
Synthesis and Crystallographic Characterization of Heteroleptic Ir(III) Complexes Containing the N-oxide Functional Group and Crystallographic Characterization of Ir(III) N-oxide Precursors
 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Synthesis
2.3. Single-Crystal X-ray Diffraction
3. Results and Discussion
3.1. Synthesis of Iridium(III) N-oxide Complexes
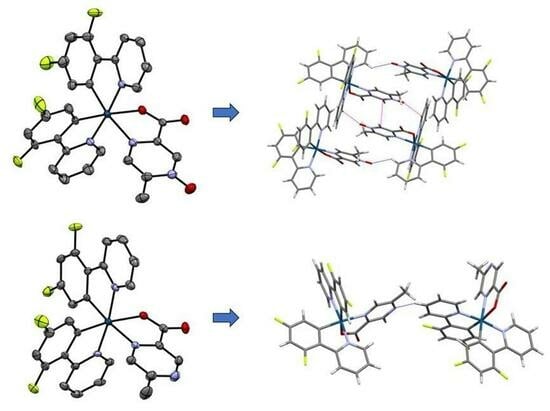
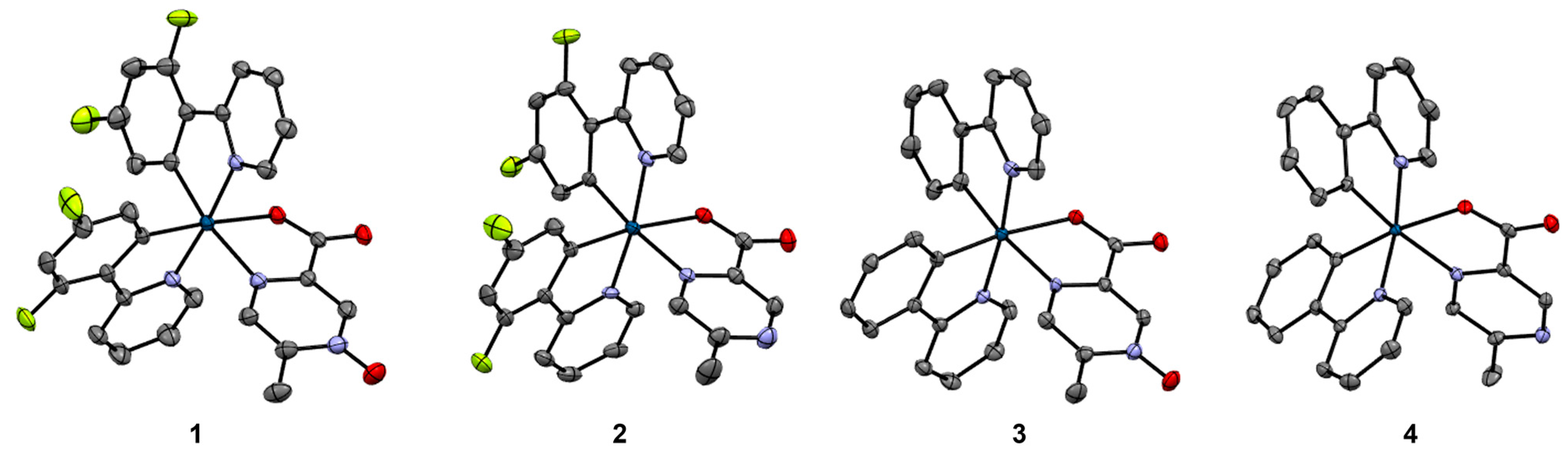
3.2. Structural Comparison of Iridium(III) N-oxide Complexes with Comparable Iridium(III) Complexes with Non-oxidized Ligands, 1–4
3.3. Electronic Characterization of 1–4
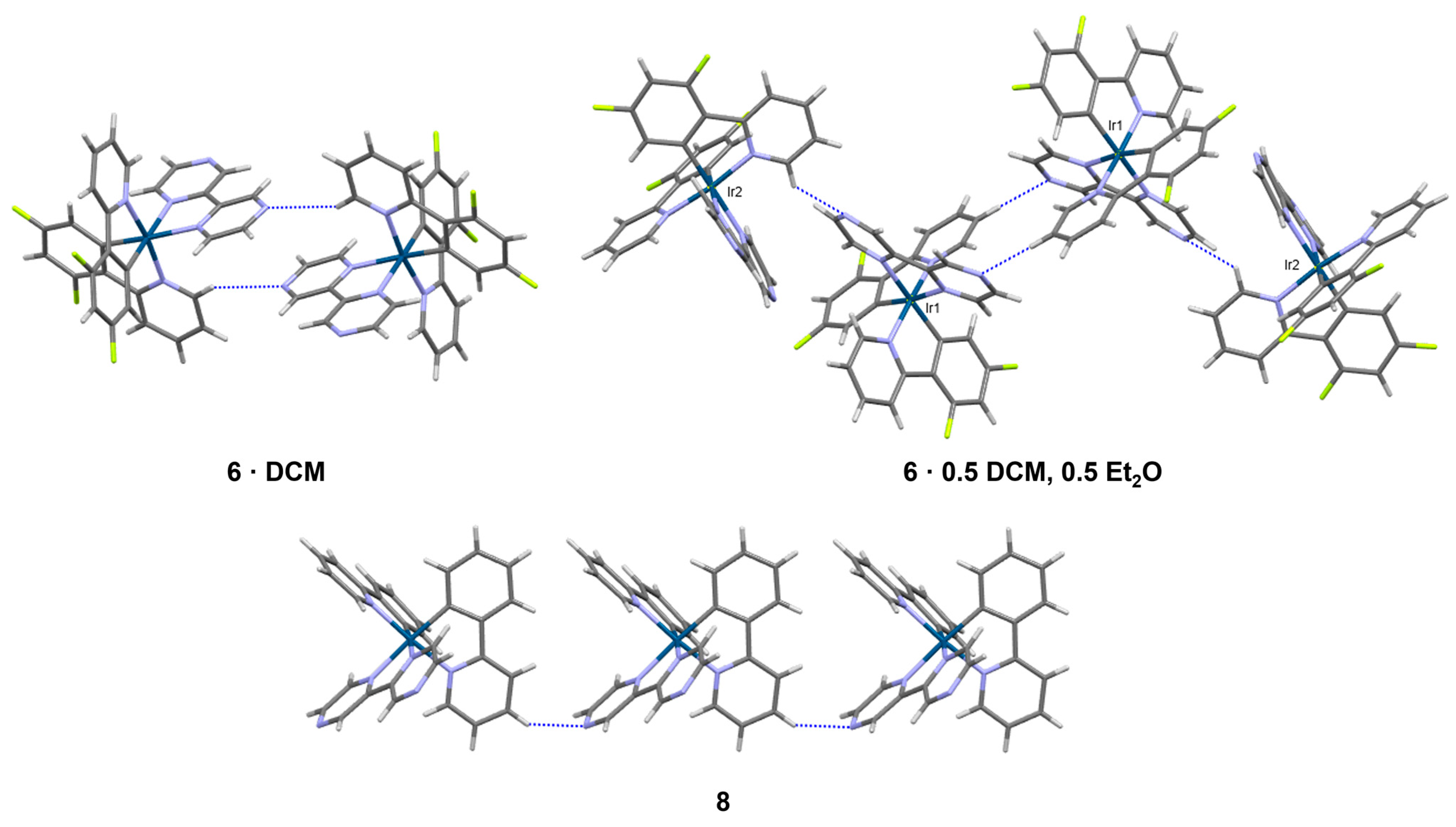
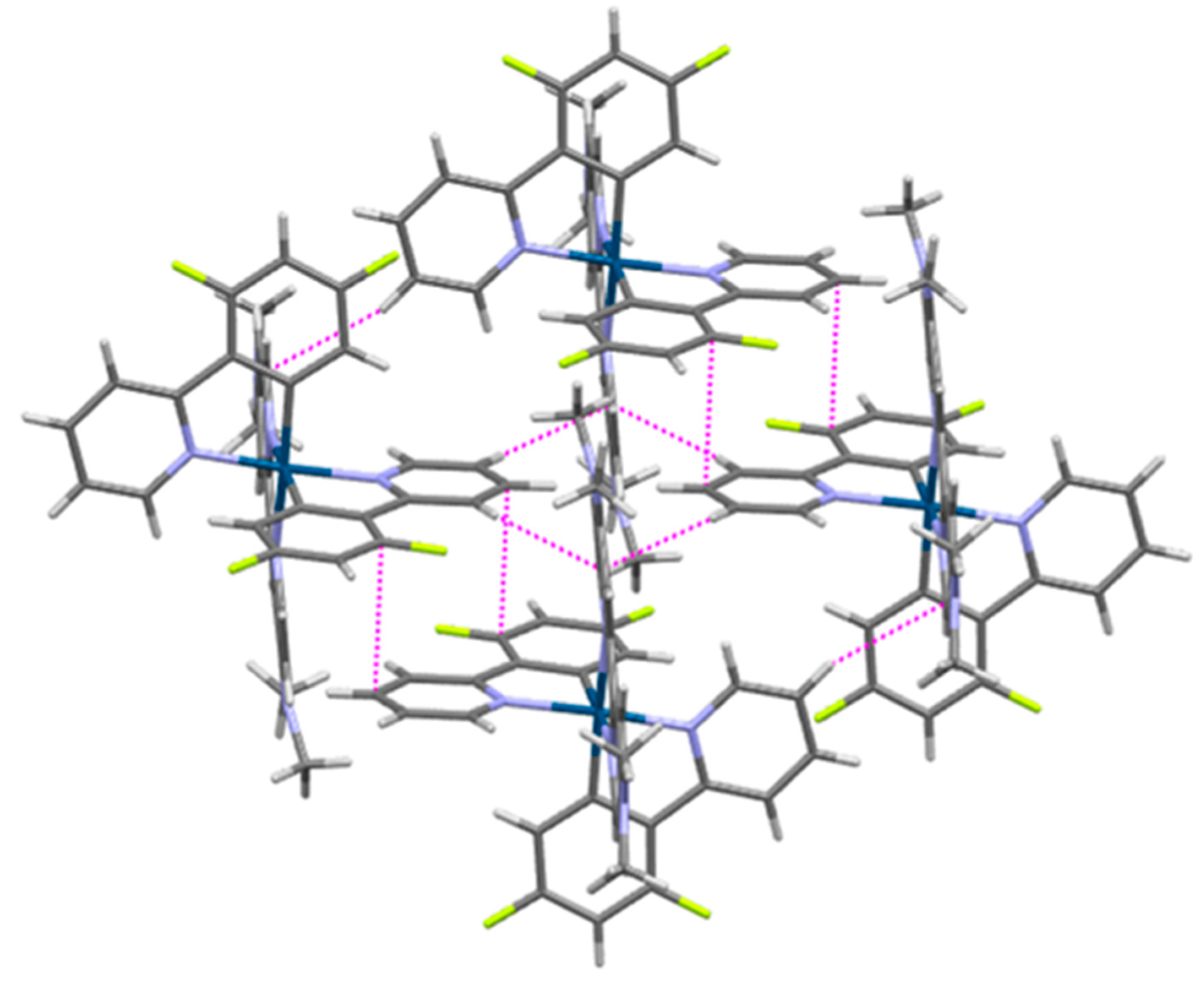
3.4. Structural Characterization of Iridium(III) Complexes with Additional Exposed Nitrogen Atoms 5–8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bull, J.A.; Mousseau, J.J.; Pelletier, G.; Charette, A.B. Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev. 2012, 112, 2642–2713. [Google Scholar] [CrossRef]
- Mfuh, A.M.; Larionov, O.V. Heterocyclic N-Oxides—An Emerging Class of Therapeutic Agents. Curr. Med. Chem. 2015, 22, 2819–2857. [Google Scholar] [CrossRef]
- Kang, D.; Cheung, S.T.; Wong-Rolle, A.; Kim, J. Enamine N-Oxides: Synthesis and Application to Hypoxia-Responsive Prodrugs and Imaging Agents. ACS Cent. Sci. 2021, 7, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Knox, H.J.; Hedhli, J.; Kim, T.W.; Khalili, K.; Dobrucki, L.W.; Chan, J. A bioreducible N-oxide-based probe for photoacoustic imaging of hypoxia. Nat. Commun. 2017, 8, 1794. [Google Scholar] [CrossRef]
- Knox, H.J.; Kim, T.W.; Zhu, Z.; Chan, J. Photophysical Tuning of N-Oxide-Based Probes Enables Ratiometric Photoacoustic Imaging of Tumor Hypoxia. ACS Chem. Biol. 2018, 13, 1838–1843. [Google Scholar] [CrossRef]
- Delahoussaye, Y.M.; Evans, J.W.; Brown, J.M. Metabolism of tirapazamine by multiple reductases in the nucleus11Abbreviations: TPZ, tirapazamine; P450 reductase, NADPH:cytochrome P450 reductase; PMSF, phenylmethylsulfonyl fluoride; DTT, dithiothreitol; DHR123, dihydrorhodamine 123; DAPI, 4′,6-diamidino-2-phenylindole, dihydrochloride; SN, supernatant from nuclei; LS, low salt; HS, high salt; and NM, nuclear matrix. Biochem. Pharmacol. 2001, 62, 1201–1209. [Google Scholar] [CrossRef]
- Skerritt, J.H.; Johnston, G.A. Enhancement of GABA binding by benzodiazepines and related anxiolytics. Eur. J. Pharmacol. 1983, 89, 193–198. [Google Scholar] [CrossRef]
- Lo, K.K.-W. Luminescent Rhenium(I) and Iridium(III) Polypyridine Complexes as Biological Probes, Imaging Reagents, and Photocytotoxic Agents. Acc. Chem. Res. 2015, 48, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Baggaley, E.; Weinstein, J.A.; Williams, J.A.G. Lighting the way to see inside the live cell with luminescent transition metal complexes. Coord. Chem. Rev. 2012, 256, 1762–1785. [Google Scholar] [CrossRef]
- Fernández-Moreira, V.; Thorp-Greenwood, F.L.; Coogan, M.P. Application of d6 transition metal complexes in fluorescence cell imaging. Chem. Commun. 2010, 46, 186–202. [Google Scholar] [CrossRef]
- Zhang, S.; Hosaka, M.; Yoshihara, T.; Negishi, K.; Iida, Y.; Tobita, S.; Takeuchi, T. Phosphorescent Light–Emitting Iridium Complexes Serve as a Hypoxia-Sensing Probe for Tumor Imaging in Living Animals. Cancer Res. 2010, 70, 4490–4498. [Google Scholar] [CrossRef]
- Venkatesh, V.; Berrocal-Martin, R.; Wedge, C.J.; Romero-Canelón, I.; Sanchez-Cano, C.; Song, J.-I.; Coverdale, J.P.C.; Zhang, P.; Clarkson, G.J.; Habtemariam, A.; et al. Mitochondria-targeted spin-labelled luminescent iridium anticancer complexes. Chem. Sci. 2017, 8, 8271–8278. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Kim, J.; Jeong, J.; Chang, S. Regioselective Introduction of Heteroatoms at the C-8 Position of Quinoline N-Oxides: Remote C–H Activation Using N-Oxide as a Stepping Stone. J. Am. Chem. Soc. 2014, 136, 10770–10776. [Google Scholar] [CrossRef]
- Hetterscheid, D.G.H.; Kaiser, J.; Reijerse, E.; Peters, T.P.J.; Thewissen, S.; Blok, A.N.J.; Smits, J.M.M.; de Gelder, R.; de Bruin, B. IrII(ethene): Metal or Carbon Radical? J. Am. Chem. Soc. 2005, 127, 1895–1905. [Google Scholar] [CrossRef]
- Theron, M.; Purcell, W.; Basson, S.S. ([eta]4-Cycloocta-1,5-diene)(2-pyridinethiolato N-oxide-[kappa]O,[kappa]S)iridium(I). Acta Crystallogr. Sect. C 1996, 52, 336–338. [Google Scholar] [CrossRef]
- Turlington, C.R.; White, P.S.; Brookhart, M.; Templeton, J.L. Oxygen Atom Transfer to a Half-Sandwich Iridium Complex: Clean Oxidation Yielding a Molecular Product. J. Am. Chem. Soc. 2014, 136, 3981–3994. [Google Scholar] [CrossRef] [PubMed]
- Hernández, Y.A.; López-Serrano, J.; Paneque, M.; Poveda, M.L.; Vattier, F.; Salazar, V.; Álvarez, E.; Carmona, E. C–N Bond Formation by O2-Mediated Dehydrogenative Coupling of Phenyl and NH-pyridylidene Ligands on TpIrIII Complexes. Chem. —A Eur. J. 2011, 17, 9302–9305. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Lu, Y.; Lin, Y.-J.; Jin, G.-X. Donor–Acceptor [2]- and [3]Catenanes Assembled from Versatile Pre-Organized Cp*Rh/Ir-Directed Pseudorotaxane Tectons. Chem.-A Eur. J. 2019, 25, 14785–14789. [Google Scholar] [CrossRef]
- Shopov, D.Y.; Sharninghausen, L.S.; Sinha, S.B.; Mercado, B.Q.; Brudvig, G.W.; Crabtree, R.H. Modification of a pyridine-alkoxide ligand during the synthesis of coordination compounds. Inorg. Chim. Acta 2019, 484, 75–78. [Google Scholar] [CrossRef]
- Carlin, R.L.; De Jongh, L.J. Structural and magnetic properties of transition metal complexes of pyridine N-oxide. Chem. Rev. 1986, 86, 659–680. [Google Scholar] [CrossRef]
- Karayannis, N.M.; Pytlewski, L.L.; Mikulski, C.M. Metal complexes of aromatic amine n-oxides. Coord. Chem. Rev. 1973, 11, 93–159. [Google Scholar] [CrossRef]
- Henkelis, J.J.; Barnett, S.A.; Harding, L.P.; Hardie, M.J. Coordination Polymers Utilizing N-Oxide Functionalized Host Ligands. Inorg. Chem. 2012, 51, 10657–10674. [Google Scholar] [CrossRef]
- Puttreddy, R.; Steel, P.J. Synthesis and X-ray crystal structures of silver complexes of 2,6-dimethylpyridine N-oxide: Steric factors override electronic effects. Polyhedron 2014, 69, 25–30. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, H.; Xu, J.; Chen, C.; Pan, Y.; Luo, Z.; Zhang, Z.; Li, H.; Xu, L. Rhodium(III)-Catalyzed Oxidative Annulation of 2,2′-Bipyridine N-Oxides with Alkynes via Dual C–H Bond Activation. Org. Lett. 2018, 20, 3843–3847. [Google Scholar] [CrossRef]
- Shahsavari, H.R.; Babadi Aghakhanpour, R.; Hossein-Abadi, M.; Kia, R.; Halvagar, M.R.; Raithby, P.R. Reactivity of a new aryl cycloplatinated(ii) complex containing rollover 2,2′-bipyridine N-oxide toward a series of diphosphine ligands. New J. Chem. 2018, 42, 9159–9167. [Google Scholar] [CrossRef]
- Puttreddy, R.; Cottam, J.R.A.; Steel, P.J. Anion dependent silver(i) complexes of pyrazine mono-N-oxide. RSC Adv. 2014, 4, 22449–22454. [Google Scholar] [CrossRef]
- Dadkhah Aseman, M.; Nikravesh, M.; Abbasi, A.; Shahsavari, H.R. Oxidative Addition of a Hypervalent Iodine Compound to Cycloplatinated(II) Complexes for the C–O Bond Construction: Effect of Cyclometalated Ligands. Inorg. Chem. 2021, 60, 18822–18831. [Google Scholar] [CrossRef]
- Li, Q.-Q.; Kang, Y.-F.; Ren, C.-Y.; Yang, G.-P.; Liu, Q.L.P.; Wang, Y.-Y. Reaction-controlled assemblies and structural diversities of seven Co(ii)/Cu(ii) complexes based on a bipyridine-dicarboxylate N-oxide ligand. CrystEngComm 2015, 17, 775–786. [Google Scholar] [CrossRef]
- Chen, L.-Z.; Wang, F.-M.; Shu, H. Construction of three metal-organic frameworks based on multifunctional T-shaped tripodal ligands (4,5-dicarboxy-1H-imidazol-2-yl)pyridine-1-oxide. J. Coord. Chem. 2012, 65, 439–452. [Google Scholar] [CrossRef]
- Ravindran Durai Nayagam, B.; Jebas, S.R.; Edward Rajkumar, J.P.; Schollmeyer, D. Tetraaquabis[3-(2-pyridylsulfanyl)propionato N-oxide]nickel(II). Acta Crystallogr. Sect. E 2009, 65, m470. [Google Scholar] [CrossRef] [PubMed]
- Pailloux, S.; Binyamin, I.; Deck, L.M.; Hay, B.P.; Duesler, E.N.; Zakharov, L.N.; Scott Kassel, W.; Rheingold, A.L.; Paine, R.T. Unexpected chelation interaction for 2-hydroxy-2-(1-oxy-pyridin-2-yl)-N,N-diphenyl acetamide with La(III). Polyhedron 2009, 28, 3979–3984. [Google Scholar] [CrossRef]
- Zucker, S.P.; Wossidlo, F.; Weber, M.; Lentz, D.; Tzschucke, C.C. Palladium-Catalyzed Directed Halogenation of Bipyridine N-Oxides. J. Org. Chem. 2017, 82, 5616–5635. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.-N.; Zhang, Y.-P.; Han, S.-J.; Li, B. Synthesis, Crystal Structures, and Characterization of Two New Complexes Constructed From Acipimox Ligands: Two Three-Dimensional Networks Formed Via Hydrogen Bonding. Synth. React. Inorg. Met.-Org. Nano-Met. Chem. 2016, 46, 409–413. [Google Scholar] [CrossRef]
- Penconi, M.; Cazzaniga, M.; Panzeri, W.; Mele, A.; Cargnoni, F.; Ceresoli, D.; Bossi, A. Unraveling the Degradation Mechanism in FIrpic-Based Blue OLEDs: II. Trap and Detect Molecules at the Interfaces. Chem. Mater. 2019, 31, 2277–2285. [Google Scholar] [CrossRef]
- You, Y.; Park, S.Y. Inter-Ligand Energy Transfer and Related Emission Change in the Cyclometalated Heteroleptic Iridium Complex: Facile and Efficient Color Tuning over the Whole Visible Range by the Ancillary Ligand Structure. J. Am. Chem. Soc. 2005, 127, 12438–12439. [Google Scholar] [CrossRef] [PubMed]
- Bevernaegie, R.; Wehlin, S.A.M.; Piechota, E.J.; Abraham, M.; Philouze, C.; Meyer, G.J.; Elias, B.; Troian-Gautier, L. Improved Visible Light Absorption of Potent Iridium(III) Photo-oxidants for Excited-State Electron Transfer Chemistry. J. Am. Chem. Soc. 2020, 142, 2732–2737. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Fantacci, S.; Evans, N.; Klein, C.; Zakeeruddin, S.M.; Moser, J.-E.; Kalyanasundaram, K.; Bolink, H.J.; Grätzel, M.; Nazeeruddin, M.K. Controlling Phosphorescence Color and Quantum Yields in Cationic Iridium Complexes: A Combined Experimental and Theoretical Study. Inorg. Chem. 2007, 46, 5989–6001. [Google Scholar] [CrossRef]
- Kim, C.S.; Tinker, L.L.; DiSalle, B.F.; Gomez, E.D.; Lee, S.; Bernhard, S.; Loo, Y.-L. Altering the Thermodynamics of Phase Separation in Inverted Bulk-Heterojunction Organic Solar Cells. Adv. Mater. 2009, 21, 3110–3115. [Google Scholar] [CrossRef]
- Apex3, Bruker AXS Inc.: Madison, WI, USA, 2015.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Bernier, D.; Wefelscheid, U.K.; Woodward, S. Properties, Preparation and Synthetic Uses of Amine N-Oxides. An Update. Org. Prep. Proced. Int. 2009, 41, 173–210. [Google Scholar] [CrossRef]
- Shevlin, M.R.; Stumbo, E.E.; McMillen, C.D.; Pienkos, J.A. Bis[3,5-difluoro-2-(pyridin-2-yl)phenyl](4,4′-dimethoxy-2,2′-bipyridine)iridium(III) hexafluoridophosphate. IUCrData 2022, 7, x220830. [Google Scholar] [CrossRef]
- Li, X.; Tong, X.; Yin, Y.; Yan, H.; Lu, C.; Huang, W.; Zhao, Q. Using highly emissive and environmentally sensitive o-carborane-functionalized metallophosphors to monitor mitochondrial polarity. Chem. Sci. 2017, 8, 5930–5940. [Google Scholar] [CrossRef]
- Moriuchi, T.; Katano, C.; Hirao, T. Poly(l-glutamic acid)-modulated Emission Properties of Iridium(III) Complexes in an Aqueous Media. Chem. Lett. 2012, 41, 310–312. [Google Scholar] [CrossRef]
- Łukomska, M.; Rybarczyk-Pirek, A.J.; Jabłoński, M.; Palusiak, M. The nature of NO-bonding in N-oxide group. Phys. Chem. Chem. Phys. 2015, 17, 16375–16387. [Google Scholar] [CrossRef]
- Kim, Y.-I.; Song, Y.-K.; Kang, S.K. (5-Methylpyrazine-2-carboxylato-[kappa]2N1,O)bis[2-(4-methylpyridin-2-yl-[kappa]N)-3,5-bis(trifluoromethyl)phenyl-[kappa]C1]iridium(III) chloroform hemisolvate. Acta Crystallogr. Sect. E 2014, 70, m34. [Google Scholar] [CrossRef]
- Hasan, K.; Bansal, A.K.; Samuel, I.D.W.; Roldán-Carmona, C.; Bolink, H.J.; Zysman-Colman, E. Tuning the Emission of Cationic Iridium (III) Complexes Towards the Red Through Methoxy Substitution of the Cyclometalating Ligand. Sci. Rep. 2015, 5, 12325. [Google Scholar] [CrossRef]
- Thamilarasan, V.; Karunakaran, P.; Kavitha, N.; Selvaraju, C.; Sengottuvelan, N. Red emitting cyclometallated iridium(III) complexes: Synthesis, characterization and evaluation of biological activities. Polyhedron 2016, 118, 12–24. [Google Scholar] [CrossRef]
- Song, M.; Yun, S.-J.; Nam, K.-S.; Liu, H.; Gal, Y.-S.; Lee, J.W.; Jin, S.-H.; Lee, J.Y.; Kang, S.K.; Kim, Y.I. Highly efficient solution-processed pure red phosphorescent organic light-emitting diodes using iridium complexes based on 2,3-diphenylquinoxaline ligand. J. Organomet. Chem. 2015, 794, 197–205. [Google Scholar] [CrossRef]
- Li, G.-N.; Gao, C.-W.; Chen, H.-H.; Chen, T.-T.; Xie, H.; Lin, S.; Sun, W.; Chen, G.-Y.; Niu, Z.-G. Color tuning of cyclometalated 2-phenylbenzo[d]oxazole-based iridium(III) complexes through modification of different N^O ancillary ligands. Inorg. Chim. Acta 2016, 445, 22–27. [Google Scholar] [CrossRef]
- Thamilarasan, V.; Jayamani, A.; Manisankar, P.; Kim, Y.-I.; Sengottuvelan, N. Green-emitting phosphorescent iridium(III) complex: Structural, photophysical and electrochemical properties. Inorg. Chim. Acta 2013, 408, 240–245. [Google Scholar] [CrossRef]
- Seo, H.-J.; Heo, Y.-M.; Jin, S.-H.; Soo Yook, K.; Yeob Lee, J.; Kwon Kang, S.; Kim, Y.-I. New heteroleptic cyclometalated iridium(III) complexes containing 2-(2′,4′-difluorophenyl)-4-methylpyridine for organic light-emitting diode applications. J. Lumin. 2010, 130, 1694–1701. [Google Scholar] [CrossRef]
- Bevernaegie, R.; Marcélis, L.; Laramée-Milette, B.; De Winter, J.; Robeyns, K.; Gerbaux, P.; Hanan, G.S.; Elias, B. Trifluoromethyl-Substituted Iridium(III) Complexes: From Photophysics to Photooxidation of a Biological Target. Inorg. Chem. 2018, 57, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, A.M.; Housecroft, C.E.; Constable, E.C.; Zampese, J.A. CCDC 1853180: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre: Cambridge, UK, 2018. [Google Scholar] [CrossRef]
- Li, M.-J. CCDC 2073478: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre: Cambridge, UK, 2021. [Google Scholar] [CrossRef]
- Baranoff, E.; Bolink, H.J.; Constable, E.C.; Delgado, M.; Häussinger, D.; Housecroft, C.E.; Nazeeruddin, M.K.; Neuburger, M.; Ortí, E.; Schneider, G.E.; et al. Tuning the photophysical properties of cationic iridium(iii) complexes containing cyclometallated 1-(2,4-difluorophenyl)-1H-pyrazole through functionalized 2,2′-bipyridine ligands: Blue but not blue enough. Dalton Trans. 2013, 42, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Baschieri, A.; Sambri, L.; Mazzanti, A.; Carlone, A.; Monti, F.; Armaroli, N. Iridium(III) Complexes with Fluorinated Phenyl-tetrazoles as Cyclometalating Ligands: Enhanced Excited-State Energy and Blue Emission. Inorg. Chem. 2020, 59, 16238–16250. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1· 1.25(THF) | 2· 0.5(DCM),0.5(H2O) | 3· 2(DCM) | 4· 2(DCM) | |
|---|---|---|---|---|
| Formula | C33H27F4IrN4O4.25 | C28.5H19ClF4IrN4O2.5 | C30H25Cl4IrN4O3 | C30H25Cl4IrN4O2 |
| F. W. (g/mol) | 815.78 | 761.13 | 823.54 | 807.54 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Crystal system | triclinic | monoclinic | monoclinic | monoclinic |
| Space group | P-1 | P21/c | P21 | P21 |
| a (Å) | 12.0056 (9) | 20.1167 (11) | 9.3656 (5) | 9.3682 (6) |
| b (Å) | 13.9227 (10) | 16.8038 (10) | 16.9853 (8) | 16.9395 (10) |
| c (Å) | 21.5251 (15) | 17.1342 (9) | 9.5139 (5) | 9.4088 (5) |
| α (°) | 84.548 (3) | 90 | 90 | 90 |
| β (°) | 81.768 (3) | 107.127 (2) | 91.2046 (18) | 91.347 (2) |
| γ (°) | 87.722 (3) | 90 | 90 | 90 |
| Volume (Å3) | 3543.5 (4) | 5535.1 (5) | 1513.11 (13) | 1492.69 (15) |
| Z | 4 | 8 | 2 | 2 |
| D(calcd) (g/cm3) | 1.529 | 1.827 | 1.808 | 1.797 |
| μ, mm−1 | 3.829 | 4.984 | 4.804 | 4.866 |
| F(000) | 1600 | 2944 | 804 | 788 |
| Cryst. Size (mm) | 0.12 × 0.17 × 0.26 | 0.12 × 0.15 × 0.23 | 0.05 × 0.15 × 0.18 | 0.16 × 0.21 × 0.22 |
| θ range,° | 2.23 to 30.09 | 2.22 to 30.08 | 2.14 to 29.62 | 2.18 to 30.06 |
| Reflns. collected | 248152 | 137437 | 32766 | 33335 |
| Indep. reflns. | 20781 | 16232 | 8406 | 8622 |
| R(int) | 0.0388 | 0.0589 | 0.0374 | 0.0370 |
| No. of parameters | 904 | 794 | 381 | 372 |
| No. of restraints | 224 | 92 | 1 | 1 |
| R indices (I > 2σ(I)) | R1 = 0.0362 wR2 = 0.1153 | R1 = 0.0364 wR2 = 0.0778 | R1 = 0.0204 wR2 = 0.0439 | R1 = 0.0164 wR2 = 0.0376 |
| R indices (all data) | R1 = 0.0409 wR2 = 0.1205 | R1 = 0.0504 wR2 = 0.0872 | R1 = 0.0236 wR2 = 0.0463 | R1 = 0.0175 wR2 = 0.0396 |
| S | 1.109 | 1.111 | 1.038 | 0.946 |
| Abs. struct. param. (Flack) | - | - | 0.009(3) | 0.015(3) |
| Largest diff. peak/hole (eÅ−3) | 2.623, −1.480 | 2.151, −2.036 | 1.495, −1.157 | 0.769, −0.699 |
| CCDC dep. no. | 2335789 | 2335790 | 2335791 | 2335792 |
| 5 | 6 · (DCM) | 6 · 0.5(DCM), 0.5(Et2O) | 7 · (DCM) | 8 | |
|---|---|---|---|---|---|
| Formula | C27H15F4IrN4O2 | C31H20Cl2F10IrN6P | C32.5H24ClF10IrN6O0.5P | C37H32Cl2F10IrN6P | C30H22F6IrN6P |
| F. W. (g/mol) | 695.63 | 960.60 | 955.20 | 1044.75 | 803.70 |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 |
| Crystal system | triclinic | monoclinic | triclinic | triclinic | orthorhombic |
| Space group | P-1 | P21/c | P-1 | P-1 | Pbca |
| a (Å) | 8.8150 (6) | 9.6985 (8) | 13.8002 (6) | 8.6227 (9) | 10.8324 (4) |
| b (Å) | 12.0339 (10) | 24.7164 (18) | 14.3504 (6) | 13.5061 (13) | 16.0987 (6) |
| c (Å) | 22.3146 (17) | 13.7261 (9) | 17.3147 (8) | 16.6738 (15) | 31.5748 (10) |
| α (°) | 87.528 (3) | 90 | 72.193 (2) | 104.272 (3) | 90 |
| β (°) | 79.542 (3) | 93.775 (3) | 89.145 (2) | 94.651 (4) | 90 |
| γ (°) | 89.854 (3) | 90 | 89.584 (2) | 90.132 (4) | 90 |
| Volume (Å3) | 2325.6 (3) | 3283.2 (4) | 3264.3 (3) | 1875.2 (3) | 5506.3 (3) |
| Z | 4 | 4 | 4 | 2 | 8 |
| D(calcd) (g/cm3) | 1.987 | 1.943 | 1.944 | 1.850 | 1.939 |
| μ, mm−1 | 5.808 | 4.370 | 4.316 | 3.834 | 4.983 |
| F(000) | 1336 | 1856 | 1856 | 1024 | 3120 |
| Cryst. Size (mm) | 0.08 × 0.08 × 0.12 | 0.03 × 0.11 × 0.17 | 0.06 × 0.12 × 0.20 | 0.08 × 0.26 × 0.28 | 0.06 × 0.12 × 0.14 |
| θ range,° | 2.35 to 26.50 | 2.10 to 25.50 | 2.10 to 25.50 | 2.24 to 27.94 | 2.36 to 26.00 |
| Reflns. collected | 81813 | 56722 | 68954 | 63741 | 53413 |
| Indep. reflns. | 9631 | 6099 | 12132 | 8981 | 5404 |
| R(int) | 0.0475 | 0.0468 | 0.0374 | 0.0401 | 0.0576 |
| No. of parameters | 685 | 460 | 982 | 518 | 397 |
| No. of restraints | 0 | 0 | 139 | 0 | 0 |
| R indices (I > 2σ(I)) | R1 = 0.0214 wR2 = 0.0419 | R1 = 0.0409 wR2 = 0.0865 | R1 = 0.0404 wR2 = 0.0839 | R1 = 0.0242 wR2 = 0.0561 | R1 = 0.0319 wR2 = 0.0599 |
| R indices (all data) | R1 = 0.0283 wR2 = 0.0464 | R1 = 0.0498 wR2 = 0.0935 | R1 = 0.0513 wR2 = 0.0944 | R1 = 0.0274 wR2 = 0.0587 | R1 = 0.0478 wR2 = 0.0707 |
| S | 1.191 | 1.067 | 1.100 | 1.123 | 1.219 |
| Largest diff. peak/hole (eÅ−3) | 1.215, −0.938 | 1.811, −1.148 | 2.044, −1.501 | 1.534, −1.109 | 2.742, −1.650 |
| CCDC dep. no. | 2335793 | 2335794 | 2335795 | 2335796 | 2335797 |
| Bond | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Ir-O | 2.151 (3) 2.156 (3) | 2.150 (3) 2.155 (3) | 2.177 (2) | 2.1699 (18) |
| Ir-N | 2.141 (3) 2.126 (4) | 2.114 (4) 2.131 (4) | 2.138 (3) | 2.136 (3) |
| Ir-Ndfppy/ppy | 2.039 (3) 2.043 (3) 2.040 (4) 2.045 (4) | 2.021 (3) 2.052 (3) 2.045 (4) 2.045 (4) | 2.040 (3) 2.052 (4) | 2.036 (3) 2.048 (3) |
| Ir-Cdfppy/ppy (trans O) | 1.993 (4) 1.987 (5) | 1.991 (4) 1.996 (4) | 1.995 (4) | 1.994 (3) |
| Ir-Cdfppy/ppy (trans N) | 2.001 (4) 2.003 (5) | 2.002 (4) 1.999 (4) | 1.999 (4) | 2.002 (3) |
| N-O | 1.283 (5) 1.279 (8) | - | 1.283 (5) | - |
| O-Ir-C (trans) | 175.83 (13) 175.17 (16) | 172.18 (14) 174.21 (14) | 172.6 (2) | 172.89 (16) |
| N-Ir-C (trans) | 169.52 (14) 170.8 (2) | 172.54 (14) 175.37 (16) | 171.52 (15) | 171.32 (12) |
| N-Ir-N (trans) | 174.92 (13) 172.29 (16) | 172.85 (15) 174.90 (14) | 173.45 (13) | 172.40 (11) |
| Bond | 5 | 6 (DCM) | 6 0.5(DCM),0.5(Et2O) | 7 | 8 |
|---|---|---|---|---|---|
| Ir-O | 2.154 (2) 2.155 (2) | - | - | - | - |
| Ir-N | 2.138 (3) 2.136 (3) | 2.132 (5) 2.133 (5) | 2.112 (5) 2.125 (5) 2.125 (5) 2.126 (5) | 2.131 (2) 2.135 (2) | 2.135 (4) 2.141 (4) |
| Ir-Ndfppy/ppy | 2.023 (3) 2.047 (3) 2.030 (3) 2.041 (4) | 2.046 (5) 2.047 (5) | 2.054 (5) 2.057 (5) 2.048 (6) 2.053 (6) | 2.034 (3) 2.037 (2) | 2.052 (4) 2.052 (4) |
| Ir-Cdfppy/ppy | 1.993 (3) 2.012 (3) 1.995 (3) 2.002 (4) | 1.998 (7) 2.012 (7) | 1.998 (7) 2.028 (6) 2.012 (6) 2.016 (6) | 2.016 (3) 2.016 (3) | 2.016 (6) 2.022 (5) |
| O-Ir-C (trans) | 174.81 (12) 173.78 (12) | - | - | - | - |
| N-Ir-C (trans) | 171.94 (11) 172.56 (12) | 171.1 (2) 175.6 (2) | 172.6 (2) 170.8 (2) 172.4 (2) 175.4 (2) | 171.25 (10) 174.75 (10) | 172.37 (17) 175.70 (19) |
| N-Ir-N (trans) | 172.12 (11) 173.16 (12) | 171.6 (2) | 173.3 (2) 172.9 (2) | 171.97 (10) | 172.81 (16) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stumbo, E.E.; Hodge, E.K.; Williams, M.; Thornton, D.A.; McMillen, C.D.; Pienkos, J.A. Synthesis and Crystallographic Characterization of Heteroleptic Ir(III) Complexes Containing the N-oxide Functional Group and Crystallographic Characterization of Ir(III) N-oxide Precursors. Crystals 2024, 14, 281. https://doi.org/10.3390/cryst14030281
Stumbo EE, Hodge EK, Williams M, Thornton DA, McMillen CD, Pienkos JA. Synthesis and Crystallographic Characterization of Heteroleptic Ir(III) Complexes Containing the N-oxide Functional Group and Crystallographic Characterization of Ir(III) N-oxide Precursors. Crystals. 2024; 14(3):281. https://doi.org/10.3390/cryst14030281
Chicago/Turabian StyleStumbo, Emily E., Emarald K. Hodge, Matthew Williams, Diana A. Thornton, Colin D. McMillen, and Jared A. Pienkos. 2024. "Synthesis and Crystallographic Characterization of Heteroleptic Ir(III) Complexes Containing the N-oxide Functional Group and Crystallographic Characterization of Ir(III) N-oxide Precursors" Crystals 14, no. 3: 281. https://doi.org/10.3390/cryst14030281