

Hydrogen-Bonded Chain of Rings Motif in N-(4-Methoxyphenyl)piperazin-1-ium Salts with Benzoate Anions: Supramolecular Assemblies and Their Energy Frameworks

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
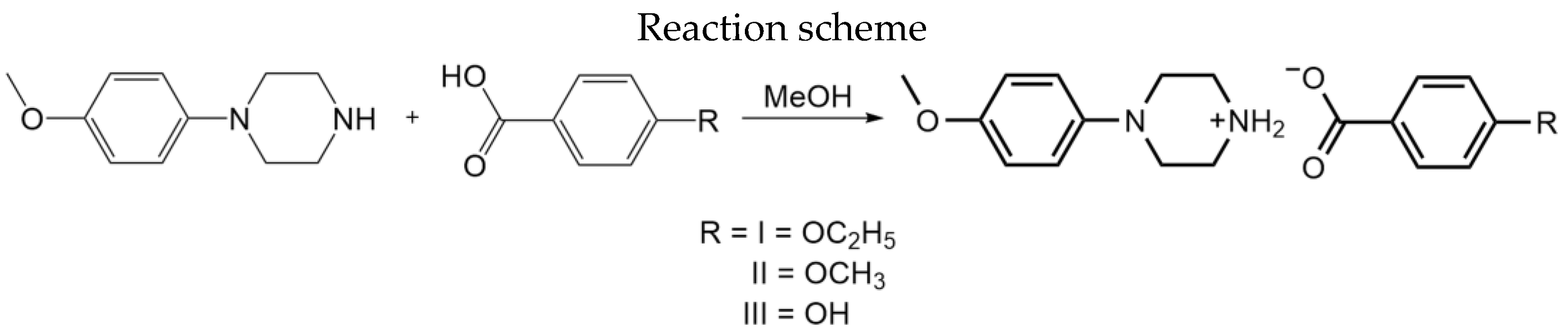
2.1. Synthesis
2.2. X-ray Single-Crystal Diffraction
2.3. Hirshfeld Surface Analysis and Energy Frameworks
2.4. Theoretical Calculations
3. Results and Discussion
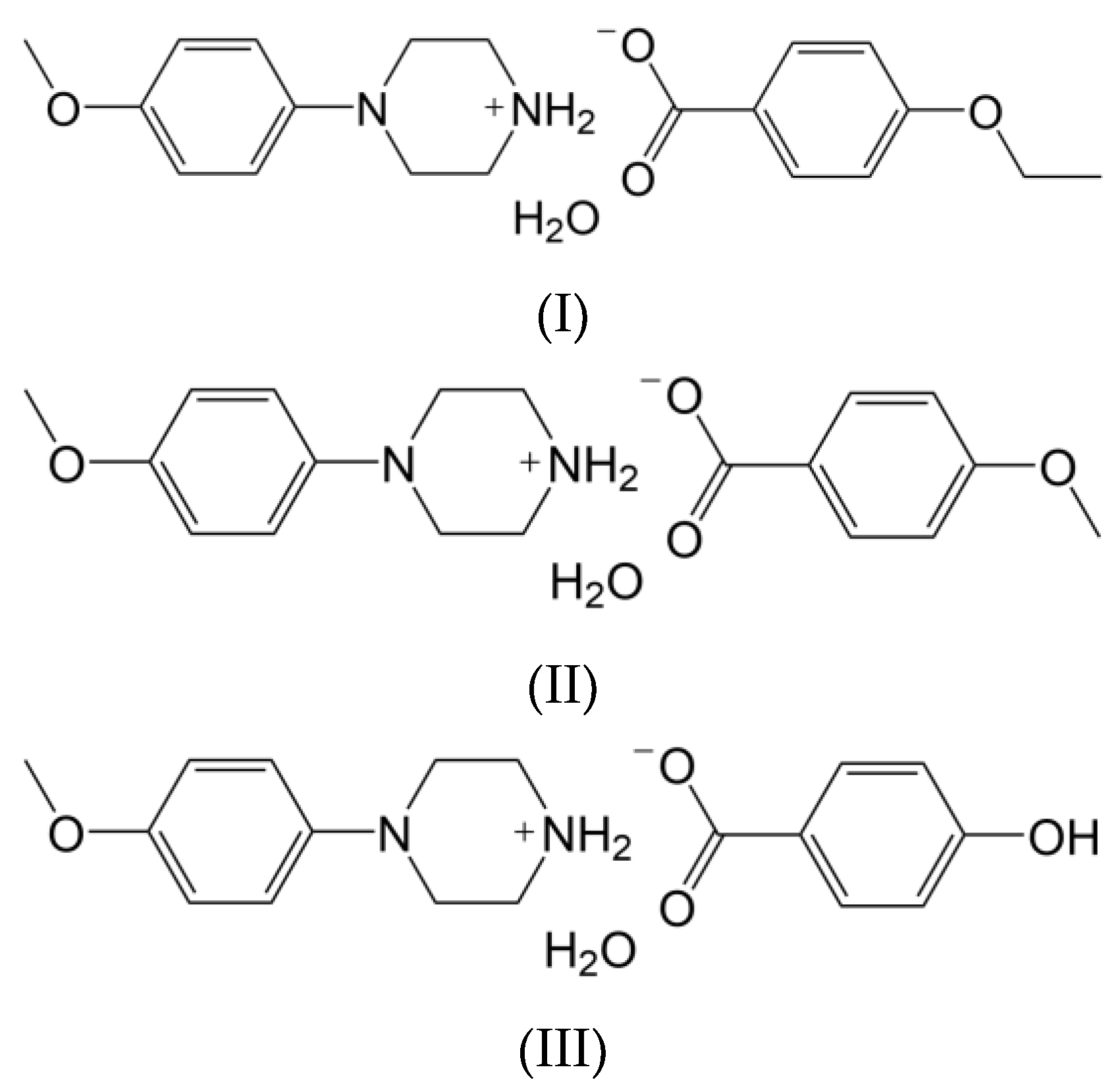
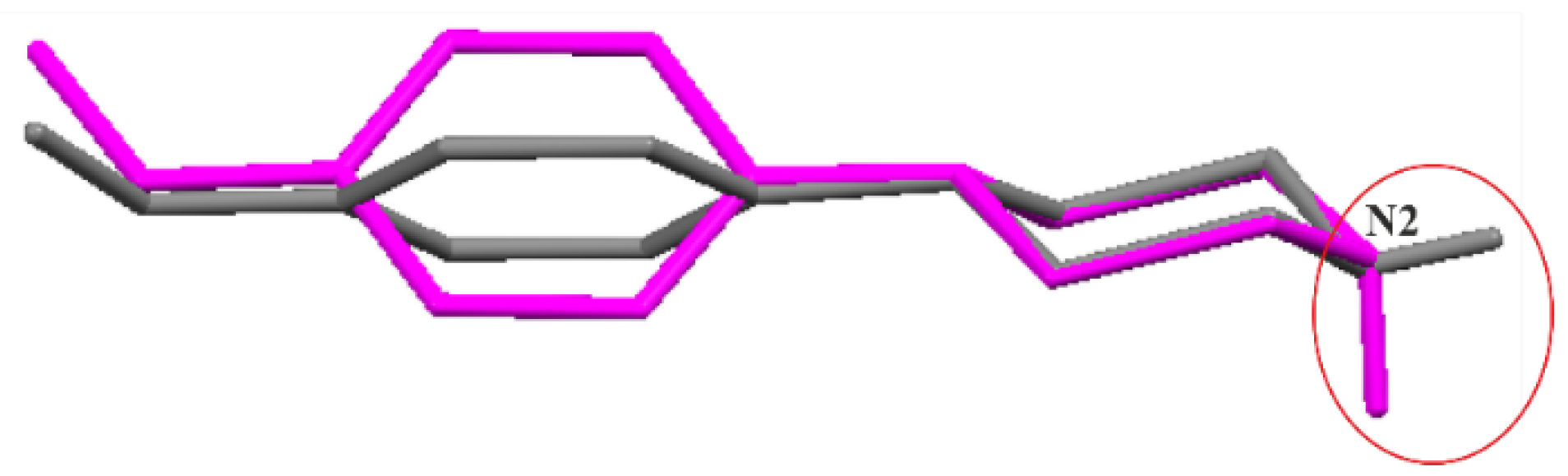
3.1. Molecular Structures
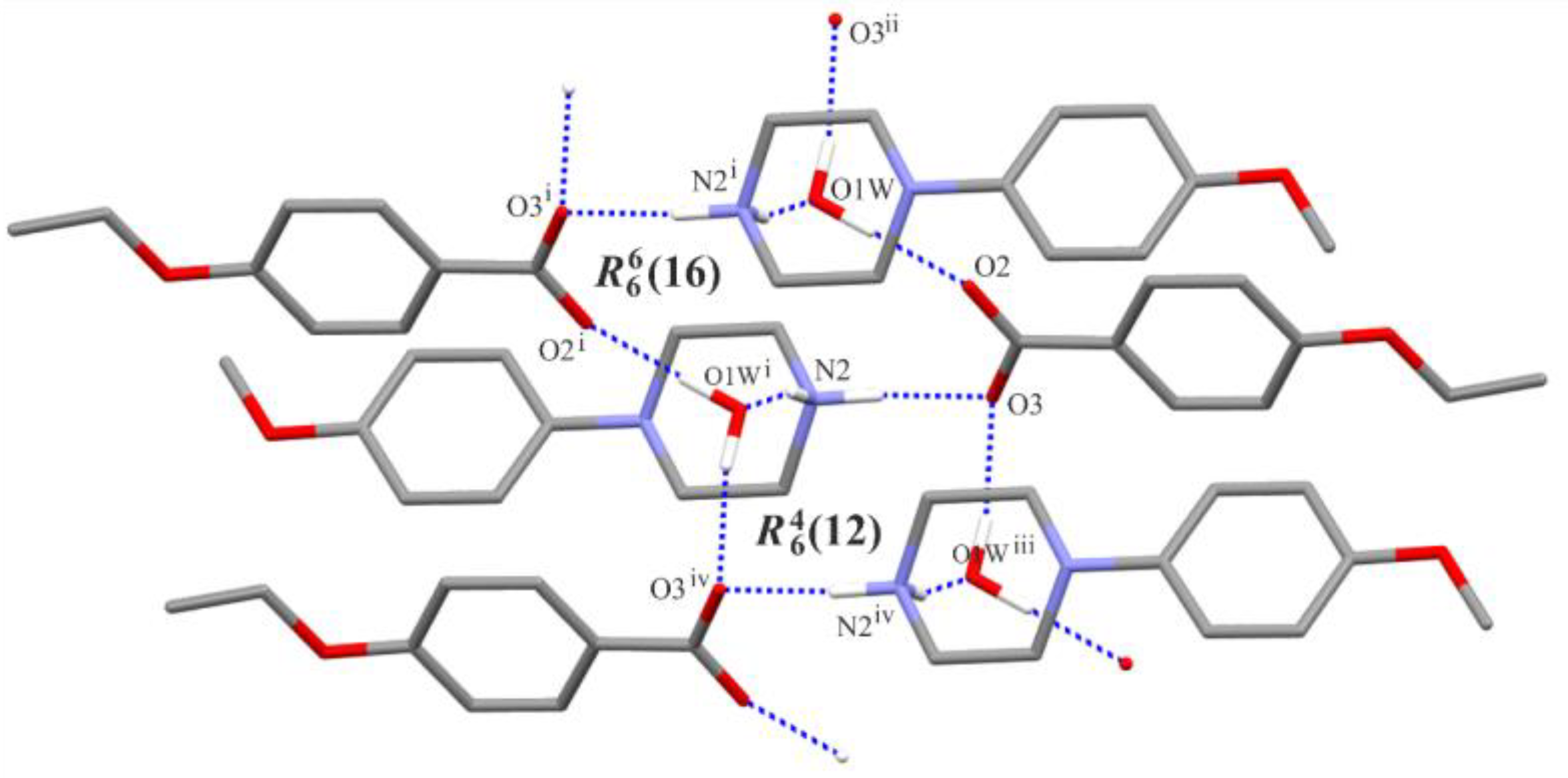
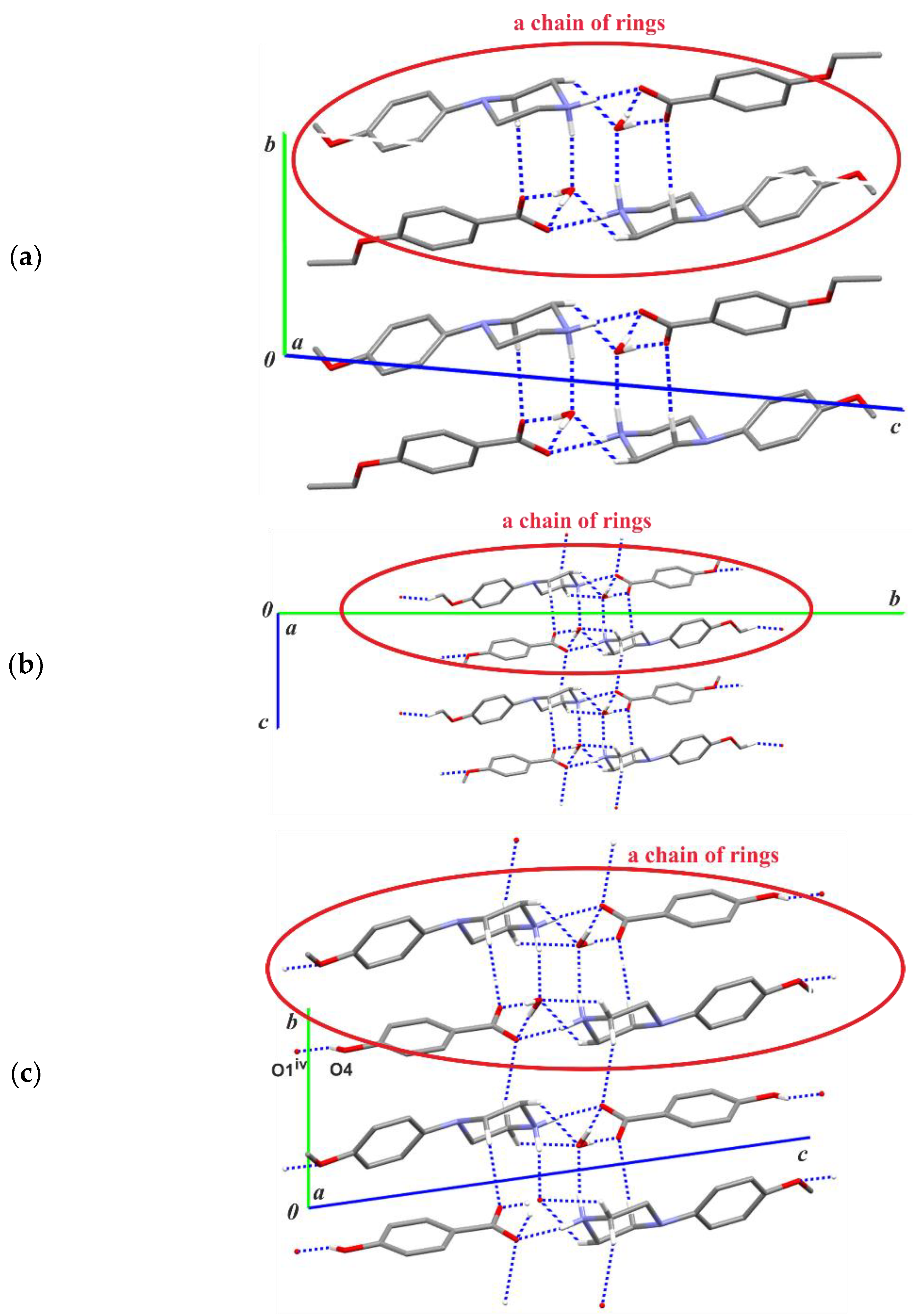
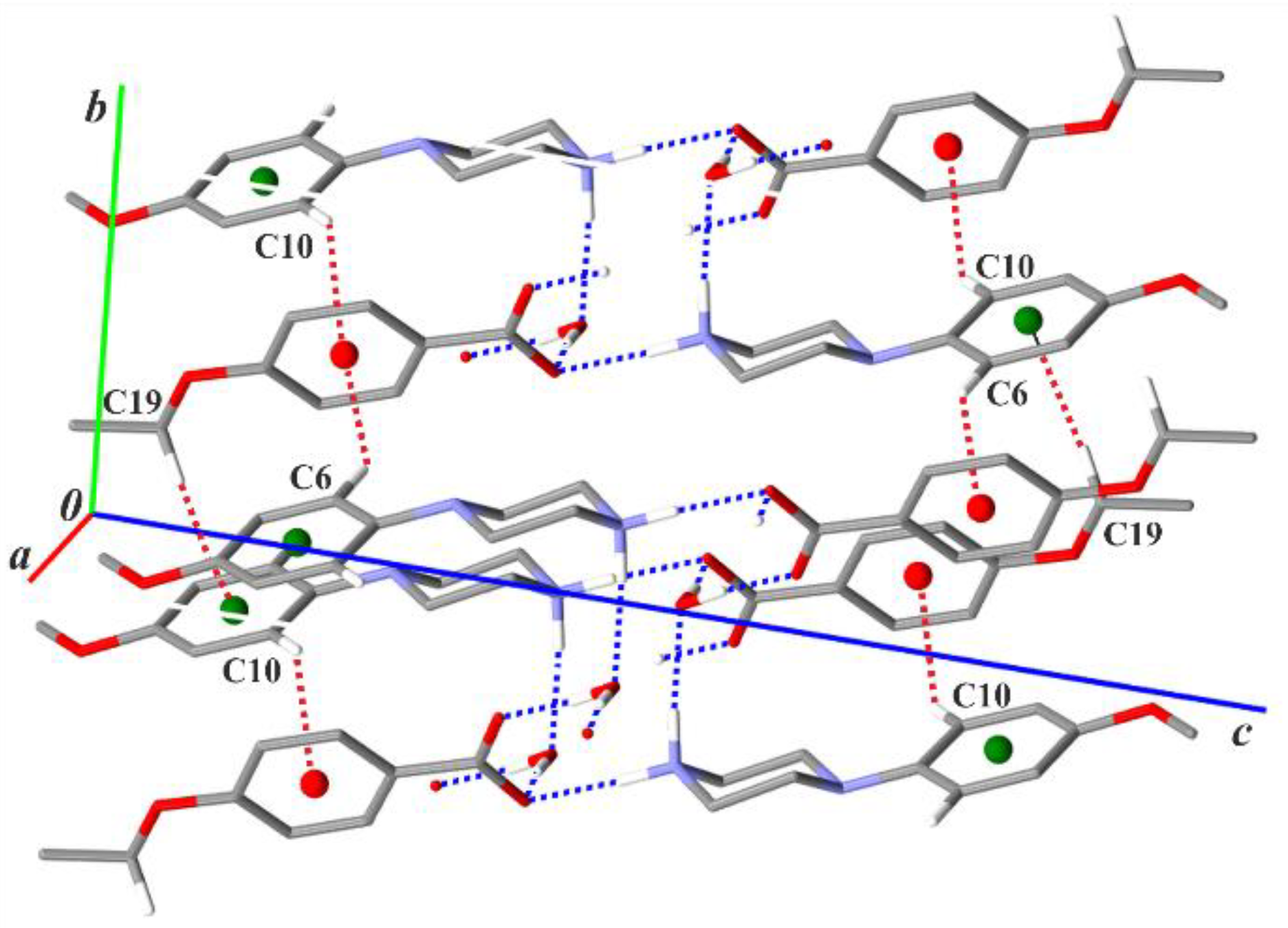
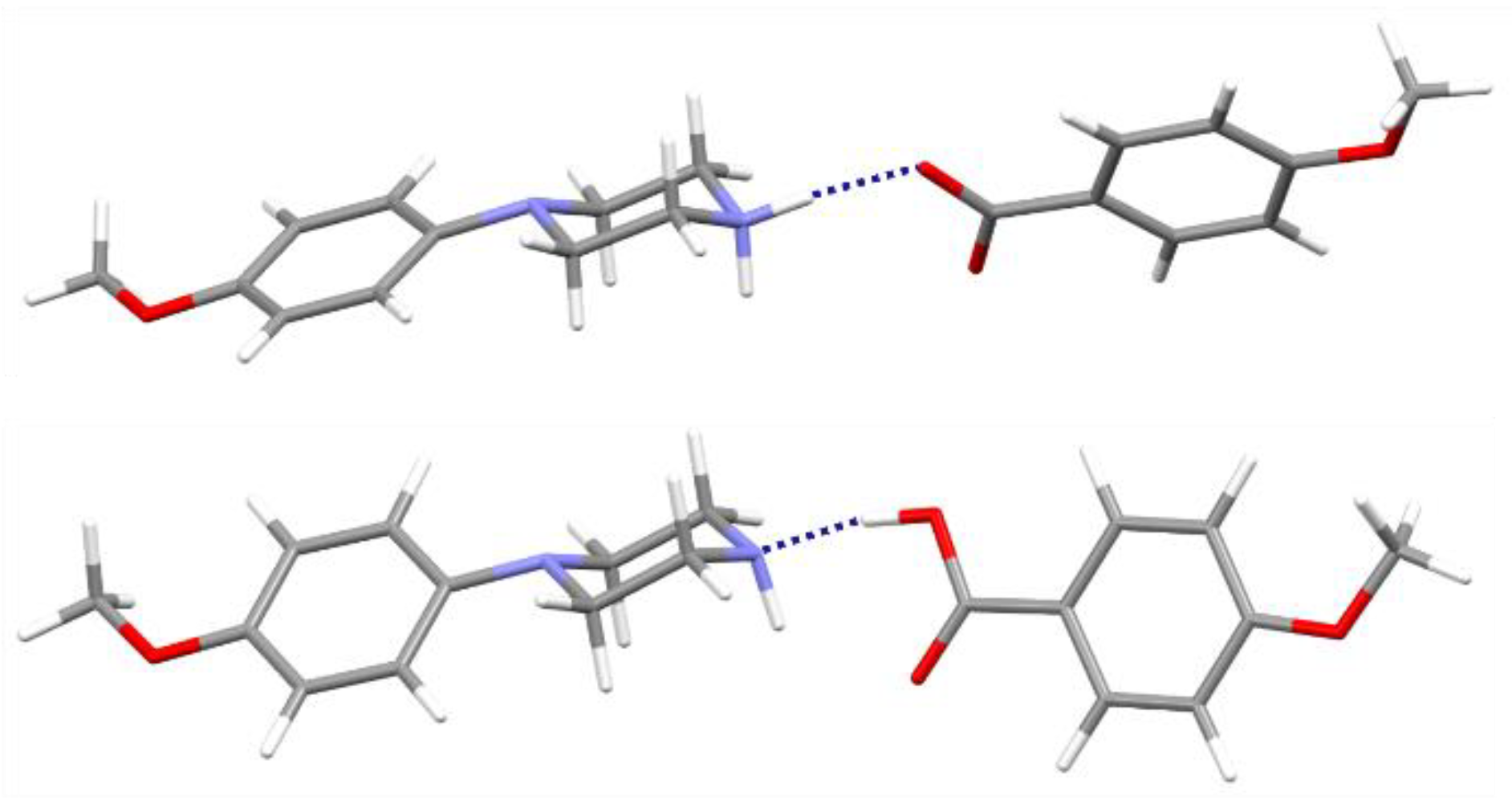
3.2. Supramolecular Features
3.3. Theoretical Considerations
3.4. Hirshfeld Surface Analysis
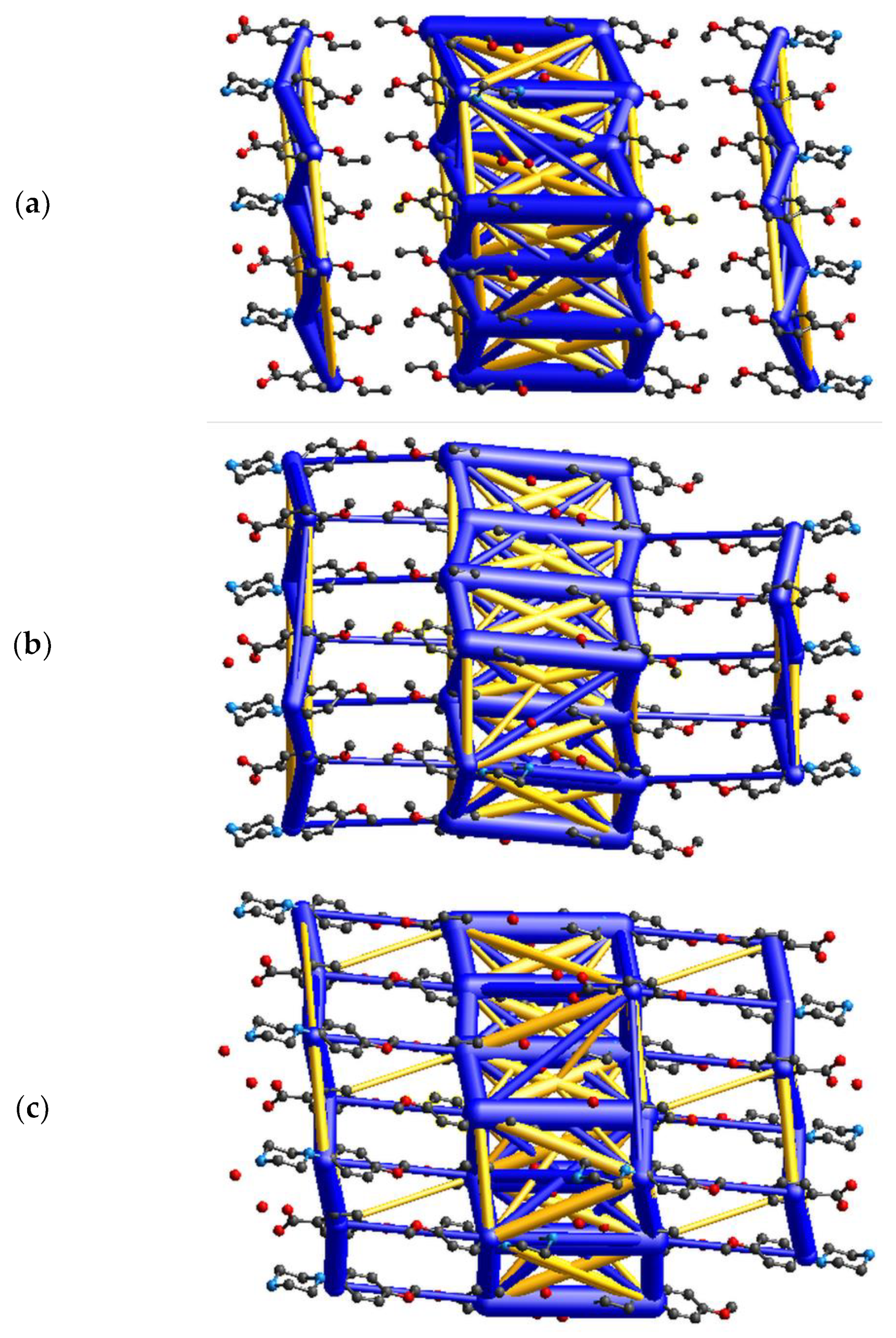
3.5. Energy Frameworks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brito, A.F.; Moreira, L.K.S.; Menegatti, R.; Costa, E.A. Piperazine derivatives with central pharmacological activity used as therapeutic tools. Fundam. Clin. Pharmacol. 2019, 33, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghorbani, M.; Bushra, B.A.; Zabiulla, S.; Mamatha, S.V.; Khanum, S.A. Piperazine and morpholine: Synthetic preview and pharmaceutical applications. J. Chem. Pharm. Res. 2015, 7, 281–301. [Google Scholar] [CrossRef]
- Asif, M. Piperazine and pyrazine containing molecules and their diverse pharmacological activities. Int. J. Adv. Sci. Res. 2015, 1, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.V.; Park, S.W. An evolving role of piperazine moieties in drug design and discovery. Mini-Rev. Med. Chem. 2013, 13, 1579–1601. [Google Scholar] [CrossRef]
- Chaudhary, P.; Kumar, R.; Verma, A.K.; Singh, D.; Yadav, V.; Chhillar, A.K.; Sharma, G.L.; Chandra, R. Synthesis and antimicrobial activity of N-alkyl and N-aryl piperazine derivatives. Bioorg. Med. Chem. 2006, 14, 1819–1826. [Google Scholar] [CrossRef]
- Jin, Y.; Huang, T.; Zhao, W.; Yang, X.; Meng, Y.; Ma, P. A study on the self-assembly mode and supramolecular framework of complexes of cucurbit[6]urils and 1-(4-methoxyphenyl)piperazine. RSC Adv. 2020, 10, 37369–37373. [Google Scholar] [CrossRef]
- Zhang, Y.; Chao, J.; Zhao, S.; Xu, P.; Wang, H.; Guo, Z.; Liu, D. Investigation on the inclusion interaction of 4-sulfonatocalix[n]arenes with 1-(4-nitrophenyl)piperazine. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 132, 44–51. [Google Scholar] [CrossRef]
- König, O.; Bürgi, H.-B.; Armbruster, T.; Hulliger, J.; Weber, T. A Study in crystal engineering: Structure, crystal growth, and physical properties of a polar perhydrotriphenylene inclusion compound. J. Am. Chem. Soc. 1997, 119, 10632–10640. [Google Scholar] [CrossRef]
- Gharbi, C.; Toumi, B.; Soudani, S.; Lefebvre, F.; Kaminsky, W.; Jelsch, C.; Nasr, C.B.; Khedhiri, L. Synthesis, structural characterization, antibacterial activity, DFT computational studies and thermal analysis of two new thiocyanate compounds based on 1-phenylpiperazine. J. Mol. Struct. 2022, 1257, 132620. [Google Scholar] [CrossRef]
- Bhati, S.; Kumar, V.; Singh, S.; Singh, J. Synthesis, biological activities and docking studies of piperazine incorporated 1,3,4-oxadiazole derivatives. J. Mol. Struct. 2019, 1191, 197–205. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Sinha, N.; Jain, S.; Kishore, N.; Chandra, R.; Arora, S.K. Optically active antifungal azoles: Synthesis and antifungal activity of (2R,3S)-2-(2,4- difluorophenyl)-3-(5-{2-[4-aryl-piperazin-1-yl]-ethyl}-tetrazol-2-yl/1-yl)-1-[1,2,4]-triazol-1-yl-butan-2-ol. Bioorg. Med. Chem. 2004, 12, 2225–2238. [Google Scholar] [CrossRef]
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef]
- Perez, M.; Fourrier, C.; Sigogneau, I.; Pauwels, P.J.; Palmier, C.; John, G.W.; Valentin, J.-P.; Halazy, S. Synthesis and serotonergic activity of arylpiperazide derivatives of serotonin: Potent agonists for 5-HT1D receptors. J. Med. Chem. 1995, 38, 3602–3607. [Google Scholar] [CrossRef]
- Arbo, M.D.; Bastos, M.L.; Carmo, H.F. Piperazine compounds as drugs of abuse. Drug Alcohol Depend. 2012, 122, 174–185. [Google Scholar] [CrossRef]
- Staack, R.F.; Maurer, H.H. Toxicological detection of the new designer drug 1-(4-methoxyphenyl)piperazine and its metabolites in urine and differentiation from an intake of structurally related medicaments using gas chromatography–mass spectrometry. J. Chromatogr. B 2003, 798, 333–342. [Google Scholar] [CrossRef]
- Staack, R.F.; Theobald, D.S.; Paul, D.; Springer, D.; Kraemer, T.; Maurer, H.H. In vivo metabolism of the new designer drug 1-(4-methoxyphenyl)piperazine (MeOPP) in rat and identification of the human cytochrome P450 enzymes responsible for the major metabolic step. Xenobiotica 2004, 34, 179–192. [Google Scholar] [CrossRef]
- Staack, R.F.; Maurer, H.H. Metabolism of designer drugs of abuse. Curr. Drug Metab. 2005, 6, 259–274. [Google Scholar] [CrossRef]
- Wohlfarth, A.; Weinmann, W.; Dresen, S. LC-MS/MS screening method for designer amphetamines, tryptamines, and piperazines in serum. Anal. Bioanal. Chem. 2010, 396, 2403–2414. [Google Scholar] [CrossRef]
- Nagai, F.; Nonaka, R.; Kamimura, K.S.H. The effects of non-medically used psychoactive drugs on monoamine neurotransmission in rat brain. Eur. J. Pharmacol. 2007, 559, 132–137. [Google Scholar] [CrossRef]
- Kiran Kumar, H.; Yathirajan, H.S.; Foro, S.; Glidewell, C. Crystal structures of the recreational drug N-(4-methoxyphenyl)piperazine (MeOPP) and three of its salts. Acta Crystallogr. Sect. E Crystallogr. Comm. 2020, 76, 488–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiran Kumar, H.; Yathirajan, H.S.; Foro, S.; Glidewell, C. Twelve 4-(4-methoxyphenyl)piperazin-1-ium salts containing organic anions: Supra-molecular assembly in one, two and three dimensions. Acta Crystallogr. Sect. E Crystallogr. Comm. 2019, 75, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Kiran Kumar, H.; Yathirajan, H.S.; Sagar, B.K.; Foro, S.; Glidewell, C. Six 1-aroyl-4-(4-methoxyphenyl)piperazines: Similar molecular structures but different patterns of supra molecular assembly Acta crystallogr. Sect. E Crystallogr. Comm. 2019, 75, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Budzikur, D.; Kinzhybalo, V.; Ślepokura, K. Crystal engineering and structural diversity of 2-aminopyridinium hypodiphosphates obtained by crystallization and dehydration. CrystEngComm 2022, 24, 4417–4429. [Google Scholar] [CrossRef]
- Barbas, R.; Portell, A.; Hunter, C.A.; Prohens, R.; Frontera, A. Combined computational/experimental investigation of new cocrystals of the drug bosentan. CrystEngComm 2022, 24, 5105–5111. [Google Scholar] [CrossRef]
- Pang, Y.; Xing, P.; Geng, X.; Zhu, Y.; Liu, F.; Wang, L. Supramolecular assemblies of 2-hydroxy-3-naphthoic acid and N-heterocycles via various strong hydrogen bonds and weak X···π (X=C–H, π) interactions. RSC Adv. 2015, 5, 40912–40923. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: From molecule to crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef]
- Zaworotko, M.J. Molecules to crystals, crystals to molecules... and back again? Cryst. Growth Des. 2007, 7, 4–9. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent interactions: A challenge for experiment and theory. Chem. Rev. 2000, 100, 143–168. [Google Scholar] [CrossRef]
- Infantes, L.; Chisholm, J.; Motherwell, S. Extended motifs from water and chemical functional groups in organicmolecular crystals. CrystEngComm 2003, 5, 480–486. [Google Scholar] [CrossRef]
- Görbitz, C.H.; Hersleth, H.-P. On the inclusion of solvent molecules in the crystal structures of organic compounds. Acta Crystallogr. B 2000, 56, 526–534. [Google Scholar] [CrossRef]
- Bruker-AXS. APEX3; Bruker-AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Crystallogr. Sect. E Crystallogr. Comm. 2020, E76, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17.5; University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and efficient model energies for exploring intermolecular interactions in molecular crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Rev. D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X—H bond lengths from neutron diffraction data. Acta Crystalogr. B 2010, 66, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Duax, L.; Norton, D.A. Atlas of Steroid Structure; IFI/Plenum: New York, NY, USA, 1975; Volume 1, pp. 16–22. [Google Scholar]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. B 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Umezawa, Y.; Tsuboyama, S.; Takahashi, H.; Uzawa, J.; Nishio, M. CHπ interaction in the conformation of organic compounds. A database study. Tetrahedron 1999, 55, 10047–10056. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873–13900. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Interaction | D−H [Å] | H···A [Å] | D···A [Å] | ∠D−H···A [°] |
|---|---|---|---|---|---|
| (I) | N2—H2NA···O1Wi | 0.93 (1) | 1.89 (1) | 2.7941 (14) | 165 (1) |
| N2—H2NB···O3 | 0.97 (1) | 1.74 (1) | 2.7064 (14) | 175 (1) | |
| C3—H3B···O1W | 0.99 | 2.42 | 3.1981 (15) | 135 | |
| C4—H4A···O2i | 0.99 | 2.54 | 3.5153 (16) | 169 | |
| O1W—H1W···O2 | 0.945 (17) | 1.705 (17) | 2.6474 (13) | 175.1 (15) | |
| O1W—H2W···O3ii | 0.877 (18) | 1.907 (18) | 2.7735 (13) | 169.3 (15) | |
| (II) | N2—H2AN···O2 | 0.98 (1) | 1.75 (1) | 2.7234 (15) | 173 (1) |
| N2—H2BN···O1W | 0.96 (1) | 1.86 (1) | 2.7967 (16) | 167 (1) | |
| C1—H1B···O3i | 0.99 | 2.53 | 3.5129 (18) | 172 | |
| C2—H2A···O1Wi | 0.99 | 2.45 | 3.2272 (17) | 135 | |
| C3—H3A···O2ii | 0.99 | 2.60 | 3.5708 (18) | 166 | |
| C3—H3B···O1Wiii | 0.99 | 2.59 | 3.3595 (17) | 135 | |
| C11—H11C···O4iv | 0.98 | 2.42 | 3.1990 (18) | 136 | |
| O1W—H1W···O3i | 0.881 (19) | 1.754 (19) | 2.6330 (15) | 175.9 (18) | |
| O1W—H2W···O2iii | 0.921 (19) | 1.857 (19) | 2.7714 (15) | 171.5 (17) | |
| (III) | N2—H2AN···O3 | 0.91 | 1.84 | 2.7462 (14) | 172 |
| N2—H2BN···O1Wi | 0.91 | 1.90 | 2.7986 (15) | 168 | |
| C2—H2A···O3ii | 0.99 | 2.50 | 3.4571 (16) | 163 | |
| C2—H2B···O1Wiii | 0.99 | 2.58 | 3.3263 (16) | 132 | |
| C3—H3C···O1W | 0.99 | 2.50 | 3.3042 (17) | 138 | |
| C4—H4B···O2i | 0.99 | 2.60 | 3.5851 (17) | 178 | |
| O4—H4···O1iv | 0.84 | 1.94 | 2.7606 (14) | 166 | |
| O1W—H1W···O2 | 0.892 (19) | 1.773 (19) | 2.6636 (14) | 176.4 (17) | |
| O1W—H2W···O3v | 0.905 (19) | 1.864 (19) | 2.7574 (14) | 168.6 (16) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Priyanka, P.; Jayanna, B.K.; Divakara, T.R.; Suresha, G.P.; Vinaya; Basavaraju, Y.B.; Yathirajan, H.S.; Parkin, S.R.; Chęcińska, L. Hydrogen-Bonded Chain of Rings Motif in N-(4-Methoxyphenyl)piperazin-1-ium Salts with Benzoate Anions: Supramolecular Assemblies and Their Energy Frameworks. Crystals 2022, 12, 1807. https://doi.org/10.3390/cryst12121807
Priyanka P, Jayanna BK, Divakara TR, Suresha GP, Vinaya, Basavaraju YB, Yathirajan HS, Parkin SR, Chęcińska L. Hydrogen-Bonded Chain of Rings Motif in N-(4-Methoxyphenyl)piperazin-1-ium Salts with Benzoate Anions: Supramolecular Assemblies and Their Energy Frameworks. Crystals. 2022; 12(12):1807. https://doi.org/10.3390/cryst12121807
Chicago/Turabian StylePriyanka, Prabhakar, Bidarur K. Jayanna, Thayamma R. Divakara, Gejjalagere P. Suresha, Vinaya, Yeriyur B. Basavaraju, Hemmige S. Yathirajan, Sean R. Parkin, and Lilianna Chęcińska. 2022. "Hydrogen-Bonded Chain of Rings Motif in N-(4-Methoxyphenyl)piperazin-1-ium Salts with Benzoate Anions: Supramolecular Assemblies and Their Energy Frameworks" Crystals 12, no. 12: 1807. https://doi.org/10.3390/cryst12121807