Activation, Deactivation and Reversibility Phenomena in Homogeneous Catalysis: A Showcase Based on the Chemistry of Rhodium/Phosphine Catalysts †

 ,
,
Abstract
:1. Introduction


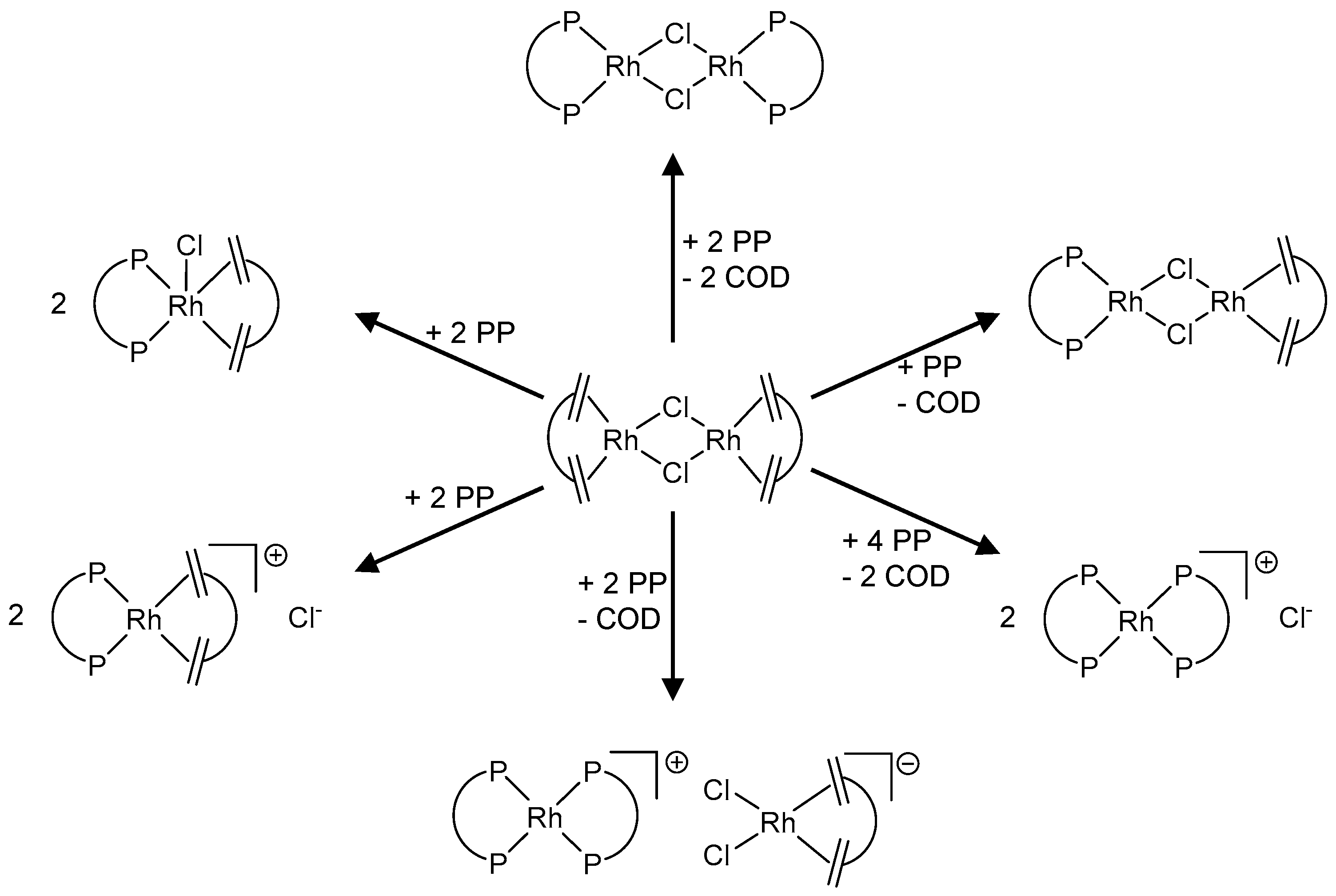
2. In Situ Generation of Precatalysts
2.1. Influence of Reaction Conditions on Outcome of In Situ Synthesis
2.2. Mechanistic Investigations into the in situ Generation of Precatalysts
3. Catalyst Activation—Induction Periods
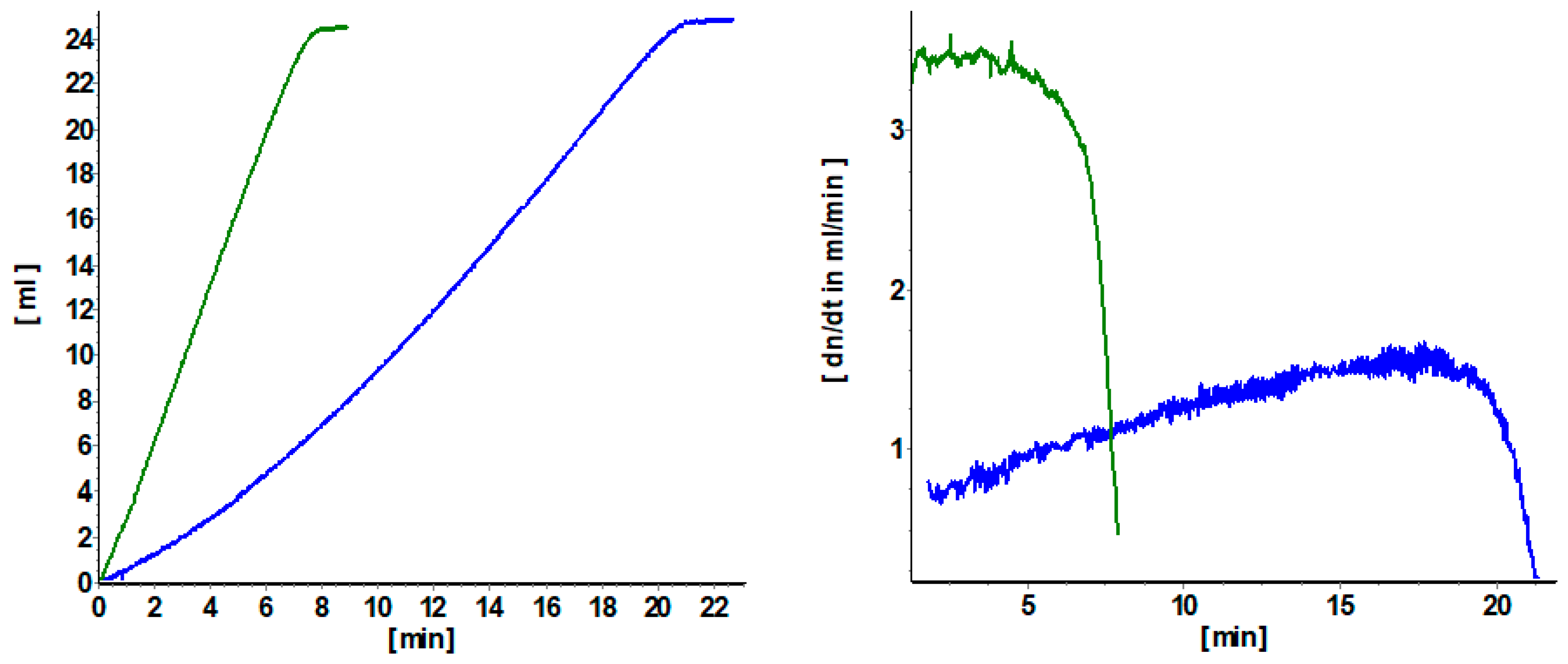
3.1. Quantification of Induction Periods
3.2. Influence of the Diolefin
3.3. Generation of Solvent Complexes
4. Catalysis in the Presence of Strongly Coordinating Ligands
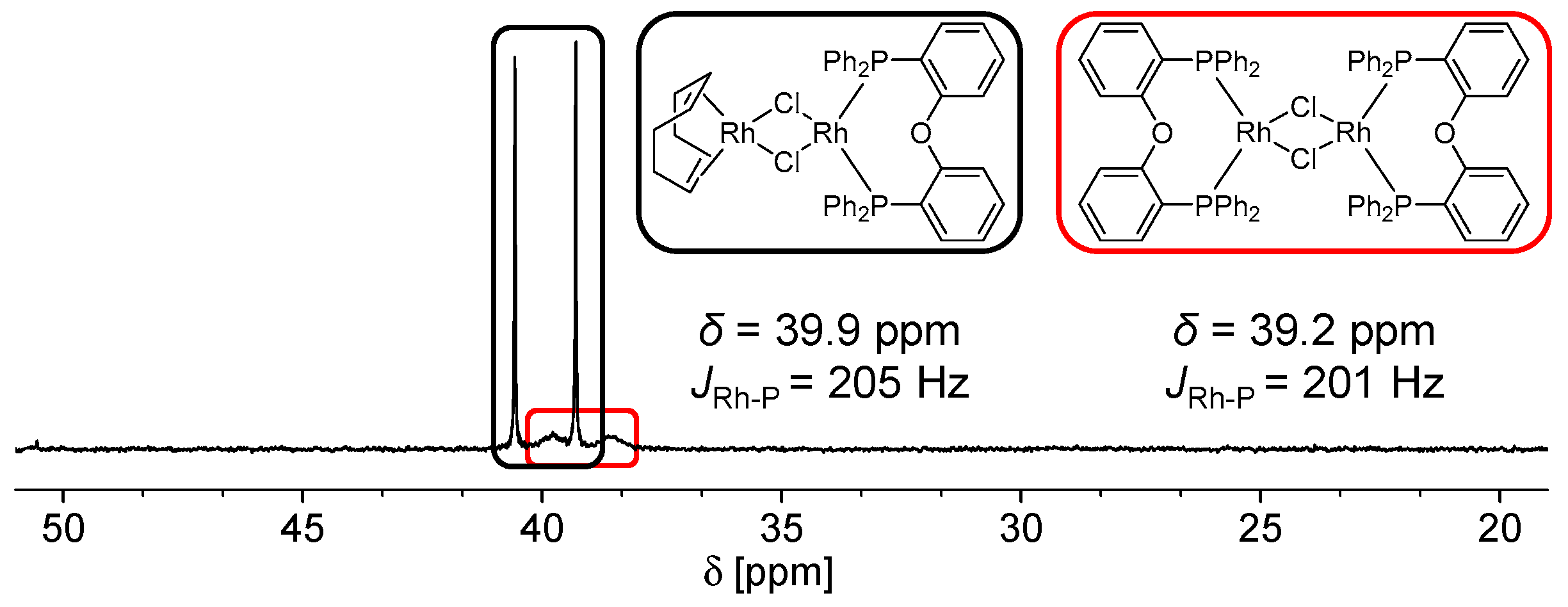
4.1. Formation of Non-Reactive, Monomeric Species
4.2. Formation of Non-Reactive, Multinuclear Species
Dinuclear Species
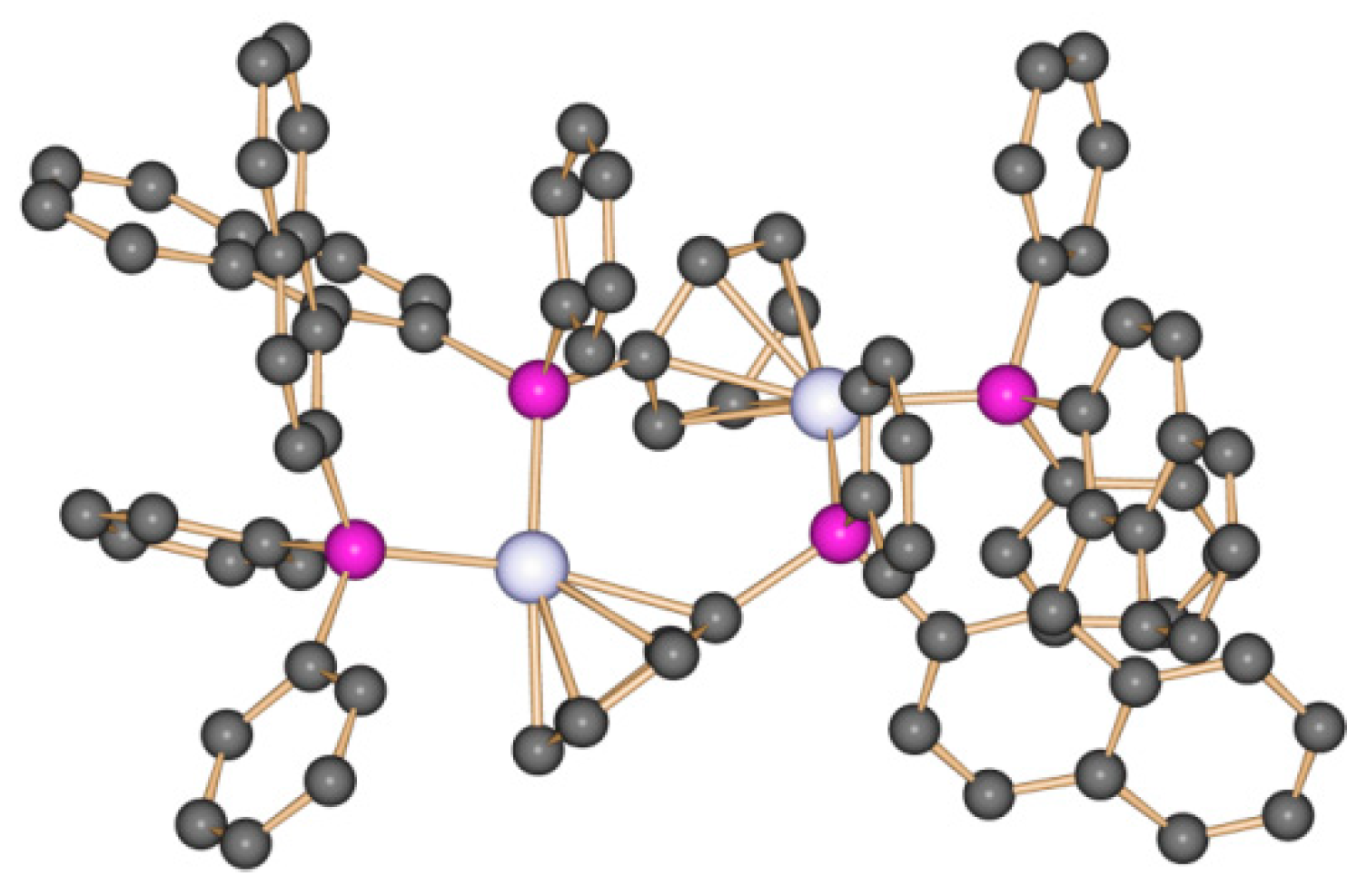
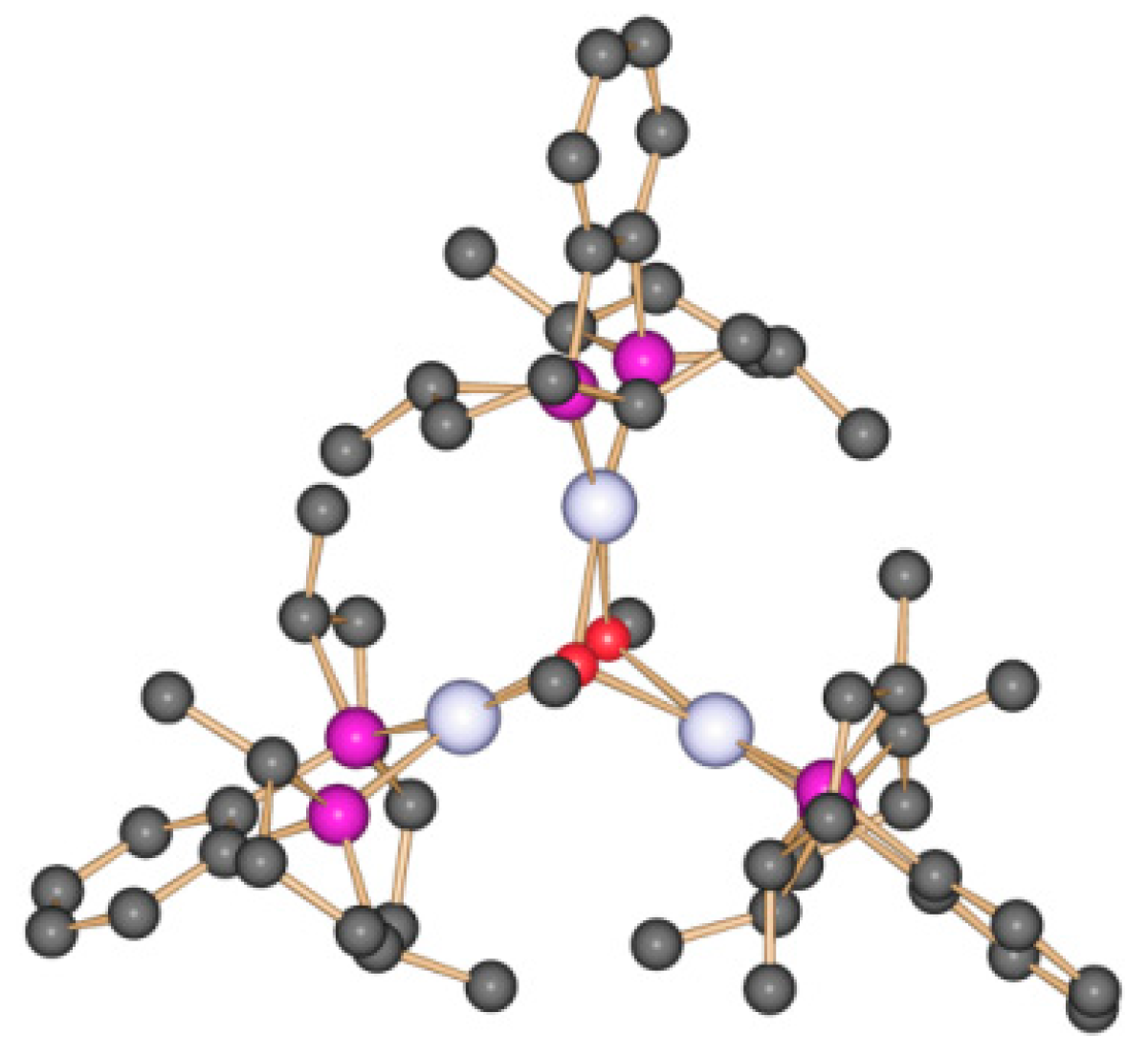
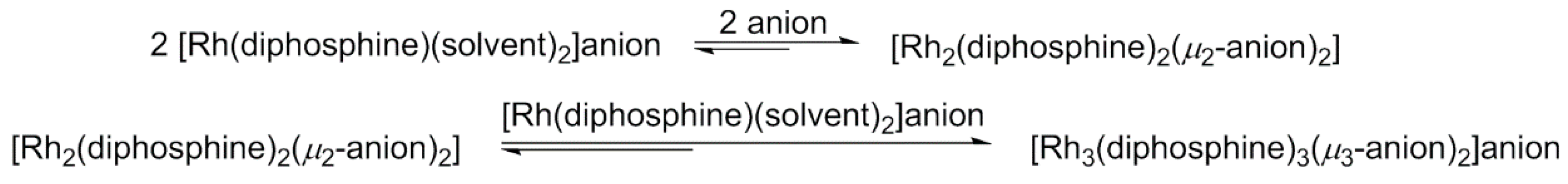
4.3. Trinuclear Complexes
5. Catalyst Deactivation due to Irreversible Reactions of the Active Catalyst
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diphosphine | Solvent | 1st Order Hydrogenation Rate Constant (1/min) | Reference | |
|---|---|---|---|---|
| COD | NBD | |||
| BINAP | 2.3∙10−1 | 26.8 | [101] | |
| THF | 2.8∙10−1 | 20.5 | [101] | |
| Propylene Carbonate | 1.4∙10−1 | 16.6 | [101] | |
| BPPM | 2.2∙10−1 | 1.2 | [101] | |
| CatASium®D(R) | ca. 1.7∙10−3 | n.d. | [108] | |
| CatASium®M(R) | 5.0∙10−2 | 25 | [101] | |
| THF | 1.5∙10−1 | 12 | [100] | |
| Propylene Carbonate | 8.5∙10−2 | 9.4 | [100] | |
| Chiraphos | 1.3∙10−3 | 3.0 | [105] | |
| Cyc-JaPhos | 1.1 | at least 700 | [105] | |
| DaniPhos | ca. 0.03 | 3.6 | [100] | |
| THF | n.d. | 4.8 | [100] | |
| DCPE | 7.5∙10−2 | 48.8 | [105] | |
| Difluorphos | 1.6∙10−1 | 28.6 | [95] | |
| DIOP | 2.3∙10−1 | 1.29 | [104] | |
| DIPAMP | ca. 2.9∙10−3 | ca. 9 | [173] | |
| EtOH | n.d. | 13 | [173] | |
| i-PrOH | n.d. | 5.9 | [173] | |
| THF | n.d. | 5.0 | [173] | |
| Trifluoroethanol | n.d. | 3.9 | [173] | |
| Propylene Carbonate | n.d. | 4.8 | [173] | |
| DPOE | 2.5∙10−1 | 16.6 | [105] | |
| DPPB | 1.6∙10−1 | 1.25 | [104] | |
| DPPE | ca. 2.0∙10−3 | 17.3 | [105] | |
| DPPP | 2.4∙10−2 | 1.55 | [105] | |
| DTBM-SEGPhos | 6.3∙10−2 | 12.0 | [95] | |
| Duanphos | 6.8∙10−1 | 53.7 | [95] | |
| Et-Butiphane | 2.9∙10−2 | n.d. | [91] | |
| Et-DuPhos | 1.2∙10−1 | 52.2 | [101,105] | |
| Et-Ferrotane | 2.0∙10−1 | 19.3 | [95] | |
| H8-BINAP | 8.5∙10−1 | 57.5 | [95] | |
| i-Pr-Butiphane | 1.0∙10−2 | n.d. | [91] | |
| JaPhos | 7.15 | 230 | [105] | |
| Josiphos | 2.3∙10−2 | ca. 34 | [100] | |
| Me-Butiphane | 1.0∙10−1 | n.d. | [91] | |
| Me-DuPhos | 1.1∙10−1 | 35.2 | [101] | |
| THF | 1.6∙10−2 | 39 | [101] | |
| Propylene Carbonate | 1.4∙10−2 | 18 | [101] | |
| Me-α-glup | 3.7∙10−1 | 13.4 | [104] | |
| Ph-β-glup-OH | 2.0∙10−1 | 9.5 | [104] | |
| Prophos | ca. 3.0∙10−3 | 9.2 | [105] | |
| Propraphosderivates | ||||
| R=2-pentyl | 3.77 | 21.9 | [104] | |
| R=3-pentyl | 4.09 | 21.4 | [104] | |
| R=cyclohexyl | 5.44 | 20.2 | [104] | |
| R=cyclopentyl | 2.94 | 18.4 | [104] | |
| R=methyl | 5.3∙10−1 | 8.2 | [104] | |
| SEGPhos | 4.7∙10−1 | 29.9 | [95] | |
| Synphos | 8.0∙10−1 | 67 | [117] | |
| Tangphos | 3.7∙10−1 | 194.4 | [120] | |
| t-Bu-BisP* | 2.1∙10−1 | 90 | [120] | |
| t-Bu-Ferrotane | 5.7∙10−1 | ca. 50 | [95] | |
| 3-Pen-SMS-Phos | n.d. | 3.8 | [23] | |
| Abbreviations | |
|---|---|
| acac | acetylacetonateanion |
| COE | cyclooctene |
| COD | cis,cis-1,5-cyclooctadiene |
| DCM IUPAC | dichloromethane International Union of Pure and Applied Chemistry |
| mac | methyl-(Z)-α -acetamidocinnamate |
| NBD SHOP | bicyclo[2.2.1]hepta-2,5-diene Shell Higher Olefin Process |
| THF | tetrahydrofuran |
| Diphosphines | |
| BINAP | 2,2′-bis(diphenylphosphino)-1,1‘-binaphthyl |
| BPPM | 2,3-bis(diphenylphosphino)-N-phenylmaleimide |
| CatASium® D(R) | N-Benzyl-(3R,4R)-bis(diphenylphosphino)pyrrolidine |
| CatASium® M(R) | 3,4-Bis[(2R,5R)-2,5-dimethyl-1-phospholanyl]furan-2,5-dione |
| Chiraphos | 2,3-bis(diphenylphosphino)butan |
| Cyc-JaPhos | 1-(2-dicyclohexylphosphinophenyl)pyrol-2-dicyclohexylphosphine |
| DaniPhos | dicyclohexyl(1-(2-(diphenylphosphanyl)phenyl)ethyl)phosphane-(tricarbonyl)chrom |
| DCPB | 1,4-bis(dicyclohexylphosphino)butane |
| DCPE | 1,2-bis(dicyclohexylphosphino)ethane |
| Difluorphos | 5,5′-bis(diphenylphosphino)-2,2,2′,2′-tetrafluoro-4,4′-bi-1,3-benzodioxole |
| DIOP | 2,3-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane) |
| DIPAMP | 1,2-bis[(2-methoxyphenyl)phenylphosphino]ethan |
| DM-SEGPhos | 5,5′−bis[di(3,5-xylyl)phosphino]-4,4′-bi-1,3-benzodioxole |
| DPEPhos | bis[(2-diphenylphosphino)phenyl]ether |
| DPOE | 1,2-bis(diphenylphosphinoxy)ethan |
| DPPB | 1,4-bis(diphenylphosphino)butane |
| DPPE | 1,2-bis(diphenylphosphino)ethan |
| DPPF | 1,1′-bis(diphenylphosphino)ferrocene |
| DPPMP | 2-[(diphenylphosphino)methyl]pyridine |
| DPPP | 1,3-bis(diphenylphosphino)propane |
| DTBM-SEGPhos | 5,5′-bis[di(3,5-di-t-butyl-4-methoxyphenyl)phosphino]-4,4′-bi-1,3-benzodioxole |
| Duanphos | (1,1′,2,2′)-2,2′-di-t-butyl-2,3,2′,3′-tetrahydro-1H,1′H(1,1′)biisophos-phindolyl |
| Et-ButiPhane | 2,3-bis(2,5-diethylphospholanyl)benzo[b]thiophene |
| Et-DuPhos | 1,2-bis-2,5-(diethylphospholano)benzene |
| Et-Ferrotane | 1,1′-bis-(2,4-diethylphosphonato)ferrocene |
| H8-BINAP | 2,2′-bis(diphenylphospino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl |
| i-Pr-Butiphane | 2,3-bis(2,5-diisopropylphospholanyl)benzo[b]thiophene |
| JaPhos | 1-(2-dicyclohexylphosphino)butane |
| Josiphos | [2-(diphenylphosphino)ferrocenyl]-ethyldicyclohexylphosphine |
| Me-BPE | 1,2-bis[(2,5)-2,5-dimethylphospholano]ethane |
| Me-Butiphane | 2,3-bis(2,5-dimethylphospholanyl)benzo[b]thiophene |
| Me-DuPhos | 1,2-bis-(2,5-dimethylphospholano)benzene |
| Me-α-glup | methyl-4,6-O-benzylidene-2,3-0-bis(diphenylphosphino)-α-D-glucopyranoside |
| MonoPhos | 3,5-dioxa-4-phosphacyclohepta[2,1-a;3,4-a′]dinaphthalen-4-yl)-dimethylamine |
| Ph-β-glup-OH | Phenyl-2,3-O-bis(diphenylphosphino)-β-D-glucopyranoside |
| PPF-P(t-Bu)2 | 1-[2-(diphenylphosphino)ferrocenyl]ethyl-di-t-butylphosphine |
| Prophos | 1,2-bis(diphenylphosphino)propane |
| SEGPhos | 5,5′-bis(diphenylphosphino)-4,4′-bi-1,3-benzodioxole |
| Synphos | [(5,6),(5′,6′)-bis(ethylenedioxy)biphenyl-2,2′-diyl]bis(diphenylphosphine) |
| Tangphos | 1,1′-di-t-butyl-(2,2′)-diphospholane |
| t-Bu-BisP* | 1,2-bis(t-butylmethylphosphanyl)ethane |
| t-Bu-Ferrotane | 1,1′-Bis-(2,4-diethylphosphonato)ferrocene |
| 3-Pen-SMS-Phos | 1,2-bis[(o-3-pentyl-O-phenyl)(phenyl)phosphino]ethane |
References and Notes
- Berzelius, J.J. Årsberättelse om Framstegen i Fysik och Kemi; Royal Swedish Academy of Sciences, P.A. Norstedt & Söner: Stockholm, Sweden, 1835. [Google Scholar]
- Berzelius, J.J. Quelques idees sur une nouvelle force agissant dans les combinaisons des corps organiques. Ann. Chim. 1836, 61, 146–151. [Google Scholar]
- Ostwald, W. Über den Wärmewert der Bestandteile der Nahrungsmittel. Z. Phys. Chem. 1894, 15, 705–706. [Google Scholar]
- Ostwald, W. Über Katalyse. Ann. Nat. 1910, 9, 1–25. [Google Scholar]
- Steinborn, D. Fundamentals of Organometallic Catalysis; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Wisniak, J. The History of Catalysis. From the Beginning to Nobel Prizes. Educ. Chim. 2010, 21, 60–69. [Google Scholar] [CrossRef]
- Lindström, B.; Pettersson, L.J. A brief history of catalysis. CATTECH 2003, 7, 130–138. [Google Scholar] [CrossRef]
- Laidler, K.J. The Development of Theories of Catalysis. Arch. Hist. Exact Sci. 1986, 35, 345–374. [Google Scholar] [CrossRef]
- Laidler, K.J. A glossary of terms used in chemical kinetics, including reaction dynamics. Pure Appl. Chem. 1996, 68, 149–192. [Google Scholar] [CrossRef]
- Kamer, P.C.J.; Vogt, D.; Thybaut, J. Contemporary Catalysis: Science, Technology, and Applications; Kamer, P.C.J., Vogt, D., Thybaut, J., Eds.; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Hagen, J. Industrial Catalysis: A Practical Approach; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Blaser, H.U. Looking Back on 35 Years of Industrial Catalysis. CHIMIA Int. J. Chem. 2015, 69, 393–406. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Cornils, B.; Herrmann, W.A.; Beller, M.; Paciello, R. (Eds.) Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, 3rd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017. [Google Scholar]
- Cornils, B.; Herrmann, W.A.; Zanthoff, H.; Wong, C.-H. (Eds.) Catalysis from A to Z: A Concise Encyclopedia, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Slaugh, L.H.; Mullineaux, R.D. Novel Hydroformylation Catalysts. J. Organomet. Chem. 1968, 13, 469–477. [Google Scholar]
- Osborn, J.A.; Jardine, F.H.; Young, J.F.; Wilkinson, G. The Preparation and Properties of Tris(triphenylphosphine)halogenorhodium(I) and Some Reactions thereof including Catalytic Homogeneous Hydrogenation of Olefins and Acetylenes and their Derivatives. J. Chem. Soc. A 1966, 1711–1732. [Google Scholar] [CrossRef]
- Scott, S.L. A Matter of Life(time) and Death. ACS Catal. 2018, 8, 8597–8599. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, R.H. Deactivation in Homogeneous Transition Metal Catalysis: Causes, Avoidance, and Cure. Chem. Rev. 2015, 115, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef] [Green Version]
- Van Leeuwen, P.W.N.M.; Chadwick, J.C. Homogeneous Catalysts Activity-Stability-Deactivation; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Heller, D.; de Vries, A.H.M.; de Vries, J.G. Catalyst Inhibition and Deactivation in Homogeneous Hydrogenation. In Handbook of Homogeneous Hydrogenation; de Vries, H.G., Elsevier, C., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; Chapter 44; pp. 1483–1516. [Google Scholar]
- Meissner, A.; Alberico, E.; Drexler, H.-J.; Baumann, W.; Heller, D. Rhodium diphosphine complexes: a case study for catalyst activation and deactivation. Catal. Sci. Technol. 2014, 4, 3409–3425. [Google Scholar] [CrossRef]
- Yuan, S.-W.; Han, H.; Li, Y.-L.; Wu, X.; Bao, X.; Gu, Z.-Y.; Xia, J.-B. Intermolecular C−H Amidation of (Hetero)arenes to Produce Amides through Rhodium-Catalyzed Carbonylation of Nitrene Intermediates. Angew. Chem. Int. Ed. 2019, 131, 8979–8984. [Google Scholar]
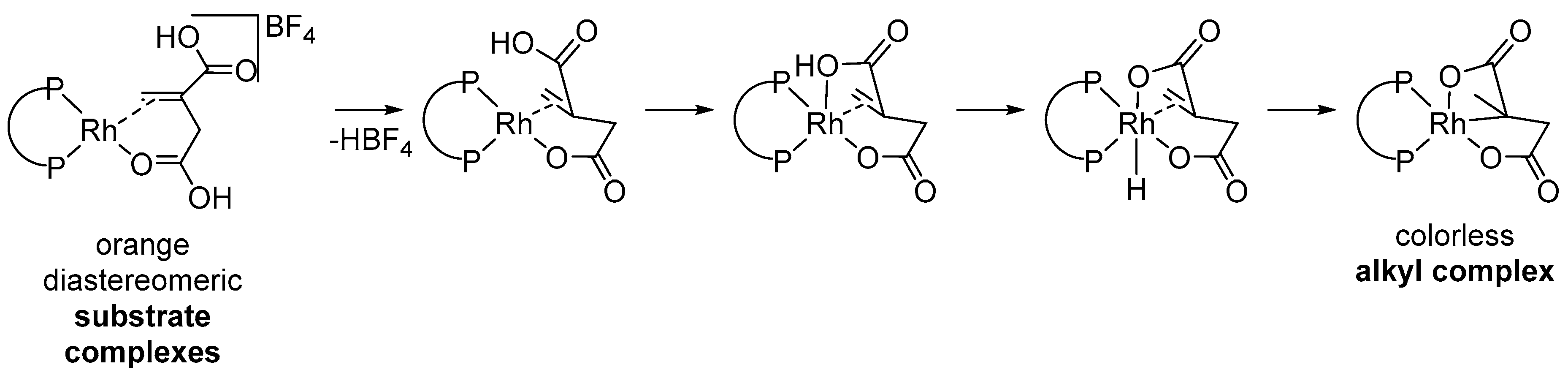
- Oonishi, Y.; Masusaki, S.; Sakamoto, S.; Sato, Y. Rhodium(I)-Catalyzed Enantioselective Cyclization of Enynes by Intramolecular Cleavage of the Rh−C Bond by a Tethered Hydroxy Group. Angew. Chem. Int. Ed. 2019, 131, 8687. [Google Scholar]
- Zheng, J.; Breit, B. Regiodivergent Hydroaminoalkylation of Alkynes and Allenes by a Combined Rhodium and Photoredox Catalytic System. Angew. Chem. Int. Ed. 2019, 131, 3430–3435. [Google Scholar] [CrossRef]
- Ohmura, T.; Sasaki, I.; Suginome, M. Catalytic Generation of Rhodium Silylenoid for Alkene–Alkyne–Silylene [2 + 2 + 1] Cycloaddition. Org. Lett. 2019, 21, 1649–1653. [Google Scholar] [CrossRef]
- Yang, X.-H.; Davison, R.T.; Dong, V.M. Catalytic Hydrothiolation: Regio- and Enantioselective Coupling of Thiols and Dienes. J. Am. Chem. Soc. 2018, 140, 10443–10446. [Google Scholar] [CrossRef]
- Wu, S.T.; Luo, S.Y.; Guo, W.J.; Wang, T.; Xie, Q.X.; Wang, J.H.; Liu, G.Y. Direct Conversion of Ethyl Ketone to Alkyl Ketone via Chelation-Assisted Rhodium(I)-Catalyzed Carbon Carbon Bond Cleavage: Ligands Play an Important Role in the Inhibition of beta-Hydrogen Elimination. Organometallics 2018, 37, 2335–2341. [Google Scholar] [CrossRef]
- Choi, K.; Park, H.; Lee, C. Rhodium-Catalyzed Tandem Addition–Cyclization–Rearrangement of Alkynylhydrazones with Organoboronic Acids. J. Am. Chem. Soc. 2018, 140, 10407–10411. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.F.; Xu, Q.J.; Kang, Q. Rhodium/Lewis Acid Catalyzed Regioselective Addition of 1,3-Dicarbonyl Compounds to Internal Alkynes. Organometallics 2017, 36, 2323–2330. [Google Scholar] [CrossRef]
- Yu, Y.; Xu, M. Chiral phosphorus-olefin ligands for asymmetric catalysis. Huaxue Xuebao 2017, 75, 655–670. [Google Scholar] [CrossRef]
- Wang, H.W.; Lu, Y.; Zhang, B.; He, J.; Xu, H.J.; Kang, Y.S.; Sun, W.Y.; Yu, J.Q. Ligand-Promoted Rhodium(III)-Catalyzed ortho-C-H Amination with Free Amines. Angew. Chem. Int. Ed. 2017, 56, 7449–7453. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Nogi, K.; Yorimitsu, H. Rh/Cu-cocatalyzed Ring-opening Diborylation of Dibenzothiophenes for Aromatic Metamorphosis via Diborylbiaryls. Chem. Lett. 2017, 46, 1131–1134. [Google Scholar] [CrossRef]
- Furusawa, T.; Tanimoto, H.; Nishiyama, Y.; Morimoto, T.; Kakiuchi, K. Rhodium-catalyzed Carbonylative Annulation of 2-Bromobenzylic Alcohols with Internal Alkynes Using Furfural via beta-Aryl Elimination. Chem. Lett. 2017, 46, 926–929. [Google Scholar] [CrossRef]
- Loh, C.C.J.; Schmid, M.; Peters, B.; Fang, X.; Lautens, M. Benzylic Functionalization of Anthrones via the Asymmetric Ring Opening of Oxabicycles Utilizing a Fourth-Generation Rhodium Catalytic System. Angew. Chem. Int. Ed. 2016, 55, 4600–4604. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Hartwig, J.F. Rhodium-Catalyzed Enantioselective Silylation of Cyclopropyl C-H Bonds. Angew. Chem. Int. Ed. 2016, 55, 8723–8727. [Google Scholar] [CrossRef]
- Meissner, A.; Preetz, A.; Drexler, H.-J.; Baumann, W.; Spannenberg, A.; Koenig, A.; Heller, D. In Situ Synthesis of Neutral Dinuclear Rhodium Diphosphine Complexes [{Rh(diphosphine)(μ2-X)}2]: Systematic Investigations. Chem. Plus. Chem. 2015, 80, 169–180. [Google Scholar]
- Meissner, A.; Koenig, A.; Drexler, H.-J.; Thede, R.; Baumann, W.; Heller, D. New Pentacoordinated Rhodium Species as Unexpected Products during the In Situ Generation of Dimeric Diphosphine-Rhodium Neutral Catalysts. Chem. Eur. J. 2014, 20, 14721–14728. [Google Scholar] [CrossRef] [PubMed]
- Preetz, A.; Kohrt, C.; Meissner, A.; Wei, S.; Buschmann, H.; Heller, D. Halide bridged trinuclear rhodium complexes and their inhibiting influence on catalysis. Catal. Sci. Technol. 2013, 3, 462–468. [Google Scholar] [CrossRef]
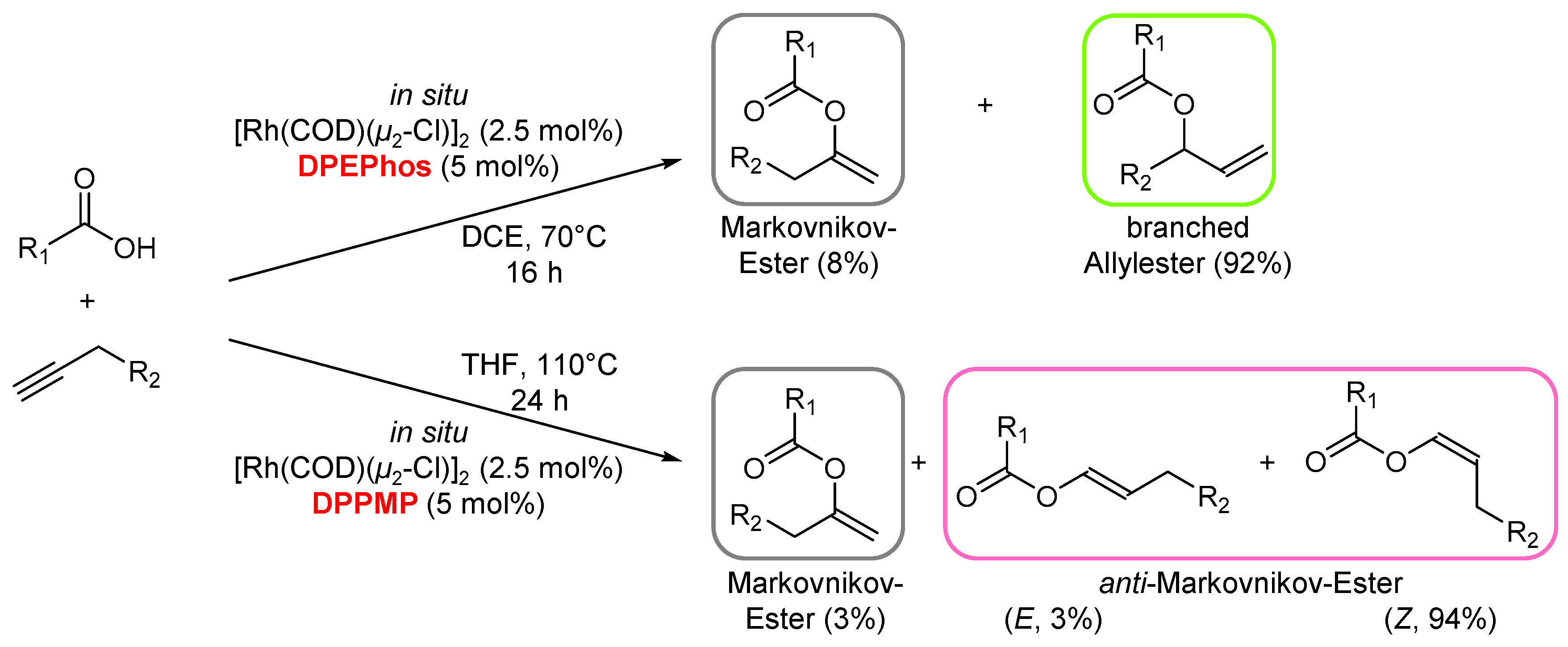
- Lumbroso, A.; Vautravers, N.R.; Breit, B. Rhodium-Catalyzed Selective anti-Markovnikov Addition of Carboxylic Acids to Alkynes. Org. Lett. 2010, 12, 5498–5501. [Google Scholar] [CrossRef] [PubMed]
- Koschker, P.; Lumbroso, A.; Breit, B. Enantioselective Synthesis of Branched Allylic Esters via Rhodium-Catalyzed Coupling of Allenes with Carboxylic Acids. J. Am. Chem. Soc. 2011, 133, 20746–20749. [Google Scholar] [CrossRef] [PubMed]
- Lumbroso, A.; Koschker, P.; Vautravers, N.R.; Breit, B. Redox-Neutral Atom-Economic Rhodium-Catalyzed Coupling of Terminal Alkynes with Carboxylic Acids Toward Branched Allylic Esters. J. Am. Chem. Soc. 2011, 133, 2386–2389. [Google Scholar] [CrossRef] [PubMed]
- Pritzius, A.B.; Breit, B. Asymmetric Rhodium-Catalyzed Addition of Thiols to Allenes: Synthesis of Branched Allylic Thioethers and Sulfones. Angew. Chem. Int. Ed. 2015, 54, 3121–3125. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.M.; Breit, B. Regio- and Enantioselective Rhodium-Catalyzed Addition of 1,3-Diketones to Allenes: Construction of Asymmetric Tertiary and Quaternary All Carbon Centers. Angew. Chem. Int. Ed. 2017, 56, 1903–1907. [Google Scholar] [CrossRef]
- Parveen, S.; Li, C.K.; Hassan, A.; Breit, B. Chemo-, Regio-, and Enantioselective Rhodium-Catalyzed Allylation of Pyridazinones with Terminal Allenes. Org. Lett. 2017, 19, 2326–2329. [Google Scholar] [CrossRef] [PubMed]
- Berthold, D.; Breit, B. Chemo-, Regio-, and Enantioselective Rhodium-Catalyzed Allylation of Triazoles with Internal Alkynes and Terminal Allenes. Org. Lett. 2018, 20, 598–601. [Google Scholar] [CrossRef]
- Wei, S.; Pedroni, J.; Meissner, A.; Lumbroso, A.; Drexler, H.-J.; Heller, D.; Breit, B. Development of an Improved Rhodium Catalyst for Z-Selective Anti-Markovnikov Addition of Carboxylic Acids to Terminal Alkynes. Chem. Eur. J. 2013, 19, 12067–12076. [Google Scholar] [CrossRef]
- Keller, E.; Pierrard, J.-S. (Eds.) All Pictures Showing X-ray Structures Were Prepared Using the program SCHAKAL; SCHAKAL99; University of Freiburg: Breisgau, Germany, 1999. [Google Scholar]
- Möller, S.; Drexler, H.-J.; Heller, D. Two Precatalysts for Application in Propargylic CH Activation. Acta Cryst. C 2019. submitted. [Google Scholar]
- Moon, S.; Nishii, Y.; Miura, M. Thioether-Directed Peri-Selective C–H Arylation under Rhodium Catalysis: Synthesis of Arene-Fused Thioxanthenes. Org. Lett. 2019, 21, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Satake, S.; Kurihara, T.; Nishikawa, K.; Mochizuki, T.; Hatano, M.; Ishihara, K.; Yoshino, T.; Matsunaga, S. Pentamethylcyclopentadienyl Rhodium(III)–Chiral Disulfonate Hybrid Catalysis for Enantioselective C–H Bond Functionalization. Nat. Catal. 2018, 1, 585–591. [Google Scholar] [CrossRef]
- Ghosh, K.; Mihara, G.; Nishii, Y.; Miura, M. Nondirected C-H Alkenylation of Arenes with Alkenes under Rhodium Catalysis. Chem. Lett. 2019, 48, 148–151. [Google Scholar] [CrossRef] [Green Version]
- Fischer, C.; Koenig, A.; Meissner, A.; Thede, R.; Selle, C.; Pribbenow, C.; Heller, D. Quantitative UV/Vis Spectroscopic Investigations of the In Situ Synthesis of Neutral μ2-Chloro-Bridged Dinuclear (Diphosphine)rhodium Complexes. Eur. J. Inorg. Chem. 2014, 34, 5849–5855. [Google Scholar] [CrossRef]
- Duan, C.-L.; Tan, Y.-X.; Zhang, J.-L.; Yang, S.; Dong, H.-Q.; Tian, P.; Lin, G.-Q. Highly Enantioselective Rhodium-Catalyzed Cross-Addition of Silylacetylenes to Cyclohexadienone-Tethered Internal Alkynes. Org. Lett. 2019, 21, 1690–1693. [Google Scholar] [CrossRef]
- Hilpert, L.J.; Sieger, S.V.; Haydl, A.M.; Breit, B. Palladium- and Rhodium-Catalyzed Dynamic Kinetic Resolution of Racemic Internal Allenes Towards Chiral Pyrazoles. Angew. Chem. Int. Ed. 2019, 131, 3416–3419. [Google Scholar] [CrossRef]
- Tanaka, K. Rhodium Catalysis in Organic Synthesis: Methods and Reactions; Wiley-VCerlag GmbH & Co. KGaA: Weinheim, Germany, 2018. [Google Scholar]
- Fairlie, D.P.; Bosnich, B. Homogeneous Catalysis - Conversion of 4-Pentenals to Cyclopentanones by Efficient Rhodium-Catalyzed Hydroacylation. Organometallics 1988, 7, 936–945. [Google Scholar] [CrossRef]
- Fischer, C.; Beweries, T.; Preetz, A.; Drexler, H.-J.; Baumann, W.; Peitz, S.; Rosenthal, U.; Heller, D. Kinetic and Mechanistic Investigations in Homogeneous Catalysis Using Operando UV/vis Spectroscopy. Catal. Today 2010, 155, 282–288. [Google Scholar] [CrossRef]
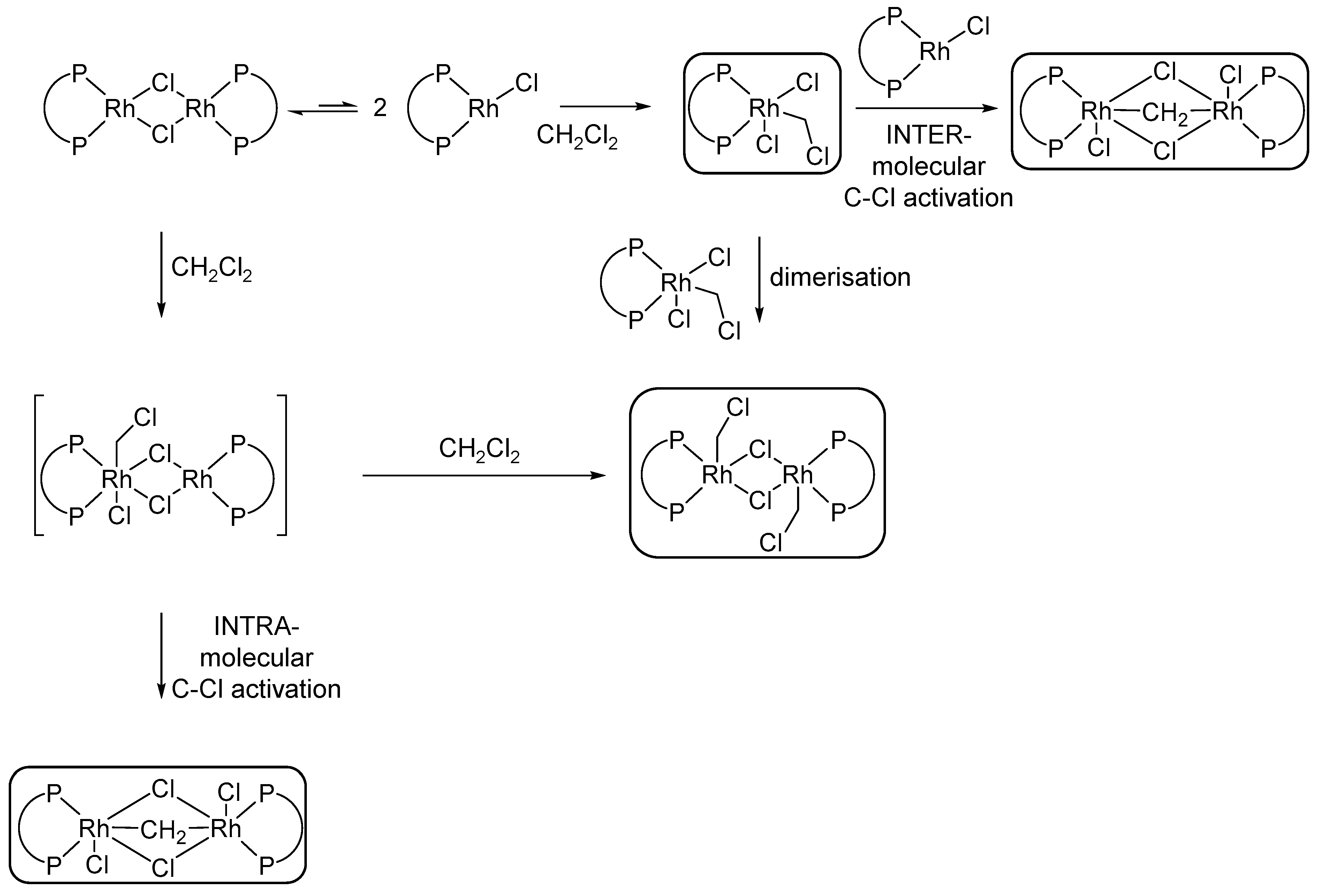
- Mannu, A.; Drexler, H.-J.; Thede, R.; Ferro, M.; Baumann, W.; Rüger, J.; Heller, D. Oxidative Addition of CH2Cl2 to Neutral Dimeric Rhodium Diphosphine Complexes. J. Organomet. Chem. 2018, 871, 178–184. [Google Scholar] [CrossRef]
- Möller, S.; Drexler, H.-J.; Kubis, C.; Alberico, E.; Heller, D. Investigations into the Mechanism of the In Situ Formation of Neutral Dinuclear Rhodium Complexes. J. Organomet. Chem. 2019. submitted. [Google Scholar]
- Time for 98 % conversion under the following reaction conditions: Ca. 1.0·10−2 mmol [Rh(diolefin)(µ2-Cl)]2 and 2.0·10−2 mmol ligand in 5 ml solvent.
- Lumbroso, A.; Cooke, M.L.; Breit, B. Catalytic Asymmetric Synthesis of Allylic Alcohols and Derivatives and their Applications in Organic Synthesis. Angew. Chem. Int. Ed. 2013, 52, 1890–1932. [Google Scholar] [CrossRef] [PubMed]
- Gellrich, U.; Meissner, A.; Steffani, A.; Kahny, M.; Drexler, H.J.; Heller, D.; Plattner, D.A.; Breit, B. Mechanistic Investigations of the Rhodium Catalyzed Propargylic CH Activation. J. Am. Chem. Soc. 2014, 136, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Breit, B. Rhodium-Catalyzed Chemo- and Regioselective Decarboxylative Addition of β-Ketoacids to Allenes: Efficient Construction of Tertiary and Quaternary Carbon Centers. J. Am. Chem. Soc. 2014, 136, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Koschker, P.; Kaehny, M.; Breit, B. Enantioselective Redox-Neutral Rh-Catalyzed Coupling of Terminal Alkynes with Carboxylic Acids Toward Branched Allylic Esters. J. Am. Chem. Soc. 2015, 137, 3131–3137. [Google Scholar] [CrossRef] [PubMed]
- James, B.R.; Mahajan, D. Bis(ditertiaryphosphine) Complexes of Rhodium(I). Synthesis, Spectroscopy, and Activity for Catalytic Hydrogenation. Can. J. Chem. 1979, 57, 180–187. [Google Scholar] [CrossRef]
- Castellanos-Paez, A.; Thayaparan, J.; Castillon, S.; Claver, C. Reactivity of Tetracarbonyl Dithiolate-Bridged Rhodium(I) Complexes with Diphosphines. J. Organomet. Chem. 1998, 551, 375–381. [Google Scholar] [CrossRef]
- Slack, D.A.; Baird, M.C. Investigations of Olefin Hydrogenation Catalysts. The Major Species Present in Solutions Containing Rhodium(I) Complexes of Chelating Diphosphines. J. Organomet. Chem. 1977, 142, C69–C72. [Google Scholar] [CrossRef]
- Crosman, A.; Hoelderich, W.F. Enantioselective Hydrogenation over Immobilized Rhodium Diphosphine Complexes on Aluminated SBA-15. J. Catal. 2005, 232, 43–50. [Google Scholar] [CrossRef]
- Van Haaren, R.J.; Zuidema, E.; Fraanje, J.; Goubitz, K.; Kamer, P.C.J.; van Leeuwen, P.W.N.M.; van Strijdonck, G.P.F. Probing the Mechanism of Rhodium (I) Catalyzed Dehydrocoupling of di-n-hexylsilane. C. R. Chim. 2002, 5, 431–440. [Google Scholar] [CrossRef]
- Wagner, H.H.; Hausmann, H.; Hoelderich, W.F. Immobilization of Rhodium Diphosphine Complexes on Mesoporous Al-MCM-41 Materials: Catalysts for Enantioselective Hydrogenation. J. Catal. 2001, 203, 150–156. [Google Scholar] [CrossRef]
- Sinou, D.; Bakos, J. (S,S)-2,3-Bis[Di(m-Sodiumsulfonatophenyl)-Phosphino]Butane (Chiraphosts) and (S,S)-2,4-Bis[Di(m-Sodiumsulfonatophenyl)- Phosphino]Pentane (BDPPts). Inorg. Synth. 1998, 32, 36–40. [Google Scholar]
- Baxley, G.T.; Weakley, T.J.R.; Miller, W.K.; Lyon, D.K.; Tyler, D.R. Synthesis and Catalytic Chemistry of Two New Water-soluble Chelating Phosphines. Comparison of Ionic and Nonionic Functionalities. J. Mol. Catal. A Chem. 1997, 116, 191–198. [Google Scholar] [CrossRef]
- Bartik, T.; Bunn, B.B.; Bartik, B.; Hanson, B.E. Synthesis, Reactions, and Catalytic Chemistry of the Water-Soluble Chelating Phosphine l,2-Bis[bis(m-sodiosulfonatophenyl)phosphino]ethane (DPPETS). Complexes with Nickel, Palladium, Platinum, and Rhodium. Inorg. Chem. 1994, 33, 164–169. [Google Scholar] [CrossRef]
- Bakos, J.; Tóth, I.; Heil, B.; Szalontai, G.; Párkányi, L.; Fülöp, V. Catalytic and Structural Studies of RhI Complexes of (−)-(2S,4S)-2,4-bis(diphenylphosphino)pentane. Asymmetric Hydrogenation of Acetophenonebenzylimine and Acetophenone. J. Organomet. Chem. 1989, 370, 263–276. [Google Scholar] [CrossRef]
- Meng, G.; Szostak, M. Rhodium-Catalyzed C–H Bond Functionalization with Amides by Double C–H/C–N Bond Activation. Org. Lett. 2016, 18, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, A.; Ise, Y.; Kimachi, T.; Inamoto, K. Rhodium-Catalyzed Cyclization of 2-Ethynylanilines in the Presence of Isocyanates: Approach toward Indole-3-carboxamides. Org. Lett. 2016, 18, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, L.-C.; Wang, J.; Jiang, C.; Zhang, Q.-W.; He, W. Rh(I)-Catalyzed Insertion of Allenes into C–C Bonds of Benzocyclobutenols. Org. Lett. 2016, 18, 328–331. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.; Qi, W.-Y.; Xu, B.; Xu, M.-H. Rhodium(I)-Catalyzed Asymmetric Carbene Insertion into B–H Bonds: Highly Enantioselective Access to Functionalized Organoboranes. J. Am. Chem. Soc. 2015, 137, 5268–5271. [Google Scholar] [CrossRef]
- Lim, D.S.W.; Lew, T.T.S.; Zhang, Y. Direct Amidation of N-Boc- and N-Cbz-Protected Amines via Rhodium-Catalyzed Coupling of Arylboroxines and Carbamates. Org. Lett. 2015, 17, 6054–6057. [Google Scholar] [CrossRef]
- Gopula, B.; Yang, S.-H.; Kuo, T.-S.; Hsieh, J.-C.; Wu, P.-Y.; Henschke, J.P.; Wu, H.-L. Direct Synthesis of Chiral 3-Arylsuccinimides by Rhodium-Catalyzed Enantioselective Conjugate Addition of Arylboronic Acids to Maleimides. Chem. Eur. J. 2015, 21, 11050–11055. [Google Scholar] [CrossRef]
- Lee, S.; Lee, W.L.; Yun, J. Rhodium-Catalyzed Addition of Alkyltrifluoroborate Salts to Imines. Adv. Synth. Catal. 2015, 357, 2219–2222. [Google Scholar] [CrossRef]
- Rao, H.; Yang, L.; Shuai, Q.; Li, C.-J. Rhodium-Catalyzed Aerobic Coupling between Aldehydes and Arenesulfinic Acid Salts: A Novel Synthesis of Aryl Ketones. Adv. Synth. Catal. 2011, 353, 1701–1706. [Google Scholar] [CrossRef]
- Morimoto, T.; Yamasaki, K.; Hirano, A.; Tsutsumi, K.; Kagawa, N.; Kakiuchi, K.; Harada, Y.; Fukumoto, Y.; Chatani, N.; Nishioka, T. Rh(I)-Catalyzed CO Gas-Free Carbonylative Cyclization Reactions of Alkynes with 2-Bromophenylboronic Acids Using Formaldehyde. Org. Lett. 2009, 11, 1777–1780. [Google Scholar] [CrossRef] [PubMed]
- Cornils, B.; Herrmann, W.A.; Schlögl, R.; Wong, C.-H. (Eds.) Catalysis from A to Z, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Temkin, O.N. Mechanisms of Formation of Catalytically Active Metal Complexes. In Homogeneous Catalysis with Metal Complexes: Kinetic Aspects and Mechanisms, 1st ed.; Temkin, O.N., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2012; Chapter 5; pp. 453–544. [Google Scholar]
- Evans, D.; Osborn, J.A.; Wilkinson, G. Hydroformylation of Alkenes by Use of Rhodium Complex Catalysts. J. Chem. Soc. A 1968, 3133–3142. [Google Scholar] [CrossRef]
- Brown, C.K.; Wilkinson, G. Homogeneous Hydroformylation of Alkenes with Hydridocarbonyltris-(triphenylphosphine)rhodium(i) as Catalyst. J. Chem. Soc. A 1970, 2753–2764. [Google Scholar] [CrossRef]
- Adams, G.M.; Ryan, D.E.; Beattie, N.A.; McKay, A.I.; Lloyd-Jones, G.C.; Weller, A.S. Dehydropolymerization of H3B·NMeH2 Using a [Rh(DPEphos)]+ Catalyst: The Promoting Effect of NMeH2. ACS Catal. 2019, 9, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Schulz, S.; Drexler, H.-J.; Selle, C.; Lotz, M.; Sawall, M.; Neymeyr, K.; Heller, D. The Influence of Substituents in Diphosphine Ligands on the Hydrogenation Activity and Selectivity of the Corresponding Rhodium Complexes as Exemplified by ButiPhane. ChemCatChem 2012, 4, 81–88. [Google Scholar] [CrossRef]
- 0.01 mmol rhodium complex and 1.0 mmol prochiral olefin in 15.0 ml MeOH at 25.0 °C and 1 bar total pressure
- Nagel, U.; Kinzel, E.; Andrade, J.; Prescher, G. Enantioselektive Katalyse, 4. Synthese N-substituierter (R,R)-3,4-Bis(diphenylphosphino)-pyrrolidine und Anwendung ihrer Rhodiumkomplexe zur Asymmetrischen Hydrierung von α-(Acylamino)acrylsäure-Derivaten. Chem. Ber. 1986, 119, 3326–3343. [Google Scholar] [CrossRef]
- Nagel, U.; Krink, T. Catalytic Hydrogenation with Rhodium Complexes Containing dipamp-pyrphos Hybrid Ligands. Angew. Chem. Int. Ed. Engl. 1993, 32, 1052–1054. [Google Scholar] [CrossRef]
- Thiel, I.; Horstmann, M.; Jungk, P.; Keller, S.; Fischer, F.; Drexler, H.-J.; Heller, D.; Hapke, M. Insight into the Activation of In Situ-Generated Chiral Rh(I)-Catalysts and their Application in Cyclotrimerizations. Chem. Eur. J. 2017, 23, 17048–17057. [Google Scholar] [CrossRef]
- Alberico, E.; Baumann, W.; de Vries, J.G.; Drexler, H.-J.; Gladiali, S.; Heller, D.; Henderickx, H.J.W.; Lefort, L. Unravelling the Reaction Path of Rhodium–MonoPhos-Catalysed Olefin Hydrogenation. Chem. Eur. J. 2011, 17, 12683–12695. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, M. Rhodium-Catalyzed Asymmetric Hydrogenation Using Phosphoramidite Ligands. Ph.D. Thesis, University of Groningen, Groningen, The Netherlands, 2006. Available online: http://hdl.handle.net/11370/c6d065b0-5bc1-4a2a-a452-fb237d5a4e6e (accessed on 5 June 2019).
- Esteruelas, M.A.; Herrero, J.; Martin, M.; Oro, M.L.A.; Real, V.M. Mechanism of the Hydrogenation of 2,5-Norbornadiene Catalyzed by [Rh(NBD)(PPh3)2]BF4 in Dichloromethane: a Kinetic and Spectroscopic Investigation. J. Organomet. Chem. 2000, 599, 178–184. [Google Scholar] [CrossRef]
- Heller, D.; Kortus, K.; Selke, R. Kinetische Untersuchungen zur Ligandenhydrierung in Katalysatorvorstufen für die Asymmetrische Reduktion prochiraler Olefine. Liebigs Ann. 1995, 575–581. [Google Scholar] [CrossRef]
- Braun, W.; Salzer, A.; Drexler, H.-J.; Spannenberg, A.; Heller, D. Investigations into the Hydrogenation of Diolefins and Prochiral Olefins Employing the “Daniphos”-type Ligands. Dalton Trans. 2003, 1606–1613. [Google Scholar] [CrossRef]
- Preetz, A.; Drexler, H.-J.; Fischer, C.; Dai, Z.; Börner, A.; Baumann, W.; Spannenberg, A.; Thede, R.; Heller, D. Rhodium Complex Catalyzed Asymmetric Hydrogenation - Transfer of Pre-Catalysts Into Active Species. Chem. Eur. J. 2008, 14, 1445–1451. [Google Scholar] [CrossRef]
- Selent, D.; Heller, D. In-Situ Techniques for Homogeneous Catalysis, in Catalysis From Principle to Application; Beller, M., Renken, A., van Santen, R., Eds.; Wiley-VCH: Weinheim, Germany, 2012; Chapter 23; pp. 465–492. [Google Scholar]
- Drexler, H.-J.; Preetz, A.; Schmidt, T.; Heller, D. Kinetics of Homogeneous Hydrogennations: Measurement and Interpretation. In Handbook of Homogeneous Hydrogenation; de Vries, H.G., Elsevier, C., Eds.; Wiley-VCH: Weinheim, Germany, 2007; Chapter 10; pp. 257–293. [Google Scholar]
- Heller, D.; Borns, S.; Baumann, W.; Selke, R. Kinetic Investigations of the Hydrogenation of Diolefin Ligands in Catalyst Precursors for the Asymmetric Reduction of Prochiral Olefins, II. Chem. Ber. 1996, 129, 85–89. [Google Scholar] [CrossRef]
- Drexler, H.-J.; Baumann, W.; Spannenberg, A.; Fischer, C.; Heller, D. COD- versus NBD-Precatalysts. Dramatic Difference in the Asymmetric Hydrogenation of Prochiral Olefins with Five Membered Diphosphine Rh-Hydrogenation Catalysts. J. Organomet. Chem. 2001, 621, 89–102. [Google Scholar] [CrossRef]
- Baseda Krüger, M.; Selle, C.; Heller, D.; Baumann, W. Determination of Gas Concentrations in Liquids by Nuclear Magnetic Resonance: Hydrogen in Organic Solvents. J. Chem. Eng. Data 2012, 57, 1737–1744. [Google Scholar] [CrossRef]
- Krüger, M.B. Bestimmung von Gaskonzentrationen in Flüssigen Medien Mittels NMR-Spektroskopie: Eine Methode für Kinetische und in-situ Studien. Ph.D. Thesis, University of Rostock, Rostock, Germany, 2013. [Google Scholar]
- Greiner, L.; Ternbach, M.B. Kinetic Study of Homogeneous Alkene Hydrogenation by Model Discrimination. Adv. Synth. Catal. 2004, 346, 1392–1396. [Google Scholar] [CrossRef]
- Drexler, H.-J.; Zhang, S.; Sun, A.; Spannenberg, A.; Arrieta, A.; Preetz, A.; Heller, D. Cationic Rh-Bisphosphane-Diolefin Complexes as Precatalysts for Enantioselective Catalysis - What Informations Do Single Crystal Structures Contain Regarding Product Chirality? Tetrahedron Asymmetry 2004, 15, 2139–2150. [Google Scholar] [CrossRef]
- Brown, J.M.; Chaloner, P.A. The Mechanism of Asymmetric Hydrogenation Catalysed by Rhodium (I) Dipamp Complexes. Tetrahedron Lett. 1978, 19, 1877–1880. [Google Scholar] [CrossRef]
- Brown, J.M.; Chaloner, P.A. The Mechanism of Asymmetric Homogeneous Hydrogenation. Rhodium (I) Complexes of Dehydroamino Acids Containing Asymmetric Ligands Related to Bis(1,2-diphenylphosphino)ethane. J. Am. Chem. Soc. 1980, 102, 3040–3048. [Google Scholar] [CrossRef]
- De Vries, J.G.; Lefort, L. High-Throughput Experimentation and Ligand Libraries. In Handbook of Homogeneous Hydrogenation; de Vries, H.G., Elsevier, C., Eds.; Wiley-VCH: Weinheim, Germany, 2007; Chapter 36; pp. 1245–1278. [Google Scholar]
- Preetz, A.; Drexler, H.-J.; Schulz, S.; Heller, D. BINAP: Rhodium-Diolefin Complexes in Asymmetric Hydrogenation. Tetrahedron Asymm. 2010, 21, 1226–1231. [Google Scholar] [CrossRef]
- Cobley, C.J.; Lennon, I.C.; McCague, R.; Ramsden, J.A.; Zanotti-Gerosa, A. On the Economic Application of DuPHOS Rhodium(I) Catalysts: A Comparison of COD versus NBD Precatalysts. Tetrahedron Lett. 2001, 42, 7481–7483. [Google Scholar] [CrossRef]
- Moxham, G.L.; Randell-Sly, H.E.; Brayshaw, S.K.; Woodward, R.L.; Weller, A.S.; Willis, M.C. A Second-Generation Catalyst for Intermolecular Hydroacylation of Alkenes and Alkynes Using β-S-Substituted Aldehydes: The Role of a Hemilabile P-O-P Ligand. Angew. Chem. Int. Ed. 2006, 45, 7618–7622. [Google Scholar] [CrossRef] [PubMed]
- Preetz, A.; Fischer, C.; Kohrt, C.; Drexler, H.-J.; Baumann, W.; Heller, D. Cationic Rhodium-BINAP Complexes: Full Characterization of Solvate- and Arene Bridged Dimeric Species. Organometallics 2011, 30, 5155–5159. [Google Scholar] [CrossRef]
- Meißner, A.; Drexler, H.-J.; Keller, S.; Selle, C.; Ratovelomanana-Vidal, V.; Heller, D. Synthesis and Characterisation of Cationic Synphos-Rhodium Complexes. Eur. J. Inorg. Chem. 2014, 4836–4842. [Google Scholar] [CrossRef]
- Webster, R.; Bçing, C.; Lautens, M. Reagent-Controlled Regiodivergent Resolution of Unsymmetrical Oxabicyclic Alkenes Using a Cationic Rhodium Catalyst. J. Am. Chem. Soc. 2009, 131, 444–445. [Google Scholar] [CrossRef]
- Preetz, A.; Kohrt, C.; Drexler, H.-J.; Torrens, A.; Buschmann, H.; Lopez, M.G.; Heller, D. Asymmetric Ring Opening of Oxabicyclic Alkenes With Cationic Rhodium Complexes. Adv. Synth. Catal. 2010, 352, 2073–2080. [Google Scholar] [CrossRef]
- Fischer, C.; Kohrt, C.; Drexler, H.-J.; Baumann, W.; Heller, D. Trinuclear Hydride Complexes of Rhodium. Dalton Trans. 2011, 40, 4162–4166. [Google Scholar] [CrossRef]
- Kohrt, C.; Hansen, S.; Drexler, H.-J.; Rosenthal, U.; Schulz, A.; Heller, D. Molecular Vibration Spectroscopy Studies on Novel Trinuclear Rhodium-7-Hydride Complexes of the General Type {[Rh(PP*)X]3(µ2-X)3(µ3-X)}(BF4)2 (X = H, D). Inorg. Chem. 2012, 51, 7377–7383. [Google Scholar] [CrossRef] [PubMed]
- Kohrt, C.; Baumann, W.; Spannenberg, A.; Drexler, H.-J.; Gridnev, I.; Heller, D. Formation of Trinuclear Rhodium-Hydride Complexes {[Rh(PP*)H]3(µ2-H)3(µ3-H)}(Anion)2 - During Asymmetric Hydrogenation? Chem. Eur. J. 2013, 19, 7443–7451. [Google Scholar] [CrossRef] [PubMed]
- Kohrt, C. Mehrkernige Hydridkomplexe des Rhodiums mit Bisphosphanliganden: Charakterisierung, Bildung und Anwendung. Ph.D. Thesis, University of Rostock, Rostock, Germany, 2013. [Google Scholar]
- Gridnev, I.D.; Imamoto, T. Mechanism of Enantioselection in Rh-Catalyzed Asymmetric Hydrogenation. The origin of Utmost Catalytic performance. Chem. Comm. 2009, 7447–7464. [Google Scholar] [CrossRef] [PubMed]
- Indeed a mechanism for the formation of the trinuclear rhodium (III) polyhydride species {[Rh(diphosphine)H]3(µ2-H)3(µ3-H)}2+ from the rhodium(I) solvate dihydride [Rh(III)(diphosphine)(H)2(solvent)2]+ has been put forward, based on experimental evidence, which includes the intermediacy of a dinuclear rhodium (III) polyhydride species of general formula {[Rh(diphosphine)H]2(µ2-H)3}[X]. The equilibria, the species present in solution and their relative concentrations, depends on the properties of the diphosphine ligand, the temperature and the solvent.
- The hydrogenation of diolefins in complexes with diphosphines which do not contain aryl groups in nonpolar solvents such as DCM is even more complicated: With the ligand t-BuBisP* 1H-103Rh-HMQC-NMR spectra show the formation of three polyhydrido species. One of them has been assigned the structure {[Rh(t-Bu-BisP*)H]3(µ2-H)3(µ3-Cl)}2+, while that of the other two remain unclear.
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics, 4th ed.; Wiley-VCH Verlag & Co. KgaA: Weinheim, Germany, 2012. [Google Scholar]
- Kollár, L.; Törös, S.; Heil, B.; Markó, L. Phosphinerhodium Complexes as Homogeneous Catalysts: XI. Decarbonylation of Primary Alcohols Used as Solvents Under Conditions of Olefin Hydrogenation; a Side Reaction Leading to Catalyst Deactivation. J. Organomet. Chem. 1980, 192, 253–256. [Google Scholar]
- Benn, R.; Rufińska, A. High-Resolution Metal-NMR Spectroscopy of Organometallic Compounds. Angew. Chem. Int. Ed. Engl. 1986, 25, 861–881. [Google Scholar] [CrossRef]
- Von Philipsborn, W. Probing Organometallic Structure and Reactivity by Transition Metal NMR Spectroscopy. Chem. Soc. Rev. 1999, 28, 95–105. [Google Scholar] [CrossRef]
- Ernsting, J.M.; Elsevier, C.J.; de Lange, W.G.J.; Timmer, K. Inverse Two-Dimensional 31P, 103Rh{1H} NMR of Cationic Rhodium (1) Complexes Containing Chelating Diphosphines. Magn. Reson. Chem. 1991, 29, S118–S124. [Google Scholar] [CrossRef]
- Leitner, W.; Bühl, M.; Fornika, R.; Six, C.; Baumann, W.; Dinjus, E.; Kessler, M.; Krüger, C.; Rufińska, A. 103Rh Chemical Shifts in Complexes Bearing Chelating Bidentate Phosphine Ligands. Organometallics 1999, 18, 1196–1206. [Google Scholar] [CrossRef]
- Halpern, J.; Riley, D.P.; Chan, A.S.C.; Pluth, J.J. Novel Coordination Chemistry and Catalytic Properties of Cationic 1,2-Bisjdiphenylphosphino)ethanerhodium(I) Complexes. J. Am. Chem. Soc. 1977, 99, 8055–8057. [Google Scholar] [CrossRef]
- Halpern, J.; Chan, A.S.C.; Riley, D.P.; Pluth, J.J. Some Aspects of the Coordination Chemistry and Catalytic Properties of Cationic Rhodium-Phosphine Complexes. Adv. Chem. Ser. 1979, 173, 16–25. [Google Scholar]
- Landis, C.R.; Halpern, J. Homogeneous Catalysis of Arene Hydrogenation by Cationic Rhodium Arene Complexes. Organometallics 1983, 2, 840–842. [Google Scholar] [CrossRef]
- Fischer, C.; Thede, R.; Drexler, H.-J.; König, A.; Baumann, W.; Heller, D. Investigations into the Formation and Stability of Cationic Rhodium Diphosphane η6-Arene Complexes. Chem. Eur. J. 2012, 18, 11920–11928. [Google Scholar] [CrossRef] [PubMed]
- Polster, J.; Lachmann, H. Spectrometric Titrations: Analysis of Chemical Equilibria; Wiley-VCH: Weinheim, Germany, 1989. [Google Scholar]
- Benesi, H.A.; Hildebrand, J.H. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Scott, R.L. Some Comments on the Benesi-Hildebrand Equation. Rec. Trav. Chim. 1956, 75, 787–789. [Google Scholar] [CrossRef]
- Scatchard, G. The Attractions of Proteins for Small Molecules and Ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of Equilibrium Constants from Multiwavelength Spectroscopic Data—I: Mathematical Considerations. Talanta 1985, 32, 95–101. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of Equilibrium Constants from Multiwavelength Spectroscopic data—II132, 95.: Specfit: Two User-Friendly Programs in Basic and Standard Fortran 77. Talanta 1985, 32, 257–264. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of Equilibrium Constants From Multiwavelength Spectroscopic Data—III: Model-Free Analysis of Spectrophotometric and ESR Titrations. Talanta 1985, 32, 1133–1139. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of Equilibrium Constants from Multiwavelength Spectroscopic Data—IV: Model-Free Least-Squares Refinement by Use of Evolving Factor Analysis. Talanta 1986, 33, 943–951. [Google Scholar] [CrossRef]
- ReactLABTM Equilibria. Available online: http://jplusconsulting.com/products/reactlab-equilibria/ (accessed on 5 June 2019).
- Fischer, C. UV-vis-Spektroskopie in der homogenen Katalyse – Komplexchemische Untersuchungen an Rhodium-Katalysatoren. Ph.D. Thesis, University of Rostock, Rostock, Germany, 2010. [Google Scholar]
- Gridnev, I.D.; Higashi, N.; Asakura, K.; Imamoto, T. Mechanism of Asymmetric Hydrogenation Catalyzed by a Rhodium Complex of (S,S)-1,2-Bis(tert-butylmethylphosphino)ethane. Dihydride Mechanism of Asymmetric Hydrogenation. J. Am. Chem. Soc. 2000, 122, 7183–7194. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Yasutake, M.; Higashi, N.; Imamoto, T. Asymmetric Hydrogenation of Enamides with Rh-BisP* and Rh-MiniPHOS Catalysts. Scope, Limitations, and Mechanism. J. Am. Chem. Soc. 2001, 123, 5268–5276. [Google Scholar] [CrossRef] [PubMed]
- Heller, D.; Drexler, H.-J.; Spannenberg, A.; Heller, B.; You, J.; Baumann, W. The Inhibiting Influence of Aromatic Solvents on the Activity of Asymmetric Hydrogenations. Angew. Chem. Int. Ed. 2002, 41, 777–780. [Google Scholar] [CrossRef]
- Sablong, R.; van der Vlugt, J.I.; Thormann, R.; Mecking, S.; Vogt, D. Disperse Amphiphilic Submicron Particles as Non-Covalent Supports for Cationic Homogeneous Catalysts. Adv. Synth. Catal. 2005, 347, 633–636. [Google Scholar] [CrossRef]
- Balué, J.; Bayón, J.C. Hydroformylation of Styrene Catalyzed by a Rhodium Thiolate Binuclear Catalyst Supported on a Cationic Exchange Resin. J. Mol. Catal. A 1999, 137, 193–203. [Google Scholar] [CrossRef]
- Šebesta, R. (Ed.) Enantioselective Homogeneous Supported Catalysis; RSC: Cambridge, UK, 2012. [Google Scholar]
- Pugin, B.; Blaser, H.-U. The Immobilization of Rhodium-4-(diphenylphosphino)-2- (diphenylphosphinomethyl)-pyrrolidine (Rh-PPM) Complexes: A Systematic Study. Adv. Synth. Catal. 2006, 348, 1743–1751. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Marco, M. Preparation of Polymer-Supported Ligands and Metal Complexes for Use in Catalysis. Chem. Rev. 2002, 102, 3217–3274. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, C.; Barthe, M.; de Saint Laumer, J.-Y. Synthesis of Citronellal by RhI-Catalysed Asymmetric Isomerization of N,N-Diethyl-Substituted Geranyl- and Nerylamines or Geraniol and Nerol in the Presence of Chiral Diphosphino Ligands, under Homogeneous and Supported Conditions. Helv. Chim. Acta 2001, 84, 230–242. [Google Scholar] [CrossRef]
- Bayston, D.J.; Fraser, J.L.; Ashton, M.R.; Baxter, A.D.; Polywka, M.E.C.; Moses, E. Preparation and Use of a Polymer Supported BINAP Hydrogenation Catalyst. J. Org. Chem. 1998, 63, 3137–3140. [Google Scholar] [CrossRef]
- Renaud, E.; Baird, M.C. Effects of Catalyst Site Accessibility on Catalysis by Rhodium(I) Complexes of Amphiphilic Ligands [Ph2P(CH2)nPMe3]+(n = 2, 3, 6 or 10) tethered to a cation-exchange resin. J. Chem. Soc. Dalton Trans. 1992, 2905–2906. [Google Scholar] [CrossRef]
- Kalck, P.; de Oliveira, E.L.; Queau, R.; Peyrille, B.; Molinier, J. Dinuclear Rhodium Complexes Immobilized on Functionalized Diphenylphosphino-(Styrene-Divinylbenzene) Resins Giving High Selectivities for Linear Aldehydes in Hydroformylation reactions. J. Organomet. Chem. 1992, 433, C4–C8. [Google Scholar] [CrossRef]
- Fischer, C.; Thede, R.; Baumann, W.; Drexler, H.-J.; König, A.; Heller, D. Investigations into Metal Leaching from Polystyrene-Supported Rhodium Catalysts. ChemCatChem 2016, 8, 352–356. [Google Scholar] [CrossRef]
- Schwarze, M.; Milano-Brusco, J.-S.; Strempel, V.; Hamerla, T.; Wille, S.; Fischer, C.; Baumann, W.; Arlt, W.; Schomäcker, R. Rhodium Catalyzed Hydrogenation Reactions in Aqueous Micellar Systems as Green Solvents. RSC Adv. 2011, 1, 474–483. [Google Scholar]
- Mikami, K.; Kataoka, S.; Yusa, Y.; Aikawa, K. Racemic but Tropos (Chirally Flexible) BIPHEP Ligands for Rh(I)-Complexes: Highly Enantioselective Ene-Type Cyclization of 1,6-Enynes. Org. Lett. 2004, 6, 3699–3701. [Google Scholar] [CrossRef] [PubMed]
- Mikami, K.; Yusa, Y.; Hatano, M.; Wakabayashi, K.; Aikawa, K. Highly Enantioselective Spiro Cyclization of 1,6-Enynes Catalyzed by Cationic Skewphos Rhodium(I) Complex. Chem. Commun. 2004, 98–99. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, R.W.; Wang, X.; Noheda, P.; Bergens, S.H.; Whelan, J.; Bosnich, B. Asymmetric Catalysis. Asymmetric Catalytic Intramolocular Hydrosilation and Hydroacylation. Tetrahedron 1994, 50, 4335–4346. [Google Scholar] [CrossRef]
- Allen, D.G.; Wild, S.B.; Wood, D.L. Catalytic Asymmetric Hydrogenation of Prochiral Enamides by Rhodium(I) Complexes Containing the Enantiomers of (R*,R*)-(.+-.)-1,2-Phenylenebis(methylphenyl-phosphine) and its Arsenic Isosteres. Organometallics 1986, 5, 1009–1015. [Google Scholar] [CrossRef]
- Miyashita, A.; Takaya, H.; Souchi, T.; Noyori, R. 2, 2′-Bis(diphenylphosphino)-1, 1′-binaphthyl(binap): A New Atropisomeric Bis(triaryl)phosphine. Synthesis and its Use in the Rh(l)-Catalyzed Asymmetric Hydrogenation of α-(Acylamino)acrylic Acids. Tetrahedron 1984, 40, 1245–1253. [Google Scholar] [CrossRef]
- Riley, D.P.J. Solution Studies of the Asymmetric Hydrogenation Catalyst System Derived from The [Rhodium (1,2-Bis(diphenylphosphino)-1-cyclohexylethane)] Moiety. Organomet. Chem. 1982, 234, 85–97. [Google Scholar] [CrossRef]
- Miyashita, A.; Yasuda, A.; Takaya, H.; Toriumi, K.; Ito, T.; Souchi, T.; Noyori, R. Synthesis of 2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), an Atropisomeric Chiral Bis(triaryl)phosphine, and its Use in the Rhodium(I)-Catalyzed Asymmetric Hydrogenation of Alpha-(acylamino)acrylic Acids. J. Am. Chem. Soc. 1980, 102, 7932–7934. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Alberico, E.; Gladiali, S. Captured at Last: a Catalyst–Substrate Adduct and a Rh-Dihydride Solvate in the Asymmetric Hydrogenation by a Rh-Monophosphine Catalyst. Chem. Commun. 2012, 48, 2186–2188. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Fan, C.; Pringle, P.G. New Insights Into the Mechanism of Asymmetric Hydrogenation Catalysed by Monophosphonite–Rhodium Complexes. Chem. Commun. 2007, 1319–1321. [Google Scholar] [CrossRef] [PubMed]
- Rifat, A.; Patmore, N.J.; Mahon, M.F.; Weller, A.S. Rhodium Phosphines Partnered with the Carborane Monoanions [CB11H6Y6]− (Y = H, Br). Synthesis and Evaluation as Alkene Hydrogenation Catalysts. Organometallics 2002, 21, 2856–2865. [Google Scholar] [CrossRef]
- Marcazzan, P.; Ezhova, M.B.; Patrick, B.O.; James, B.R. Synthesis and Structure of Dimeric Rh-Bis(tertiary phosphine) Complexes, Exceptionally Useful Synthetic Precursors. C. R. Chim. 2002, 5, 373–378. [Google Scholar] [CrossRef]
- Singewald, E.T.; Mirkin, C.A.; Stern, C.L. A Redox-Switchable Hemilabile Ligand: Electrochemical Control of the Coordination Environment of a RhI Complex. Angew. Chem. Int. Ed. 1995, 35, 1624–1627. [Google Scholar] [CrossRef]
- Preetz, A.; Baumann, W.; Fischer, C.; Drexler, H.-J.; Schmidt, T.; Thede, R.; Heller, D. Asymmetric Hydrogenation. Dimerization of Solvate Complexes: Synthesis and Characterization of Dimeric [Rh(DIPAMP)]22+, a Valuable Catalyst Precursor. Organometallics 2009, 28, 3673–3677. [Google Scholar] [CrossRef]
- Fairlie, D.P.; Bosnich, B. Homogeneous Catalysis. Mechanism of Catalytic Hydroacylation: the Conversion of 4-Pentenals to Cyclopentanones. Organometallics 1988, 7, 946–954. [Google Scholar] [CrossRef]
- Barnhart, R.W.; Xianqi, W.; Noheda, P.; Bergens, S.H.; Whelan, J.; Bosnich, B. Asymmetric Catalysis. Asymmetric Catalytic Intramolecular Hydroacylation of 4-Pentenals Using Chiral RhodiumDiphosphine Catalysts. J. Am. Chem. Soc. 1994, 116, 1821–1830. [Google Scholar] [CrossRef]
- Masutomi, K.; Sugiyama, H.; Uekusa, H.; Shibata, Y.; Tanaka, K. Asymmetric Synthesis of Protected Cyclohexenylamines and Cyclohexenols by Rhodium-Catalyzed [2+2+2] Cycloaddition. Angew. Chem. Int. Ed. 2016, 55, 15373–15376. [Google Scholar] [CrossRef]
- Aida, Y.; Sugiyama, H.; Uekusa, H.; Shibata, Y.; Tanaka, K. Rhodium-Catalyzed Asymmetric [2 + 2 + 2] Cycloaddition of α,ω-Diynes with Unsymmetrical 1,2-Disubstituted Alkenes. Org. Lett. 2016, 18, 2672–2675. [Google Scholar] [CrossRef]
- Yoshizaki, S.; Nakamura, Y.; Masutomi, K.; Yoshida, T.; Noguchi, K.; Shibata, Y.; Tanaka, K. Rhodium-Catalyzed Asymmetric [2 + 2 + 2] Cycloaddition of 1,6-Enynes with Cyclopropylideneacetamides. Org. Lett. 2016, 18, 388–391. [Google Scholar] [CrossRef]
- Shintani, R.; Takano, R.; Nozaki, K. Rhodium-Catalyzed Asymmetric Synthesis of Silicon-Stereogenic Silicon-Bridged Arylpyridinones. Chem. Sci. 2016, 7, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Roglans, A.; Pla-Quintana, A. An Enantioselective Cascade Cyclopropanation Reaction Catalyzed by Rhodium(I): Asymmetric Synthesis of Vinylcyclopropanes. Adv. Synth. Catal. 2016, 358, 3512–3516. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, M.; Parera, M.; Parella, T.; Lledj, A.; le Bras, J.; Muzart, J.; Pla-Quintana, A.; Roglans, A. Rhodium-Catalyzed [2+2+2] Cycloadditions of Diynes with Morita–Baylis–Hillman Adducts: A Stereoselective Entry to Densely Functionalized Cyclohexadiene Scaffolds. Adv. Synth. Catal. 2016, 358, 1848–1853. [Google Scholar] [CrossRef]
- Amatore, M.; Leboeuf, D.; Malacria, M.; Gandon, V.; Aubert, C. Highly Enantioselective Rhodium-Catalyzed [2+2+2] Cycloaddition of Diynes to Sulfonimines. J. Am. Chem. Soc. 2013, 135, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Noguchi, K.; Tanaka, K. Enantioselective Synthesis of Planar-Chiral Carba-Paracyclophanes: Rhodium-Catalyzed [2+2+2] Cycloaddition of Cyclic Diynes with Terminal Monoynes. Angew. Chem. Int. Ed. 2013, 52, 5617–5621. [Google Scholar] [CrossRef] [PubMed]
- Augé, M.; Barbazanges, M.; Tran, A.T.; Simonneau, A.; Elley, P.; Amouri, H.; Aubert, C.; Fensterbank, L.; Gandon, V.; Malacria, M.; et al. Atroposelective [2+2+2] Cycloadditions Catalyzed by a Rhodium(I)–Chiral Phosphate System. Chem. Commun. 2013, 49, 7833–7835. [Google Scholar] [CrossRef]
- Lu, Y.; Woo, S.K.; Krische, M.J. Total Synthesis of Bryostatin 7 via C–C Bond-Forming Hydrogenation. J. Am. Chem. Soc. 2011, 133, 13876–13879. [Google Scholar] [CrossRef]
- Kong, J.R.; Krische, M.J. Catalytic Carbonyl Z-Dienylation via Multicomponent Reductive Coupling of Acetylene to Aldehydes and α-Ketoesters Mediated by Hydrogen: Carbonyl Insertion into Cationic Rhodacyclopentadienes. J. Am. Chem. Soc. 2006, 128, 16040–16041. [Google Scholar] [CrossRef]
- Kong, J.-R.; Cho, C.-W.; Krische, M.J. Hydrogen-Mediated Reductive Coupling of Conjugated Alkynes with Ethyl (N-Sulfinyl)iminoacetates: Synthesis of Unnatural α-Amino Acids via Rhodium-Catalyzed C−C Bond Forming Hydrogenation. J. Am. Chem. Soc. 2005, 127, 11269–11276. [Google Scholar] [CrossRef]
- Rhee, J.U.; Krische, M.J. Highly Enantioselective Reductive Cyclization of Acetylenic Aldehydes via Rhodium Catalyzed Asymmetric Hydrogenation. J. Am. Chem. Soc. 2006, 128, 10674–10675. [Google Scholar] [CrossRef]
- Jang, H.-Y.; Hughes, F.W.; Gong, H.; Zhang, J.; Brodbelt, J.S.; Krische, M.J. Enantioselective Reductive Cyclization of 1,6-Enynes via Rhodium-Catalyzed Asymmetric Hydrogenation: C−C Bond Formation Precedes Hydrogen Activation. J. Am. Chem. Soc. 2005, 127, 6174–6175. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Sakiyama, N.; Noguchi, K.; Tanaka, K. Rhodium-Catalyzed Regio-, Diastereo-, and Enantioselective [2+2+2] Cycloaddition of 1,6-Enynes with Acrylamides. Angew. Chem. Int. Ed. 2012, 51, 13031–13035. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, N.; Noguchi, K.; Tanaka, K. Rhodium-Catalyzed Intramolecular Cyclization of Naphthol- or Phenol-Linked 1,6-Enynes Through the Cleavage and Formation of sp2 C-O Bonds. Angew. Chem. Int. Ed. 2012, 51, 5976–5980. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Shibata, Y.; Noguchi, K.; Tanaka, K. Rhodium-Catalyzed Asymmetric [2+2+2] Cyclization of 1,6-Enynes and Aldehydes. Chem. Eur. J. 2011, 17, 12578–12581. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, Y.; Kobayashi, M.; Tanaka, K. Rhodium-Catalyzed Intermolecular [2+2+2] Cross-Trimerization of Aryl Ethynyl Ethers and Carbonyl Compounds To Produce Dienyl Esters. Angew. Chem. Int. Ed. 2011, 50, 10922–10926. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-Y.; Krische, M.J. Rhodium-Catalyzed Reductive Cyclization of 1,6-Diynes and 1,6-Enynes Mediated by Hydrogen: Catalytic C−C Bond Formation via Capture of Hydrogenation Intermediates. J. Am. Chem. Soc. 2004, 126, 7875–7880. [Google Scholar] [CrossRef] [PubMed]
- Preetz, A. Rhodium-Präkatalysatoren in der asymmetrischen Katalyse. Ph.D. Thesis, University of Rostock, Rostock, Germany, 2008. [Google Scholar]
- König, A.; Fischer, C.; Alberico, E.; Selle, C.; Drexler, H.-J.; Baumann, W.; Heller, D. Oxidative Addition of Aryl Halides to Cationic Bis(phosphane)rhodium Complexes: Application in C–C Bond Formation. Eur. J. Inorg. Chem. 2017, 2040–2047. [Google Scholar] [CrossRef]
- Jiao, Y.; Brennessel, W.W.; Jones, W.D. Oxidative Addition of Chlorohydrocarbons to a Rhodium Tris(pyrazolyl)borate Complex. Organometallics 2015, 34, 1552–1566. [Google Scholar] [CrossRef]
- Townsend, N.S.; Chaplin, A.B.; Naser, M.A.; Thompson, A.L.; Rees, N.H.; Macgregor, S.A.; Weller, A.S. Reactivity of the Latent 12-Electron Fragment [Rh(PiBu3)2]+ with Aryl Bromides: Aryl-Br and Phosphine Ligand C-H Activation. Chem. Eur. J. 2010, 16, 8376–8389. [Google Scholar] [CrossRef]
- Chen, S.; Li, Y.; Zhao, J.; Li, X. Chelation-Assisted Carbon-Halogen Bond Activation by a Rhodium(I) Complex. Inorg. Chem. 2009, 48, 1198–1206. [Google Scholar] [CrossRef]
- Douglas, T.M.; Chaplin, A.B.; Weller, A.S. Dihydrogen Loss from a 14-Electron Rhodium(III) Bis-Phosphine Dihydride To Give a Rhodium(I) Complex That Undergoes Oxidative Addition with Aryl Chlorides. Organometallics 2008, 27, 2918–2921. [Google Scholar] [CrossRef]
- Pike, S.D.; Weller, A.S. C–Cl activation of the weakly coordinating anion [B(3,5-Cl2C6H3)4]− at a Rh(I) centre in solution and the solid-state. Dalton Trans. 2013, 42, 12832–12835. [Google Scholar] [CrossRef] [PubMed]
- Curto, S.G.; Esteruelas, M.A.; Olivan, M.; Onate, E.; Velez, A. Selective C–Cl Bond Oxidative Addition of Chloroarenes to a POP–Rhodium Complex. Organometallics 2017, 36, 114–128. [Google Scholar] [CrossRef]
- Puri, M.; Gatard, S.; Smith, D.A.; Ozerov, O.V. Competition Studies of Oxidative Addition of Aryl Halides to the (PNP)Rh Fragment. Organometallics 2011, 30, 2472–2482. [Google Scholar] [CrossRef]
- Gatard, S.; Guo, C.; Foxman, B.M.; Ozerov, O.V. Thioether, Dinitrogen, and Olefin Complexes of (PNP)Rh: Kinetics and Thermodynamics of Exchange and Oxidative Addition Reactions. Organometallics 2007, 26, 6066–6075. [Google Scholar] [CrossRef]
- Gatard, S.; Celenligil-Cetin, R.; Guo, C.; Foxman, B.M.; Ozerov, O.V. Carbon−Halide Oxidative Addition and Carbon−Carbon Reductive Elimination at a (PNP)Rh Center. J. Am. Chem. Soc. 2006, 128, 2808–2809. [Google Scholar] [CrossRef]
- Timpa, S.D.; Pell, C.J.; Zhou, J.; Ozerov, O.V. Fate of Aryl/Amido Complexes of Rhodium(III) Supported by a POCOP Pincer Ligand: C–N Reductive Elimination, β-Hydrogen Elimination, and Relevance to Catalysis. Organometallics 2014, 33, 5254–5262. [Google Scholar] [CrossRef]
- Timpa, S.D.; Fafard, C.M.; Herberta, D.E.; Ozerov, O.V. Catalysis of Kumada–Tamao–Corriu Coupling by a (POCOP)Rh Pincer Complex. Dalton Trans. 2011, 40, 5426–5429. [Google Scholar] [CrossRef]
- Ito, J.-I.; Miyakawa, T.; Nishiyama, H. Amine-Assisted C−Cl Bond Activation of Aryl Chlorides by a (Phebox)Rh-Chloro Complex. Organometallics 2008, 27, 3312–3315. [Google Scholar] [CrossRef]
- Qian, Y.Y.; Lee, M.H.; Yang, W.; Chan, K.S. Aryl Carbon–Chlorine (Ar–Cl) and Aryl Carbon–Fluorine (Ar–F) Bond Cleavages by Rhodium Porphyrins. J. Organomet. Chem. 2015, 791, 82–89. [Google Scholar] [CrossRef]
- Willems, S.T.H.; Budzelaar, P.H.M.; Moonen, N.N.P.; de Gelder, R.; Smits, J.M.M.; Gal, A.W. Coordination and Oxidative Addition at a Low-Coordinate Rhodium(I) β-Diiminate Centre. Chem. Eur. J. 2002, 8, 1310–1320. [Google Scholar] [CrossRef]
- Yamagata, T.; Tani, K.; Tatsuno, Y.; Saito, T. A New Rhodium Trinuclear Complex Containing Highly Protected Hydroxo Groups, [{Rh(binap)}3(µ3-OH)2]CIO4, Responsible for Deactivation of the 1,3-Hydrogen Migration Catalyst of Allylamine [Binap = 2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl]. J. Chem. Soc. Chem. Commun. 1988, 466–468. [Google Scholar] [CrossRef]
- Preetz, A.; Baumann, W.; Drexler, H.-J.; Fischer, C.; Sun, J.; Spannenberg, A.; Zimmer, O.; Hell, W.; Heller, D. Trinuclear Rhodium Complexes and Their Relevance for Asymmetric Hydrogenation. Chem. Asian. J. 2008, 3, 1979–1982. [Google Scholar] [CrossRef] [PubMed]
- Horstmann, M.; Drexler, H.-J.; Baumann, W.; Heller, D. Ammine and Amido Complexes of Rhodium: Synthesis, Application and Contributions to Analytics. Unpublished work. 2019. [Google Scholar]
- [2-(3-Methoxy-phenyl)-cyclohex-1-enylmethyl]-dimethylamine, which can be transformed by a stereoselective hydrogenation to Tramadol, an important analgesic drug used world-wide, is a member of a class of 1,2-disubstituded cyclohexene derivatives which are important intermediates in the synthesis of pharmaceutically active compounds acting on the central nervous system.
- The hydrogenation product is a key intermediate in the synthesis of 4-amino-2-(R)-methylbutan-1-ol en route towards the receptor antagonist TAK-637.
- Cobley, C.J.; Lennon, I.C.; Praquin, C.; Zanotti-Gerosa, A. Highly Efficient Asymmetric Hydrogenation of 2-Methylenesuccinamic Acid Using a Rh-DuPHOS Catalyst. Org. Process Res. Dev. 2003, 7, 407–411. [Google Scholar] [CrossRef]
- The prochiral olefin (Z)-3-[1-(dimethylamino)-2-methylpent-2-en-3-yl]phenol is an important precursor of the new analgesic drug Tapentadol which is as an open-chain analogue of Tramadol. The (R,R) stereoisomer of Tapentadol is a novel, centrally acting analgesic with a dual mode of action: M-opioid receptor (MOR) agonism and norepinephrine reuptake inhibition.
- Christopfel, W.C.; Vineyard, B.D. Catalytic Asymmetric Hydrogenation With a Rhodium(I) Chiral Bisphosphine System. A Study of Itaconic Acid and Some of its Derivatives and Homologs. J. Am. Chem. Soc. 1979, 101, 4406–4408. [Google Scholar] [CrossRef]
- Schmidt, T.; Drexler, H.-J.; Sun, J.; Dai, Z.; Baumann, W.; Preetz, A.; Heller, D. Unusual Deactivation in the Asymmetric Hydrogenation of Itaconic Acid. Adv. Synth. Catal. 2009, 351, 750–754. [Google Scholar] [CrossRef]
- Schmidt, T.; Baumann, W.; Drexler, H.-J.; Heller, D. Unusual Deactivation in the Asymmetric Hydrogenation of Itaconic Acid. J. Organomet. Chem. 2011, 696, 1760–1767. [Google Scholar] [CrossRef]
- Schmidt, M.; Schreiber, S.; Franz, L.; Langhoff, H.; Farhang, A.; Horstmann, M.; Drexler, H.-J.; Heller, D.; Schwarze, M. Hydrogenation of Itaconic Acid in Micellar Solutions: Catalyst Recycling with Cloud Point Extraction? Ind. Eng. Chem. Res. 2019, 58, 2445–2453. [Google Scholar] [CrossRef]
- Other possibilities are extraction with organic solvents, micellar-enhanced ultrafiltration (MEUF) and phase separation.
- Friedrich, H.B.; Moss, J.R. Halogenoalkyl Complexes of Transition Metals. Adv. Organomet. Chem. 1991, 33, 235–290. [Google Scholar]
- Ball, G.E.; Cullen, W.R.; Fryzuk, M.D.; James, B.R.; Rettig, S.J. Oxidative Addition of Dichloromethane to [(dppe)Rh]2(μ-Cl)2 (dppe = Ph2PCH2CH2PPh2). X-ray structure of [(dppe)RhCl]2(μ-Cl)2(μ-CH2). Organometallics 1991, 10, 3767–3769. [Google Scholar] [CrossRef]
- Fennis, P.J.; Budzelaar, P.H.M.; Frijns, J.H.G. Dichloromethane Addition to Rhodium-β-Diketonate Complexes of Diphosphines and Pyridyl-Substituted Diphosphines. J. Organomet. Chem. 1990, 393, 287–298. [Google Scholar] [CrossRef]
- Marder, T.B.; Fultz, W.C.; Calabrese, J.C.; Harlow, R.L.; Milstein, D. Activation of Dichloromethane by Basic Rhodium(I) and Iridium(I) Phosphine Complexes. Synthesis and Structures of fac-[Rh(PMe3)3Cl2(CH2PMe3)]Cl·CH2Cl2 and trans-[Rh(Me2PCH2CH2PMe2)2Cl(CH2Cl)]Cl. J. Chem. Soc. Chem. Commun. 1987, 1543–1545. [Google Scholar] [CrossRef]
- Blank, B.; Glatz, G.; Kempe, R. Single and Double C-Cl-Activation of Methylene Chloride by P,N-ligand Coordinated Rhodium Complexes. Chem. Asian J. 2009, 4, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Ciriano, M.A.; Tena, M.A.; Oro, A. Reactions of Chloroform and gem-Dichlorocarbons with Binuclear Rhodium Complexes Leading to Functionalized Methylene-Bridged Compounds. J. Chem. Soc. Dalton Trans. 1992, 2123–2124. [Google Scholar] [CrossRef]
- Mannu, A.; Ferro, M.; Möller, S.; Heller, D. Monomerisation of [Rh2(1,3-Bis-(Diphenylphosphino)-Propane)2(μ2-Cl)2] Detected by Pulsed Gradient Spin Echo Spectroscopy and 31P Nmr Monitoring of Metathesis Experiments. J. Chem. Res. 2018, 42, 402–404. [Google Scholar] [CrossRef]
- This reaction has been known for a long time and is used in the synthesis of mononuclear cationic complexes from neutral µ2-chloro brigded precursors by chloride abstraction with a silver salt.
- Anastas, P.T.; Kirchhoff, M.M.; Williamson, T.C. Catalysis as a Foundational Pillar of Green Chemistry. Appl. Catal. A Gen. 2001, 221, 3–13. [Google Scholar] [CrossRef]













































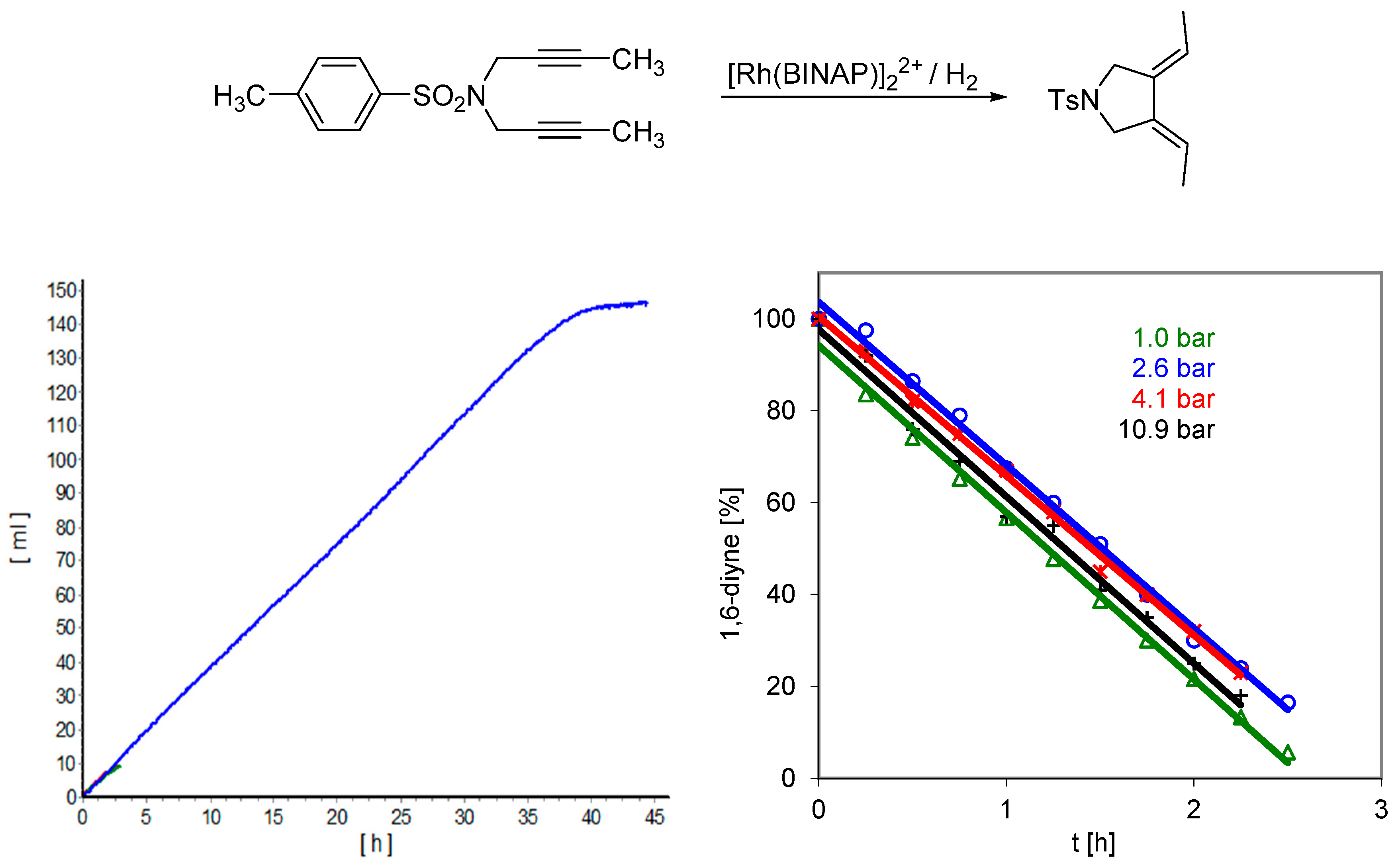
| Ligand | k1 (L·mol−1·s−1) | k2 (L·mol−1·s−1) | k3 (L·mol−1·s−1) | k4 (L·mol−1·s−1) | t98% conv. (min) |
|---|---|---|---|---|---|
| BINAP | 1790 | 18 | 39.3 | 1.10 | 22 |
| SEGPhos | 10,617 | 121 | 16.7 | 1.43 | 3 |
| DM-SEGPhos | 13,466 | 14 | 0.3 | 0.60 | 28 |
| Difluorphos | 8080 | 220 | - | - | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alberico, E.; Möller, S.; Horstmann, M.; Drexler, H.-J.; Heller, D. Activation, Deactivation and Reversibility Phenomena in Homogeneous Catalysis: A Showcase Based on the Chemistry of Rhodium/Phosphine Catalysts. Catalysts 2019, 9, 582. https://doi.org/10.3390/catal9070582
Alberico E, Möller S, Horstmann M, Drexler H-J, Heller D. Activation, Deactivation and Reversibility Phenomena in Homogeneous Catalysis: A Showcase Based on the Chemistry of Rhodium/Phosphine Catalysts. Catalysts. 2019; 9(7):582. https://doi.org/10.3390/catal9070582
Chicago/Turabian StyleAlberico, Elisabetta, Saskia Möller, Moritz Horstmann, Hans-Joachim Drexler, and Detlef Heller. 2019. "Activation, Deactivation and Reversibility Phenomena in Homogeneous Catalysis: A Showcase Based on the Chemistry of Rhodium/Phosphine Catalysts" Catalysts 9, no. 7: 582. https://doi.org/10.3390/catal9070582