New Evidences. Catal. Today 2004, 98, 19–30. [Google Scholar] [CrossRef]
- Chen, W.; Long, X.; Li, M.; Nie, H.; Li, D. Influence of Active Phase Structure of CoMo/Al2O3 Catalyst on the Selectivity of Hydrodesulfurization and Hydrodearomatization. Catal. Today 2017, 292, 97–109. [Google Scholar] [CrossRef]
- Wang, L.; Hall, W.K. On the Genesis of Molybdena-Alumina Catalyst. J. Catal. 1980, 66, 251–255. [Google Scholar] [CrossRef]
- Kim, D.S.; Kurusu, Y.; Wachs, I.E.; Hardcastle, F.D.; Segawa, K. Physicochemical Properties of MoO3 TiO2 Prepared by an Equilibrium Adsorption Method. J. Catal. 1989, 120, 325–336. [Google Scholar] [CrossRef]
- Afanasiev, P.; Geantet, C.; Breysse, M. Preparation of High-Surface-Area Mo/ZrO2 Catalysts by a Molten Salt Method: Application to Hydrodesulfurization. J. Catal. 1995, 153, 17–24. [Google Scholar] [CrossRef]
- Williams, C.C.; Ekerdt, J.G.; Jehng, J.M.; Hardcastle, F.D.; Wachs, I.E. A Raman and Ultraviolet Diffuse Reflectance Spectroscopic Investigation of Alumina-Supported Molybdenum Oxide. J. Phys. Chem. 1991, 95, 8791–8797. [Google Scholar] [CrossRef]
- Xiong, G.; Li, C.; Feng, Z.; Ying, P.; Xin, Q.; Liu, J. Surface Coordination Structure of Molybdate with Extremely Low Loading on γ-Alumina Characterized by UV Resonance Raman Spectroscopy. J. Catal. 1999, 186, 234–237. [Google Scholar] [CrossRef]
- Stencel, J.M. Raman Spectroscopy for Catalysis, 1st ed.; Davis, B., Ed.; Van Nostrand Reinhold: New York, NY, USA, 1990. [Google Scholar]
- Lozano-Hernández, G.; Lozada-Ascencio, E.M.; Guevara-Lara, A. Support Composition Effect on Superficial Structures of Nickel and Molybdenum Oxides Supported on TiO2-Al2O3 Mixed Oxides. Rev. Mex. Ing. Química 2006, 5, 311–320. [Google Scholar]
- Aveston, J.; Anacker, E.W.; Johnson, J.S. Hydrolysis of Molybdenum(VI). Ultracentrifugation, Acidity Measurements, and Raman Spectra of Polymolybdates. Inorg. Chem. 1964, 3, 735–746. [Google Scholar] [CrossRef]
- Platanitis, P.; Panagiotou, G.D.; Bourikas, K.; Kordulis, C.; Fierro, J.L.G.; Lycourghiotis, A. Preparation of Un-Promoted Molybdenum HDS Catalysts Supported on Titania by Equilibrium Deposition Filtration: Optimization of the Preparative Parameters and Investigation of the Promoting Action of Titania. J. Mol. Catal. A Chem. 2016, 412, 1–12. [Google Scholar] [CrossRef]
- Vakros, J.; Lycourghiotis, A.; Voyiatzis, G.A.; Siokou, A.; Kordulis, C. CoMo/Al2O3-SiO2 Catalysts Prepared by Co-Equilibrium Deposition Filtration: Characterization and Catalytic Behavior for the Hydrodesulphurization of Thiophene. Appl. Catal. B Environ. 2010, 96, 496–507. [Google Scholar] [CrossRef]
- Malaibari, Z.O.; Croiset, E.; Amin, A.; Epling, W. Effect of Interactions between Ni and Mo on Catalytic Properties of a Bimetallic Ni-Mo/Al2O3 Propane Reforming Catalyst. Appl. Catal. A Gen. 2015, 490, 80–92. [Google Scholar] [CrossRef]
- Arnoldy, P.; Franken, M.C.; Scheffer, B.; Moulijn, J.A. Temperature-Programmed Reduction of CoO MoO3Al2O3 Catalysts. J. Catal. 1985, 96, 381–395. [Google Scholar] [CrossRef]
- Bergwerff, J.A.; Visser, T.; Weckhuysen, B.M. On the Interaction between Co- and Mo-Complexes in Impregnation Solutions Used for the Preparation of Al2O3-Supported HDS Catalysts: A Combined Raman/UV–Vis–NIR Spectroscopy Study. Catal. Today 2008, 130, 117–125. [Google Scholar] [CrossRef]
- Okamoto, Y.; Imanaka, T. Interaction Chemistry between Molybdena and Alumina: Infrared Studies of Surface Hydroxyl Groups and Adsorbed Carbon Dioxide on Aluminas Modified with Molybdate, Sulfate, or Fluorine Anions. J. Phys. Chem. 1988, 92, 7102–7112. [Google Scholar] [CrossRef]
- Topsoe, N.Y.; Topsoe, H. FTIR Studies of Mo/Al2O3-Based Catalysts: I. Morphology and Structure of Calcined and Sulfided Catalysts. J. Catal. 1993, 139, 631–640. [Google Scholar] [CrossRef]
- Besenbacher, F.; Brorson, M.; Clausen, B.S.; Helveg, S.; Hinnemann, B.; Kibsgaard, J.; Lauritsen, J.V.; Moses, P.G.; Nørskov, J.K.; Topsøe, H. Recent STM, DFT and HAADF-STEM Studies of Sulfide-Based Hydrotreating Catalysts: Insight into Mechanistic, Structural and Particle Size Effects. Catal. Today 2008, 130, 86–96. [Google Scholar] [CrossRef]
- Lauritsen, J.V.; Kibsgaard, J.; Olesen, G.H.; Moses, P.G.; Hinnemann, B.; Helveg, S.; Nørskov, J.K.; Clausen, B.S.; Topsøe, H.; Lægsgaard, E.; et al. Location and Coordination of Promoter Atoms in Co- and Ni-Promoted MoS2-Based Hydrotreating Catalysts. J. Catal. 2007, 249, 220–233. [Google Scholar] [CrossRef]
- Vrinat, M.; Letourneur, D.; Bacaud, R.; Harlé, V.; Jouguet, B.; Leclercq, C. CoMo/Al2O3 and CoMo/TiO2-Al2O3 Catalysts in Hydrodesulfurization: Relationship between the Promoting Effect of Cobalt and the Nature of the Support. In Studies in Surface Science and Catalysis: Hydrotreatment and Hydrocracking of Oil Fractions Proceedings ofthe 2nd International Symposium/7th European Workshop; Delmon, B., Froment, G.F., Grange, P., Eds.; Elsevier: Antwerpen, Belgium, 1999; Volume 127, pp. 153–160. [Google Scholar]
- Vakros, J.; Papadopoulou, C.; Lycourghiotis, A.; Kordulis, C. Hydrodesulfurization Catalyst Bodies with Various Co and Mo Profiles. Appl. Catal. A Gen. 2011, 399, 211–220. [Google Scholar] [CrossRef]
- Montoya, J.A.; del Angel, P.; Viveros, T. The Effect of Temperature on the Structural and Textural Evolution of Sol-Gel AlO-TiO Mixed Oxides. J. Mater. Chem. 2001, 11, 944–950. [Google Scholar] [CrossRef]
- Herrera, J.E.; Resasco, D.E. Loss of Single-Walled Carbon Nanotubes Selectivity by Disruption of the Co–Mo Interaction in the Catalyst. J. Catal. 2004, 221, 354–364. [Google Scholar] [CrossRef]
- Vakros, J.; Bourikas, K.; Perlepes, S.; Kordulis, C.; Lycourghiotis, A. Adsorption of Cobalt Ions on the “Electrolytic Solution/γ-Alumina” Interface Studied by Diffuse Reflectance Spectroscopy (DRS). Langmuir 2004, 20, 10542–10550. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, C.; Vakros, J.; Matralis, H.K.; Voyiatzis, G.A.; Kordulis, C. Preparation, Characterization, and Catalytic Activity of CoMo/γ-Al2O3 Catalysts Prepared by Equilibrium Deposition Filtration and Conventional Impregnation Techniques. J. Colloid Interface Sci. 2004, 274, 159–166. [Google Scholar] [CrossRef]
- Brito, J.L.; Barbosa, A.L. Effect of Phase Composition of the Oxidic Precursor on the HDS Activity of the Sulfided Molybdates of Fe(II), Co(II), and Ni(II). J. Catal. 1997, 171, 467–475. [Google Scholar] [CrossRef]
- Papadopoulou, C.; Vakros, J.; Matralis, H.K.; Kordulis, C.; Lycourghiotis, A. On the Relationship between the Preparation Method and the Physicochemical and Catalytic Properties of the CoMo/γ-Al2O3 Hydrodesulfurization Catalysts. J. Colloid Interface Sci. 2003, 261, 146–153. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Jia, L.; Wang, W.; Zhang, T.; Han, Y.; Tsubaki, N.; Tan, Y. Effects of Tetrahedral Molybdenum Oxide Species and MoOx Domains on the Selective Oxidation of Dimethyl Ether under Mild Conditions. Catal. Sci. Technol. 2016, 6, 2975–2983. [Google Scholar] [CrossRef]
- Jermwongratanachai, T.; Jacobs, G.; Shafer, W.D.; Pendyala, V.R.R.; Ma, W.; Gnanamani, M.K.; Hopps, S.; Thomas, G.A.; Kitiyanan, B.; Khalid, S.; et al. Fischer–Tropsch Synthesis: TPR and XANES Analysis of the Impact of Simulated Regeneration Cycles on the Reducibility of Co/Alumina Catalysts with Different Promoters (Pt, Ru, Re, Ag, Au, Rh, Ir). Catal. Today 2014, 228, 15–21. [Google Scholar] [CrossRef]
- Jacobs, G.; Ji, Y.; Davis, B.H.; Cronauer, D.; Kropf, A.J.; Marshall, C.L. Fischer–Tropsch Synthesis: Temperature Programmed EXAFS/XANES Investigation of the Influence of Support Type, Cobalt Loading, and Noble Metal Promoter Addition to the Reduction Behavior of Cobalt Oxide Particles. Appl. Catal. A Gen. 2007, 333, 177–191. [Google Scholar] [CrossRef]
- Yamada, M.; Kasahara, S.; Kawahara, K. Interaction between Co Species and Mo Species in C0O-M0O3/AI2O3. In Studies in Surface Science and Catalysis: New Aspects of Spillover Effect in Catalysis; lnui, T., Fujimoto, K., Uchijima, T., Masai, M., Eds.; Elsevier: Kyoto, Japan, 1993; Volume 77, pp. 349–352. [Google Scholar] [CrossRef]
- Guo, C.; Wu, Y.; Wang, X.; Yang, B. Effect of the Support Calcination Temperature on Selective Hydrodesulfurization of TiO2 Nanotubes Supported CoMo Catalysts. J. Energy Chem. 2013, 22, 517–523. [Google Scholar] [CrossRef]
- Gandubert, A.D.; Krebs, E.; Legens, C.; Costa, D.; Guillaume, D.; Raybaud, P. Optimal Promoter Edge Decoration of CoMoS Catalysts: A Combined Theoretical and Experimental Study. Catal. Today 2008, 130, 149–159. [Google Scholar] [CrossRef]
- Ninh, T.K.T.; Laurenti, D.; Leclerc, E.; Vrinat, M. Support Effect for CoMoS and CoNiMoS Hydrodesulfurization Catalysts Prepared by Controlled Method. Appl. Catal. A Gen. 2014, 487, 210–218. [Google Scholar] [CrossRef]
- Laurenti, D.; Phung-Ngoc, B.; Roukoss, C.; Devers, E.; Marchand, K.; Massin, L.; Lemaitre, L.; Legens, C.; Quoineaud, A.-A.; Vrinat, M. Intrinsic Potential of Alumina-Supported CoMo Catalysts in HDS: Comparison between Γc, ΓT, and δ-Alumina. J. Catal. 2013, 297, 165–175. [Google Scholar] [CrossRef]
- La Parola, V.; Dragoi, B.; Ungureanu, A.; Dumitriu, E.; Venezia, A.M. New HDS Catalysts Based on Thiol Functionalized Mesoporous Silica Supports. Appl. Catal. A Gen. 2010, 386, 43–50. [Google Scholar] [CrossRef]
- Brorson, M.; Carlsson, A.; Topsøe, H. The Morphology of MoS2, WS2, Co–Mo–S, Ni–Mo–S and Ni–W–S Nanoclusters in Hydrodesulfurization Catalysts Revealed by HAADF-STEM. Catal. Today 2007, 123, 31–36. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | R0 (×108mol∙(gcat·s)−1) | Product Yields at 15% of Phenol Conversion | |||
|---|---|---|---|---|---|
| Benzene (%) | O-I (%) | Cyclohexene (%) | Cyclohexane (%) | ||
| AT2 | 6.6 ± 0.6 | 3 | 4 | 6 | 2 |
| Co/AT2 | 9.1 ± 0.9 | 3 | 1 | 2 | 9 |
| Co/(Co + Mo) | Synergic Factor |
|---|---|
| 0.1 | 1.6 |
| 0.2 | 2.8 |
| 0.3 | 2.3 |
| 0.4 | 2.2 |
| Co/(Co + Mo) | DDO/HYD |
|---|---|
| 0.1 | 1.75 ± 0.1 |
| 0.2 | 1.60 ± 0.1 |
| 0.3 | 1.36 ± 0.1 |
| 0.4 | 1.27 ± 0.1 |
| Catalysts | Mo4+ | Mo5+ | Mo6+ | |||
|---|---|---|---|---|---|---|
| BE (eV) | % | BE (eV) | % | BE (eV) | % | |
| CoMo/AT2 (0.2) | 228.76 | 69 | 230.40 | 19 | 232.80 | 12 |
| CoMo/AT2 (0.4) | 228.79 | 59 | 230.28 | 30 | 232.50 | 11 |
| Catalyst | Co9S8 | CoMoS | Co2+ | |||
|---|---|---|---|---|---|---|
| BE (eV) | % | BE (eV) | % | BE (eV) | % | |
| CoMo/AT2 (0.2) | 778.16 | 13 | 778.76 | 51 | 780.7 | 36 |
| CoMo/AT2 (0.4) | 778.30 | 19 | 778.80 | 44 | 780.8 | 37 |
| Catalysts | Concentration (gmetal·gcat−1) | Fraction | |||||
|---|---|---|---|---|---|---|---|
| MoS2 | Co9S8 | CoMoS | Co2+ | fCoMos/MoS2 | fCoMoS/Co9S8 | fCoMoS/Co2+ | |
| CoMo/AT2 (0.2) | 0.104 | 0.004 | 0.014 | 0.010 | 0.14 | 3.92 | 1.42 |
| CoMo/AT2 (0.4) | 0.089 | 0.011 | 0.026 | 0.022 | 0.29 | 2.32 | 1.19 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavizón-Pozos, J.A.; Santolalla-Vargas, C.E.; Valdés-Martínez, O.U.; de los Reyes Heredia, J.A. Effect of Metal Loading in Unpromoted and Promoted CoMo/Al2O3–TiO2 Catalysts for the Hydrodeoxygenation of Phenol. Catalysts 2019, 9, 550. https://doi.org/10.3390/catal9060550
Tavizón-Pozos JA, Santolalla-Vargas CE, Valdés-Martínez OU, de los Reyes Heredia JA. Effect of Metal Loading in Unpromoted and Promoted CoMo/Al2O3–TiO2 Catalysts for the Hydrodeoxygenation of Phenol. Catalysts. 2019; 9(6):550. https://doi.org/10.3390/catal9060550
Chicago/Turabian StyleTavizón-Pozos, J. Andrés, Carlos E. Santolalla-Vargas, Omar U. Valdés-Martínez, and José Antonio de los Reyes Heredia. 2019. "Effect of Metal Loading in Unpromoted and Promoted CoMo/Al2O3–TiO2 Catalysts for the Hydrodeoxygenation of Phenol" Catalysts 9, no. 6: 550. https://doi.org/10.3390/catal9060550