
Hydrogen Bonding in Crystals of Pyrrol-2-yl Chloromethyl Ketone Derivatives and Methyl Pyrrole-2-Carboxylate
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
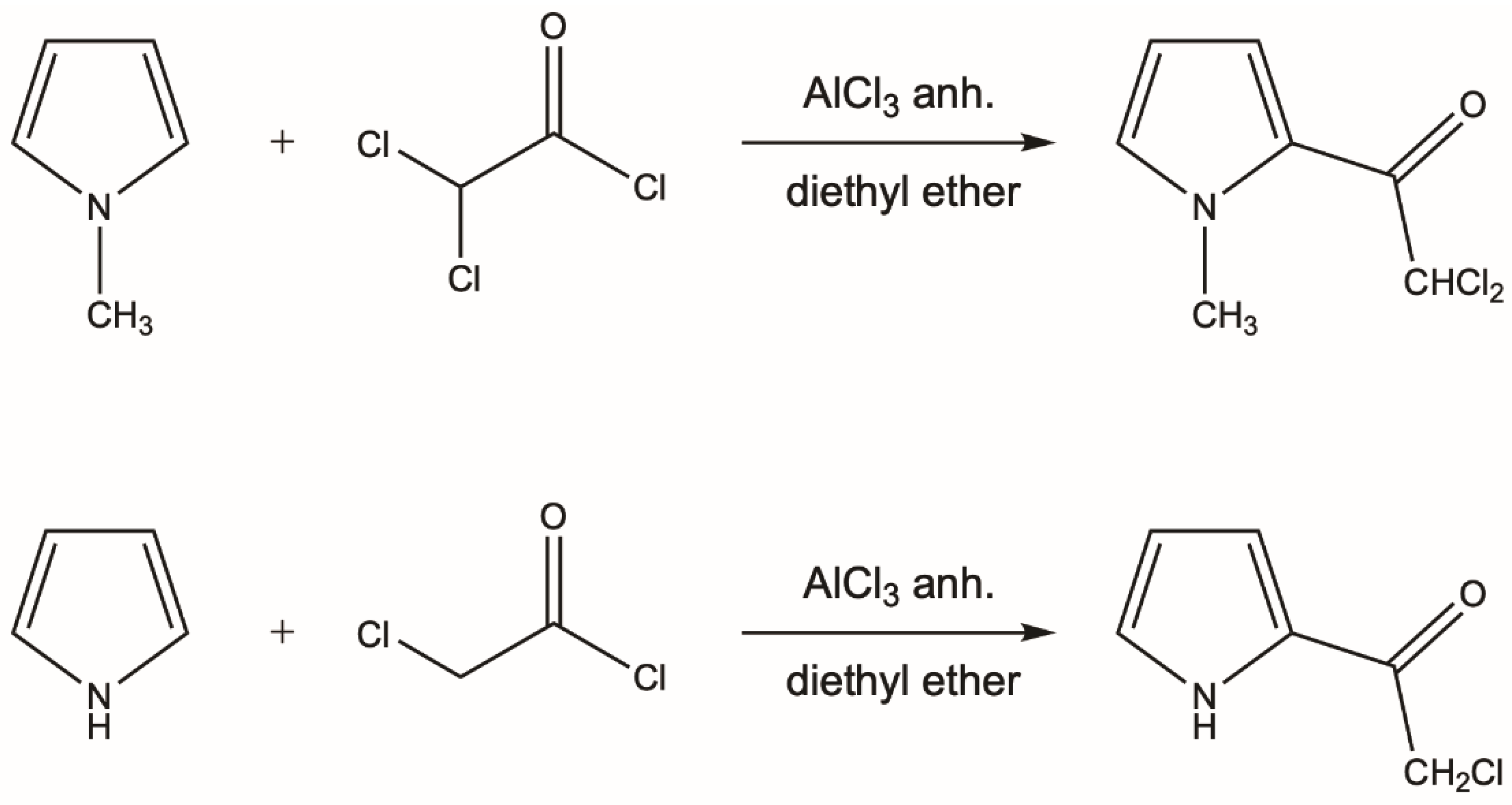
2.1. Synthesis of Pyrrol-2-yl Chloromethyl Ketones
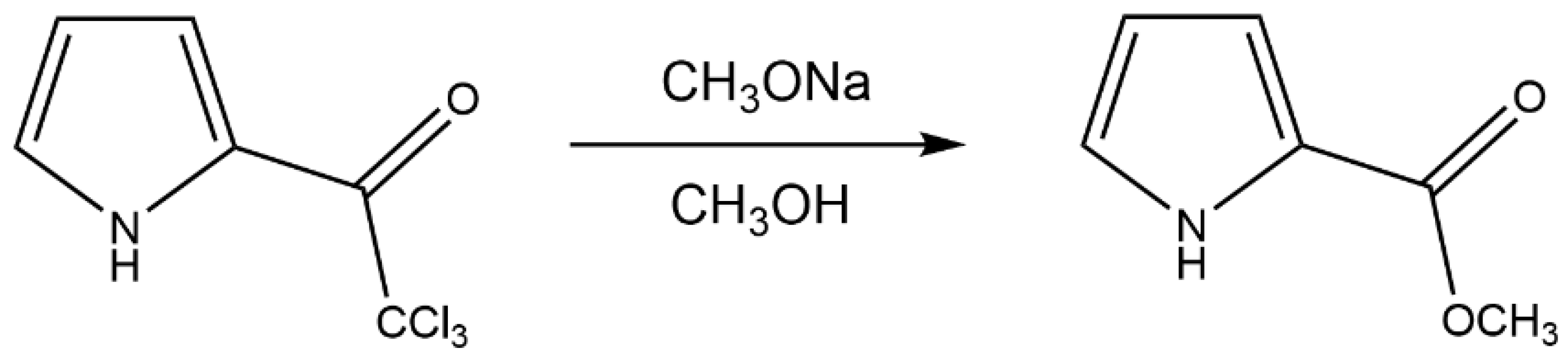
2.2. Synthesis of Methyl Pyrrole-2-Carboxylate
2.3. X-ray Diffraction Analysis
2.4. Hirshfeld Surface Analysis
2.5. Full Interaction Maps Analysis
2.6. Theoretical Calculations
3. Results and Discussion
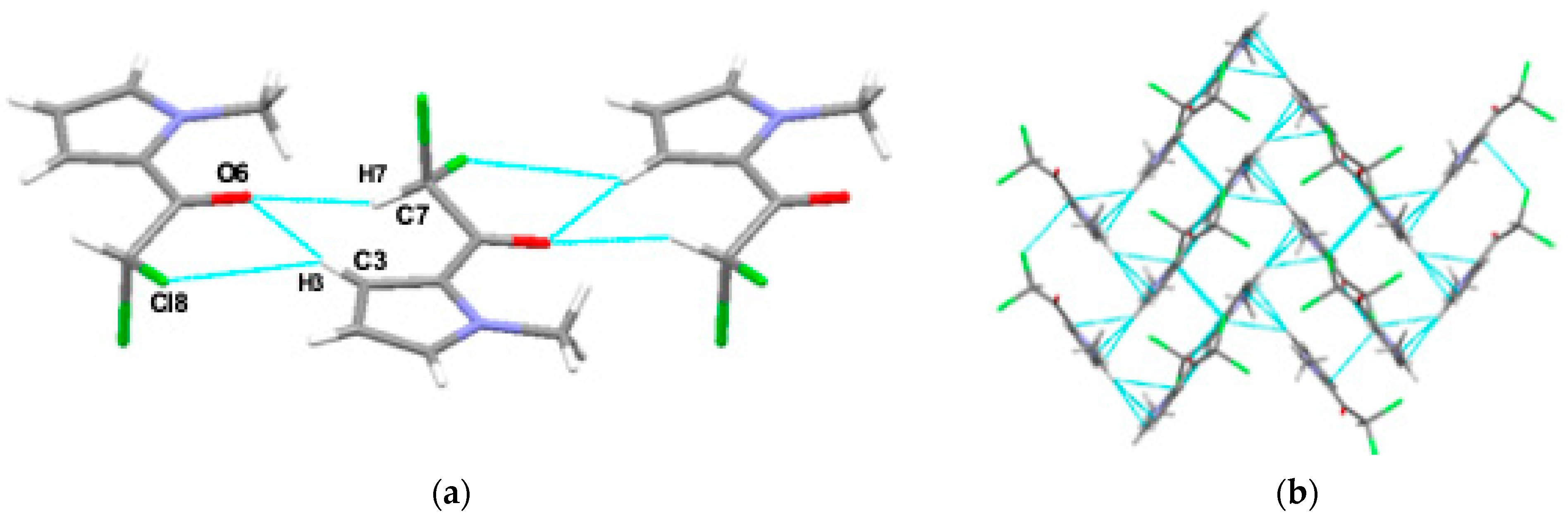
3.1. XRD Analysis
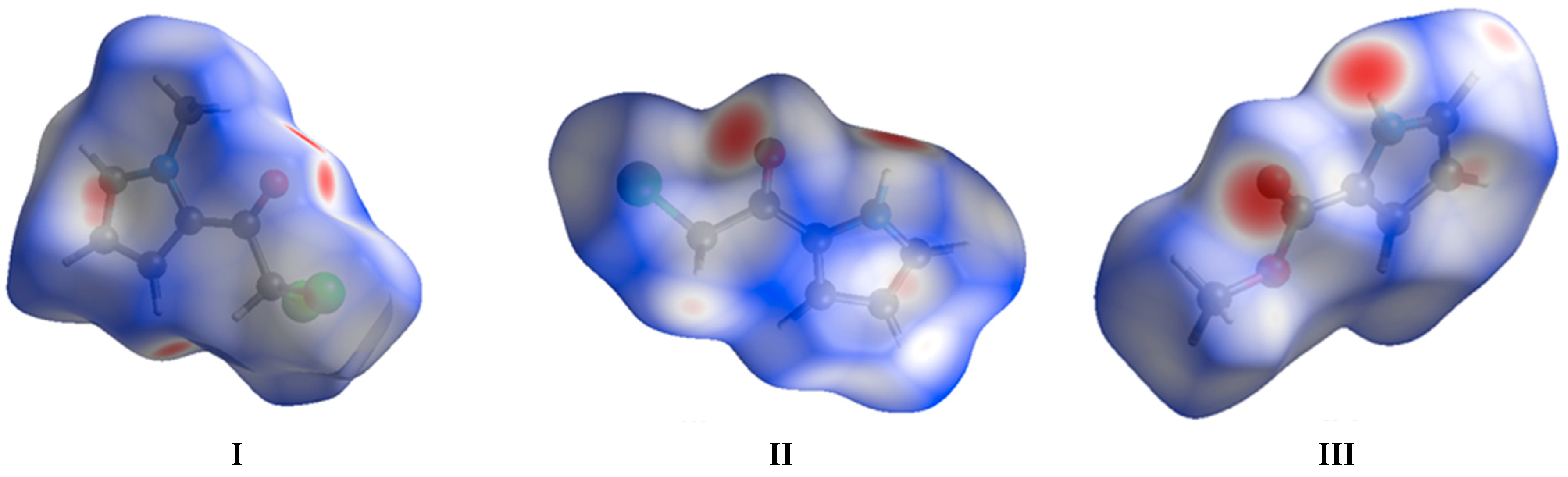
3.2. Theoretical Calculations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D-H [Å] | H⋯A [Å] | <DHA[°] | EintCP [kcal/mol] | M.p. [°] | ||
|---|---|---|---|---|---|---|
| I | C7-H7⋯O6 | 1.098 | 2.222 | 126 | −7.62 | 65.8–66.5 [60] |
| C3-H3⋯Cl8 | 1.083 | 2.843 | 152 | |||
| II | N1-H1⋯O6 | 1.030 | 1.919 | 163 | −11.02 | 120.5 [60] |
| N1-H1⋯Cl8 | 1.030 | 2.828 | 124 | |||
| III | N1-H1⋯O6 | 1.030 | 1.917 | 170 | −8.36 | 72 |
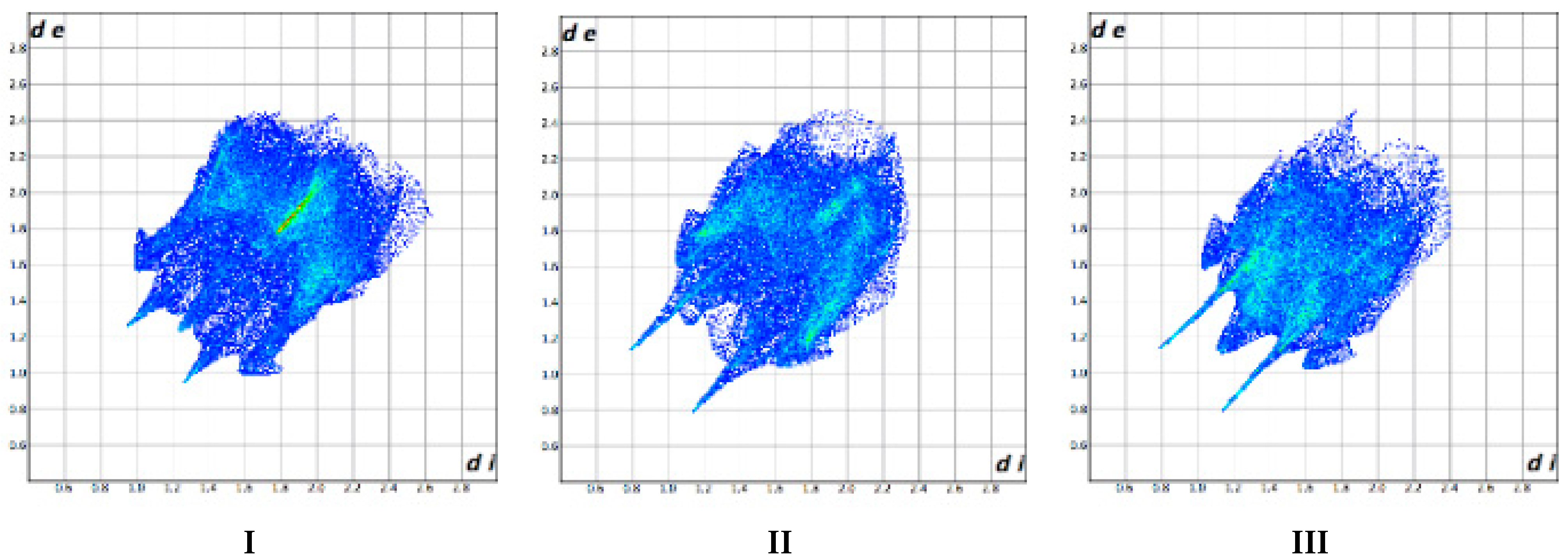
3.3. Hirshfeld Surface Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khoury, R.G.; Jaquinod, L.; Nguyen, L.T.; Smith, K.M. Macrocycles containing five pyrrole subunits: The iso-oxopentaphyrin system. Heterocycles 1998, 47, 113–119. [Google Scholar] [CrossRef]
- Tamiaki, H.; Hamada, K.; Kunieda, M. Synthesis of 3/8-carbonylated chlorophyll derivatives and regiodependent reductivity of their carbonyl substituents. Tetrahedron 2008, 64, 5721–5727. [Google Scholar] [CrossRef]
- Rowan, D.D.; Gaynor, D.L. Isolation of feeding deterrents against argentine stem weevil from ryegrass infected with the endophyte Acremonium loliae. J. Chem. Ecol. 1986, 12, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Dubis, E.N.; Brattsten, L.B.; Dungan, L.B. Effects of the endophyte-associated alkaloids peramine on Southern Armworm Microsomal Cytochrome P450. In ACS Symposium Series Molecular mechanism of Insecticide Resistance Diversity among Insects; ACS: Washington, DC, USA, 1992. [Google Scholar]
- Dubis, A.T.; Łapiński, A. Spectroscopic and theoretical study on peramine and some pyrrolopyrazinone compounds. Vib. Spec. 2009, 49, 265–273. [Google Scholar] [CrossRef]
- Łapiński, A.; Dubis, A.T. A DFT/TD-DFT study for the ground and excited states of peramine and some pyrrolopyrazinone compounds. J. Phys. Org. Chem. 2009, 22, 1058–1064. [Google Scholar] [CrossRef] [Green Version]
- Bellina, F.; Rossi, R. Synthesis and biological activity of pyrrole, pyrroline and pyrrolidine derivatives with two aryl groups on adjacent positions. Tetrahedron 2006, 62, 7213–7256. [Google Scholar] [CrossRef]
- Snavely, D.L.; Blackburn, F.R.; Ranasinghe, Y.; Walters, V.A.; Gonzalez del Riego, M. Vibrational overtone spectroscopy of pyrrole and pyrrolidine. J. Phys. Chem. 1992, 96, 3599–3605. [Google Scholar] [CrossRef] [Green Version]
- Lukeš, V.; Breza, M.; Biskupič, S. Interaction energy anisotropy of the pyrrole dimer: Ab initio theoretical study. Theor. Chem. Acc. 1999, 101, 319–324. [Google Scholar] [CrossRef]
- Dunbar, R.C. Binding of Na+, Mg+, and Al+ to the π Faces of Naphthalene and Indole: Ab Initio Mapping Study. J. Phys. Chem. A 1998, 102, 8946–8952. [Google Scholar] [CrossRef]
- Pullman, A.; Berthier, G.; Savinelli, R. Theoretical study of binding of tetramethylammonium ion with aromatics. J. Comput. Chem. 1997, 18, 2012–2022. [Google Scholar] [CrossRef]
- Pullman, A.; Berthier, G.; Savinelli, R. Interaction of the tetramethylammonium ion with the cycles of aromatic amino acids beyond the SCF ab initio level. J. Am. Chem. Soc. 1998, 120, 8553–8554. [Google Scholar] [CrossRef]
- Farnier, M.; Drakenberg, T. Nuclear magnetic resonance conformational studies of C-substituted pyrrolecarbaldehydes. Part, I. Substituent effects on aldehyde conformations as shown by long range coupling constants. J. Chem. Soc. Perkin Trans. 1975, 2, 333–337. [Google Scholar] [CrossRef]
- John, I.G.; Ritchie, G.L.D.; Radom, L. Conformations of Furan-, Pyrrole-, and Pyridine-carbaldehydes: An Ab Initio Molecular Orbital Study. J. Chem. Soc. Perkin Trans. 1977, 2, 1601–1607. [Google Scholar] [CrossRef]
- Farnier, M.; Drakenberg, T. Nuclear magnetic resonance conformational studies of C-substituted pyrrolecarbaldehydes. Part II. Barrier to internal rotation in 5-substituted pyrrole-2-carbaldehydes. J. Chem. Soc. Perkin Trans. 1975, 2, 337–340. [Google Scholar] [CrossRef]
- Dubis, A.T.; Grabowski, S.J. Infrared spectroscopic and theoretical ab initio studies on conformational isomers of methyl pyrrole-2-carboxylate. J. Mol. Struct. 2001, 562, 107–117. [Google Scholar] [CrossRef]
- Dubis, A.T.; Grabowski, S.J. Spectroscopic and theoretical studies on the monomeric and dimeric forms of methyl pyrrole-2-carboxylate. New J. Chem. 2002, 26, 165–169. [Google Scholar] [CrossRef]
- Dubis, A.T.; Grabowski, S.J. Infrared, Density-Functional Theory, and Atoms in Molecules Method Studies on Conformers of Some 2-Substituted 1H-Pyrroles. J. Phys. Chem. A 2003, 107, 8723–8729. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Dubis, A.T.; Martynowski, D.; Glówka, M.; Palusiak, M.; Leszczynski’, J. Crystal and molecular structure of pyrrole-2-carboxylic acid; pi-Electron delocalization of its dimers-DFT and MP2 calculations. J. Phys. Chem. A 2004, 108, 5815–5822. [Google Scholar] [CrossRef]
- Balaban, A.T.; Oniciu, D.C.; Katritzky, A.R. Aromaticity as a cornerstone of heterocyclic chemistry. Chem. Rev. 2004, 104, 2777–2812. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. How Aromaticity Affects the Chemical and Physicochemical Properties of Heterocycles: A Computational approach in Aromaticity in Heterocyclic Compounds. In Topics in Heterocyclic Chemistry; Krygowski, T.M., Cyrański, M.K., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 19, pp. 155–202. [Google Scholar]
- Dubis, A.T.; Wojtulewski, S.; Filipkowski, K. Spectroscopic and theoretical studies on the aromaticity of pyrrol-2-yl-carbonylconformers. J. Mol. Struct. 2013, 1041, 92–99. [Google Scholar] [CrossRef]
- Desiraju, R.; Steiner, T. The Weak Hydrogen Bond. In Structural Chemistry and Biology; Oxford University Press, Inc.: New York, NY, USA, 1999. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: The Design of Organic Solids; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar] [CrossRef] [Green Version]
- Price, S.L.; Stone, A.J.; Lucas, J.; Rowland, R.S.; Thornley, A.E. The nature of -Cl⋯Cl- intermolecular interactions. J. Am. Chem. Soc. 1994, 116, 4910–4918. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Day, G.M.; Price, S.L. A Nonempirical Anisotropic Atom-Atom Model Potential for Chlorobenzene Crystals. J. Am. Chem. Soc. 2003, 125, 16434–16443. [Google Scholar] [CrossRef] [PubMed]
- Formigué, M.; Batail, P. Activation of hydrogen- and halogen-bonding interactions in tetrathiafulvalene-based crystalline molecular conductors. Chem. Rev. 2004, 104, 5379–5418. [Google Scholar] [CrossRef]
- Zordan, F.; Brammer, L.; Sherwood, P. Supramolecular chemistry of halogens: Complementary features of inorganic (M-X) and organic (C-X′) halogens applied to M-X⋯X′-C halogen bond formation. J. Am. Chem. Soc. 2005, 127, 5979–5989. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Legon, A.C. The halogen bond: An interim perspective. Phys. Chem. Chem. Phys. 2010, 12, 7736–7747. [Google Scholar] [CrossRef]
- Bilewicz, E.; Rybarczyk-Pirek, A.; Dubis, A.T.; Grabowski, S.J. Halogen bonding in crystal structure of 1-methylpyrrol-2-yl trichloromethyl ketone. J. Mol. Struct. 2007, 829, 208–211. [Google Scholar] [CrossRef]
- Dubis, A.; Grabowski, S.J.; Romanowska, D.B.; Misiaszek, T.; Leszczynski, J. Pyrrole-2-carboxylic Acid and Its Dimers: Molecular Structures and Vibrational Spectrum. J. Phys. Chem. A 2002, 106, 10613–10621. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C.; Bernstein, J.B.; McDonald, J.C. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. Sect. B Struct. Sci. 1990, 46, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.F.L.O.M.; Riberio da Silva, M.A.V. Experimental and Computational Study on the Molecular Energetics of 2-Pyrrolecarboxylic Acid and 1-Methyl-2-pyrrolecarboxylic Acid. J. Phys. Chem. A 2009, 113, 9741–9750. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Nicolau, I.; Demopoulus, V.J. A study of the friedel-crafts acylation of 1-benzenesulfonyl-1H-pyrrole in the preparation of 3-aroylpyrroles. J. Heterocycl. Chem. 1998, 35, 1345–1348. [Google Scholar] [CrossRef]
- Dubis, A.T.; Domagała, M.; Grabowski, S.J. Spectroscopic and theoretical studies on some new pyrrol-2-yl-chloromethyl ketones. New J. Chem. 2010, 34, 556–566. [Google Scholar] [CrossRef]
- Bailey, D.M.; Johnson, R.E.; Albertson, N.F. Ethyl pyrrole-2-carboxylate. Org. Synth. 1971, 51, 100–104. [Google Scholar] [CrossRef]
- STOE IPDS-Software, Version 2.89; STOE & CIE GmbH: Darmstadt, Germany, 1998.
- North, A.C.T.; Phillips, D.C.; Mathews, F.S. A semi-empirical method of absorption correction. Acta Cryst. A 1968, 24, 351–359. [Google Scholar] [CrossRef]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Cryst. A 1995, 51, 887–897. [Google Scholar] [CrossRef] [Green Version]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. Structure Validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III.† The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Domagała, M.; Lutyńska, A.; Palusiak, M. Extremely Strong Halogen Bond. The Case of a Double-Charge-Assisted Halogen Bridge. J. Phys. Chem. A 2018, 122, 5484–5492. [Google Scholar] [CrossRef]
- Domagała, M.; Matczak, P.; Palusiak, M. Halogen bond, hydrogen bond and N⋯C interaction—On interrelation among these three noncovalent interactions. Comput. Theor. Chem. 2012, 998, 26–33. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X—H bond lengths from neutron diffraction data. Acta Crystallogr. B 2010, 66, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Dubis, A.T.; Palusiak, M.; Leszczyński, J. Heteronuclear Intermolecular Resonance-Assisted Hydrogen Bonds. The Structure of Pyrrole-2-Carboxamide (PyCa). J. Phys. Chem. B 2006, 110, 5875–5882. [Google Scholar] [CrossRef]
- Kerscher, T.; Mayer, P.; Klufers, P. Methyl 1H-pyrrole-2-carboxylate. Acta Cystallogr. Sect. E Struct. Rep. Online 2009, 65, o2195. [Google Scholar] [CrossRef] [Green Version]
- Essa, A.H.; Lerrick, R.I.; Ciftci, E.; Harrington, R.W.; Waddell, P.G.; Clegg, W.; Hall, M.J. Grignard-mediated reduction of 2,2,2-trichloro1-arylethanones. Org. Biomol. Chem. 2015, 13, 5793. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. Diamond—Crystal and Molecular Structure Visualization. Version 4.6.4, Bonn, Germany. Available online: https://www.crystalimpact.de/diamond (accessed on 15 December 2020).
- Grimme, S. Semi-empirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Scheiner, S. Weak H-bonds. Comparisons of CH⋯O to NH⋯O in proteins and PH⋯N to direct P⋯N interactions. Phys. Chem. Chem. Phys. 2011, 13, 13860–13872. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domagała, M.; Dubis, A.T.; Wojtulewski, S.; Zabel, M.; Pfitzner, A. Hydrogen Bonding in Crystals of Pyrrol-2-yl Chloromethyl Ketone Derivatives and Methyl Pyrrole-2-Carboxylate. Crystals 2022, 12, 1523. https://doi.org/10.3390/cryst12111523
Domagała M, Dubis AT, Wojtulewski S, Zabel M, Pfitzner A. Hydrogen Bonding in Crystals of Pyrrol-2-yl Chloromethyl Ketone Derivatives and Methyl Pyrrole-2-Carboxylate. Crystals. 2022; 12(11):1523. https://doi.org/10.3390/cryst12111523
Chicago/Turabian StyleDomagała, Małgorzata, Alina T. Dubis, Sławomir Wojtulewski, Manfred Zabel, and Arno Pfitzner. 2022. "Hydrogen Bonding in Crystals of Pyrrol-2-yl Chloromethyl Ketone Derivatives and Methyl Pyrrole-2-Carboxylate" Crystals 12, no. 11: 1523. https://doi.org/10.3390/cryst12111523