
A Comparative Computational Study of the Solidification Kinetic Coefficients for the Soft-Sphere BCC-Melt and the FCC-Melt Interfaces
 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Soft-Sphere Model Potential
2.2. Simulation Details
2.3. Measurement of
2.4. TDGL Theoretical Prediction of
3. Results and Discussion
4. Summary
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Asta, M.; Beckermann, C.; Karma, A.; Kurz, W.; Napolitano, R.; Plapp, M.; Purdy, G.; Rappaz, M.; Trivedi, R. Solidification microstructures and solid-state parallels: Recent developments, future directions. Acta Mater. 2009, 57, 941–971. [Google Scholar] [CrossRef]
- Wu, L.; Zhu, Y.; Wang, H.; Li, M. Crystal–melt coexistence in FCC and BCC metals: A molecular-dynamics study of kinetic coefficients. Model. Simul. Mater. Sci. Eng. 2021, 29, 065016. [Google Scholar] [CrossRef]
- Ghosh, G. Observation and kinetic analysis of a metastable b.c.c. phase in rapidly solidified Ni 9at.%Zr and Ni 8at.%Zr 1at.%X alloys. Mater. Sci. Eng. A 1994, 189, 277–284. [Google Scholar] [CrossRef]
- Volkmann, T.; Herlach, D.M.; Löser, W. Nucleation and phase selection in undercooled Fe-Cr-Ni melts: Part I. Theoretical analysis of nucleation behavior. Metall. Mater. Trans. A 1997, 28, 453–460. [Google Scholar] [CrossRef]
- Zambon, A.; Badan, B.; Eckler, K.; Gärtner, F.; Norman, A.; Greer, A.; Herlach, D.; Ramous, E. Microstructure and phase selection in containerless processing of Fe–Ni droplets. Acta Mater. 1998, 46, 4657–4670. [Google Scholar] [CrossRef]
- Li, M.; Lin, X.; Song, G.; Yang, G.; Zhou, Y. Microstructure evolution and metastable phase formation in undercooled Fe–30 at.% Co melt. Mater. Sci. Eng. A 1999, 268, 90–96. [Google Scholar] [CrossRef]
- Herlach, D.M. Metastable materials solidified from undercooled melts. J. Phys. Condens. Matter 2001, 13, 7737. [Google Scholar] [CrossRef]
- Greer, A.; Walker, I. Primary crystallization in (Fe, Ni)-based metallic glasses. J. Non-Cryst. Solids 2003, 317, 78–84. [Google Scholar] [CrossRef]
- Notthoff, C.; Feuerbacher, B.; Franz, H.; Herlach, D.M.; Holland-Moritz, D. Direct Determination of Metastable Phase Diagram by Synchrotron Radiation Experiments on Undercooled Metallic Melts. Phys. Rev. Lett. 2001, 86, 1038–1041. [Google Scholar] [CrossRef]
- Sun, D.Y.; Asta, M.; Hoyt, J.J. Crystal-melt interfacial free energies and mobilities in FCC and BCC Fe. Phys. Rev. B 2004, 69, 174103. [Google Scholar] [CrossRef]
- Mendelev, M.I.; Zhang, F.; Song, H.; Sun, Y.; Wang, C.Z.; Ho, K.M. Molecular dynamics simulation of the solid-liquid interface migration in terbium. J. Chem. Phys. 2018, 148, 214705. [Google Scholar] [CrossRef] [PubMed]
- Mikheev, L.; Chernov, A. Mobility of a diffuse simple crystal—melt interface. J. Cryst. Growth 1991, 112, 591–596. [Google Scholar] [CrossRef]
- David, A.; Kessler, J.K.; Levine, H. Pattern selection in fingered growth phenomena. Adv. Phys. 1988, 37, 255–339. [Google Scholar] [CrossRef]
- Wu, K.A.; Wang, C.H.; Hoyt, J.J.; Karma, A. Ginzburg–Landau theory of the BCC-liquid interface kinetic coefficient. Phys. Rev. B 2015, 91, 014107. [Google Scholar] [CrossRef]
- Xu, X.Q.; Laird, B.B.; Hoyt, J.J.; Asta, M.; Yang, Y. Kinetics of Crystallization and Orientational Ordering in Dipolar Particle Systems. Cryst. Growth Des. 2020, 20, 7862–7873. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, W.; Liang, Z.; Wang, Y.; Lv, S.; Liang, H.; Laird, B.B.; Yang, Y. Local collective dynamics at equilibrium BCC crystal–melt interfaces. J. Chem. Phys. 2022, 157, 084709. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Z.; Zhang, X.; Lu, W.; Yu, Z.; Ma, X.; Liang, H.; Yang, Y. Understanding the kinetic anisotropy of the soft-sphere BCC crystal–melt interfaces. J. Phys. Condens. Matter 2022, 34, 264004. [Google Scholar] [CrossRef]
- Cui, D.; Zhang, J.; Li, X.; Liang, Z.; Zhang, X.; Yang, Y.; Wang, H. Atomistic insights into sluggish crystal growth in an undercooled CoNiCrFe multi-principal element alloy. J. Alloy Compd. 2023, 941, 168881. [Google Scholar] [CrossRef]
- Hansen, J.P.; McDonald, I.R. Chapter 2—Statistical Mechanics, 4th ed.; Academic Press: Oxford, UK, 2013. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Laird, B.B. Crystal Structure and Interaction Dependence of the Crystal-Melt Interfacial Free Energy. Phys. Rev. Lett. 2005, 94, 086102. [Google Scholar] [CrossRef]
- Brańka, A.C.; Heyes, D.M. Thermodynamic properties of inverse power fluids. Phys. Rev. E 2006, 74, 031202. [Google Scholar] [CrossRef]
- Heyes, D.M.; Brańka, A.C. Self-diffusion coefficients and shear viscosity of inverse power fluids: From hard- to soft-spheres. Phys. Chem. Chem. Phys. 2008, 10, 4036. [Google Scholar] [CrossRef] [PubMed]
- Heyes, D.M.; Powles, J.G. Thermal conductivity of fluids with steeply repulsive potentials. Mol. Phys. 2001, 99, 1077–1089. [Google Scholar] [CrossRef]
- Powles, J.G.; Heyes, D.M. Viscoelastic behaviour of fluids with steeply repulsive potentials. Mol. Phys. 2000, 98, 917–927. [Google Scholar] [CrossRef]
- Mladek, B.M.; Gottwald, D.; Kahl, G.; Neumann, M.; Likos, C.N. Formation of Polymorphic Cluster Phases for a Class of Models of Purely Repulsive Soft Spheres. Phys. Rev. Lett. 2006, 96, 045701. [Google Scholar] [CrossRef]
- Agrawal, R.; Kofke, D.A. Solid-Fluid Coexistence for Inverse-Power Potentials. Phys. Rev. Lett. 1995, 74, 122–125. [Google Scholar] [CrossRef]
- Heyes, D.M.; Brańka, A.C. The influence of potential softness on the transport coefficients of simple fluids. J. Chem. Phys. 2005, 122, 234504. [Google Scholar] [CrossRef]
- Morris, J.R. Complete mapping of the anisotropic free energy of the crystal-melt interface in Al. Phys. Rev. B 2002, 66, 144104. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Du, H.; Liang, H.; Yang, Y. Molecular Dynamics Simulation of Monolayer Confined Ice-Water Phase Equilibrium. Acta Chim. Sin. 2018, 76, 483. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, X.; Lv, S.; Liang, H.; Yang, Y. Crystal-Melt Interface Kinetics and the Capillary Wave Dynamics of the Monolayer Confined Ice-Water Coexistence Lines. Acta Chim. Sin. 2021, 79, 108. [Google Scholar] [CrossRef]
- Yang, Y.; Humadi, H.; Buta, D.; Laird, B.B.; Sun, D.; Hoyt, J.J.; Asta, M. Atomistic Simulations of Nonequilibrium Crystal-Growth Kinetics from Alloy Melts. Phys. Rev. Lett. 2011, 107, 025505. [Google Scholar] [CrossRef]
- Monk, J.; Yang, Y.; Mendelev, M.I.; Asta, M.; Hoyt, J.J.; Sun, D.Y. Determination of the crystal-melt interface kinetic coefficient from molecular dynamics simulations. Model. Simul. Mater. Sci. Eng. 2009, 18, 015004. [Google Scholar] [CrossRef]
- Wu, K.A.; Karma, A.; Hoyt, J.J.; Asta, M. Ginzburg–Landau theory of crystalline anisotropy for BCC-liquid interfaces. Phys. Rev. B 2006, 73, 094101. [Google Scholar] [CrossRef]
- Wu, K.A.; Karma, A. Phase-field crystal modeling of equilibrium BCC-liquid interfaces. Phys. Rev. B 2007, 76, 184107. [Google Scholar] [CrossRef]
- Wu, K.A.; Lin, S.C.; Karma, A. Two-mode Ginzburg–Landau theory of crystalline anisotropy for FCC-liquid interfaces. Phys. Rev. B 2016, 93, 054114. [Google Scholar] [CrossRef]
- Shih, W.H.; Wang, Z.Q.; Zeng, X.C.; Stroud, D. Ginzburg–Landau theory for the solid-liquid interface of BCC elements. Phys. Rev. A 1987, 35, 2611–2618. [Google Scholar] [CrossRef]
- Freitas, R.; Reed, E.J. Uncovering the effects of interface-induced ordering of liquid on crystal growth using machine learning. Nat. Commun. 2020, 11, 3260. [Google Scholar] [CrossRef]
- Lu, W.L.; Liang, H.T.; Ma, X.M.; Yuan, Z.F.; Zhang, X.; Liang, Z.; Yang, Y. Atomistic Simulation Study of the FCC and BCC Crystal-Melt Interface Stresses. Surf. Interfaces 2022, 28, 101639. [Google Scholar] [CrossRef]

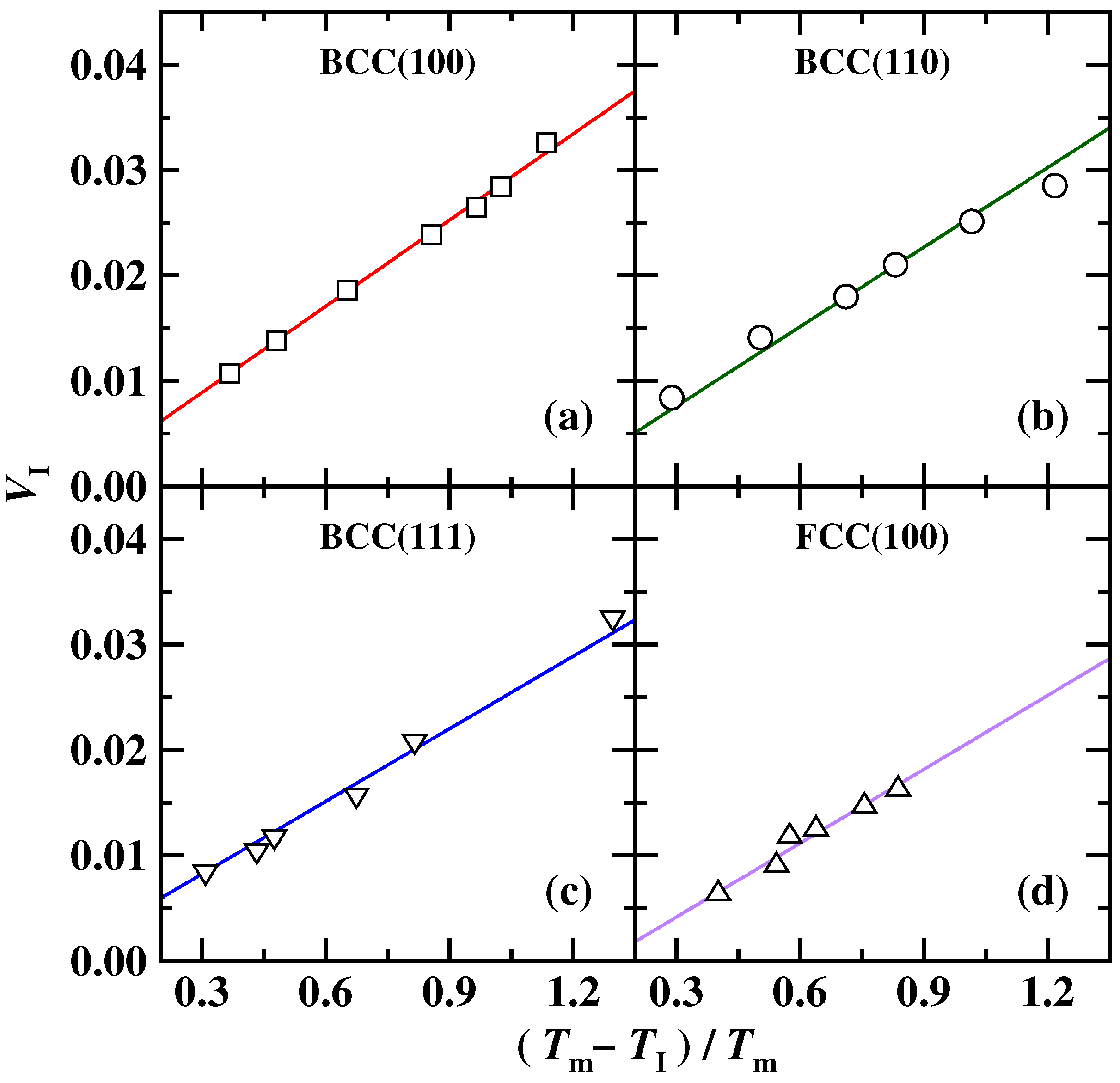
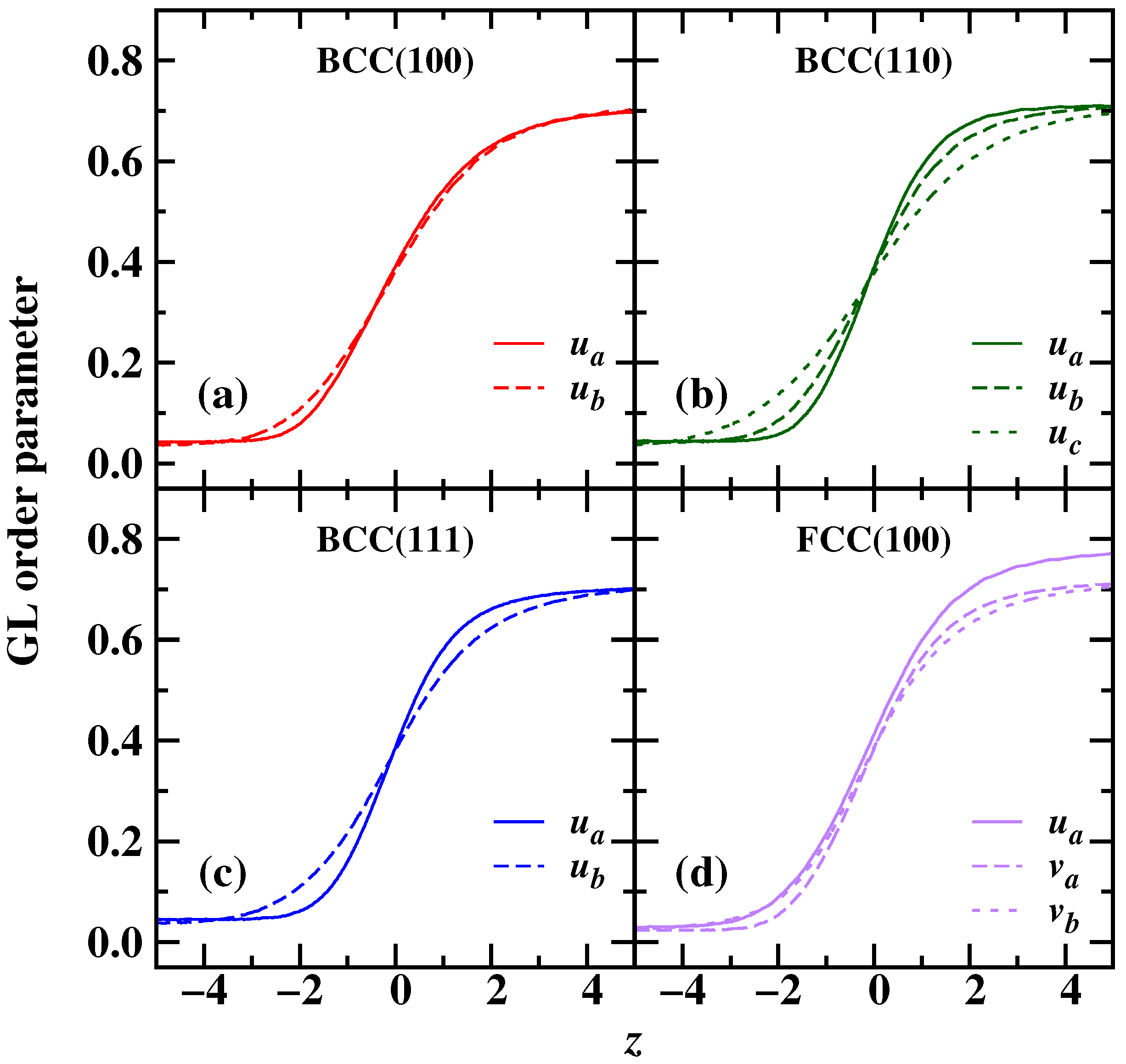

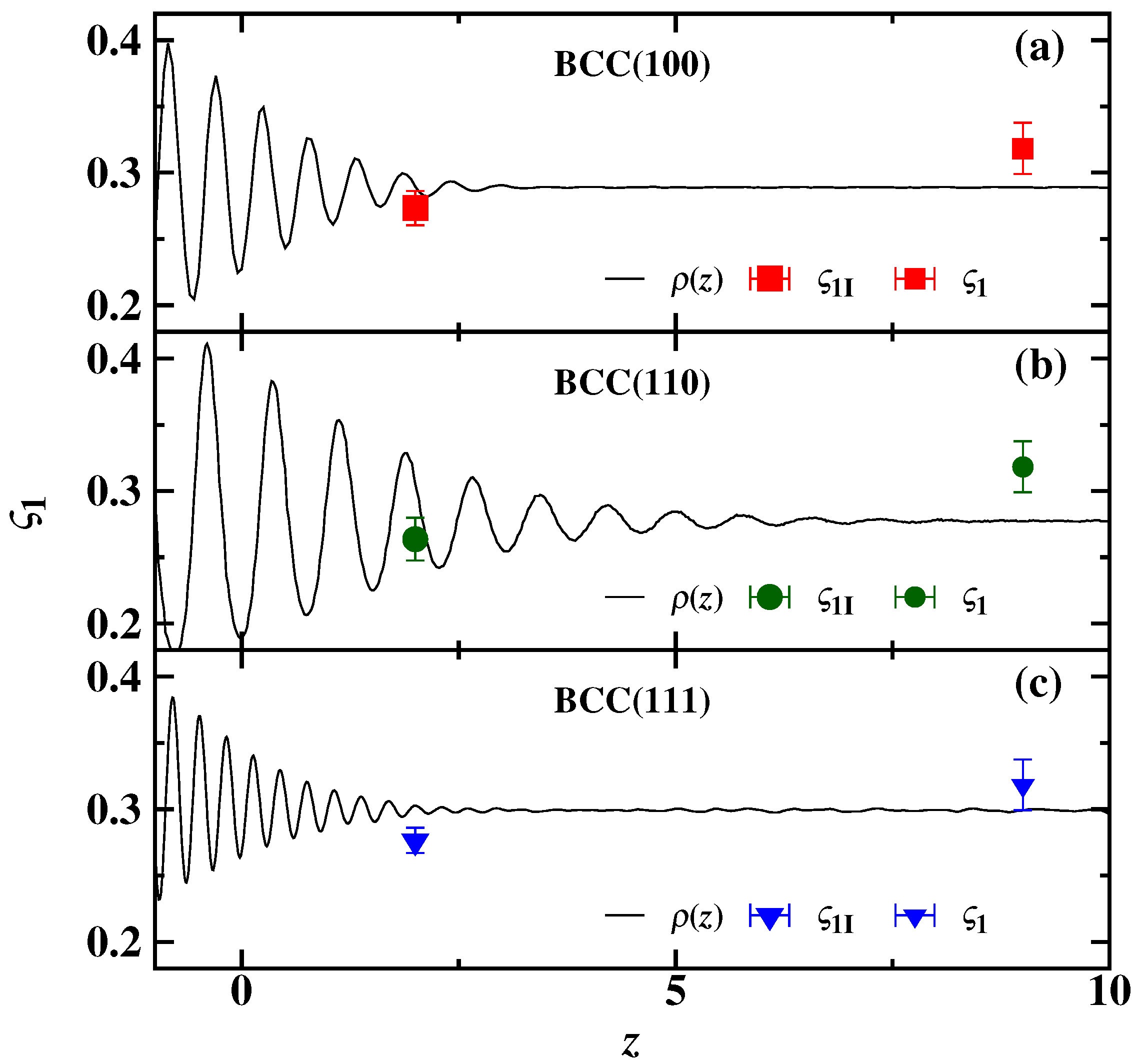
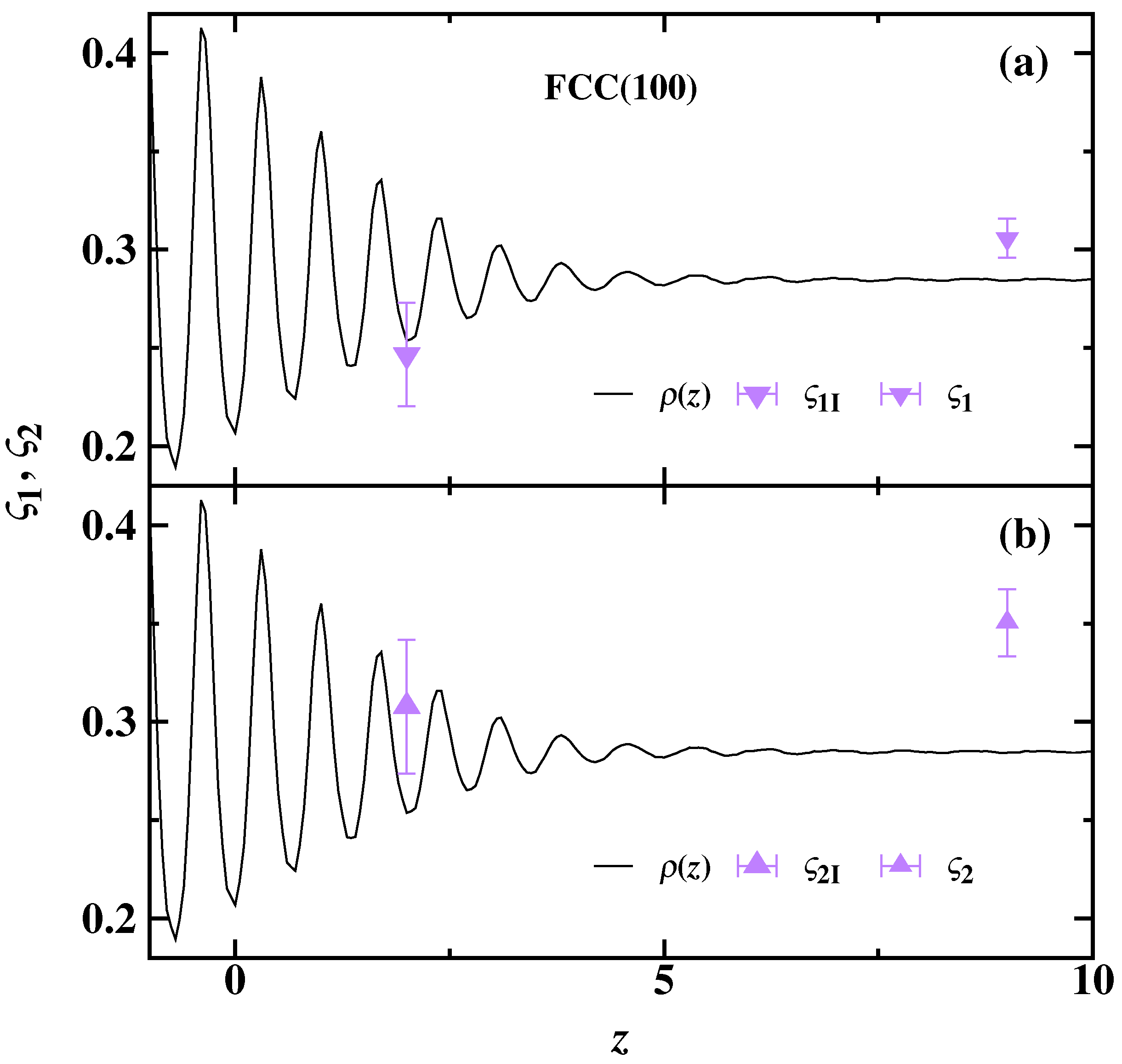

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BCC(100) | BCC(110) | BCC(111) | FCC(100) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OP category | ||||||||||
| Numbers of or | 4 | 8 | 2 | 8 | 2 | 6 | 6 | 8 | 4 | 2 |
| or | 0 | 0 | 1 | 0 | 0 | 1 | ||||
| 2.7(1) | 2.3(2) | 2.7(1) | |
| 2.5(1) | 2.1(1) | 2.4(2) | |
| 2.3(1) | 2.0(1) | 2.3(1) | |
| 2.3(2) | 1.8(1) | 2.2(2) | |
| 1.09(6) | 1.13(10) | 1.11(9) | |
| 1.19(8) | 1.16(10) | 1.19(6) | |
| 1.09(9) | 1.03(9) | 1.07(8) |
| A | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BCC(100) | 0.708 | 0.30(2) | 0.26(1) | 0.345 | 0.609 | - | - | - | 0.32(2) | - | 0.27(1) | - |
| BCC(110) | 0.708 | 0.35(2) | 0.29(2) | 0.233 | 0.728 | 0.129 | - | - | 0.32(2) | - | 0.26(2) | - |
| BCC(111) | 0.708 | 0.36(2) | 0.31(1) | 0.665 | 0.452 | - | - | - | 0.32(2) | - | 0.28(1) | - |
| FCC(100) | 0.858 | 0.47(2) | 0.39(3) | 0.860 | - | - | 0.413 | 0.172 | 0.31(1) | 0.35(4) | 0.25(3) | 0.31(3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Z.; Zhang, X.; Wang, Y.; Lv, S.; Alexandrov, D.V.; Liang, H.; Yang, Y. A Comparative Computational Study of the Solidification Kinetic Coefficients for the Soft-Sphere BCC-Melt and the FCC-Melt Interfaces. Crystals 2024, 14, 292. https://doi.org/10.3390/cryst14040292
Liang Z, Zhang X, Wang Y, Lv S, Alexandrov DV, Liang H, Yang Y. A Comparative Computational Study of the Solidification Kinetic Coefficients for the Soft-Sphere BCC-Melt and the FCC-Melt Interfaces. Crystals. 2024; 14(4):292. https://doi.org/10.3390/cryst14040292
Chicago/Turabian StyleLiang, Zun, Xin Zhang, Yashen Wang, Songtai Lv, Dmitri V. Alexandrov, Hongtao Liang, and Yang Yang. 2024. "A Comparative Computational Study of the Solidification Kinetic Coefficients for the Soft-Sphere BCC-Melt and the FCC-Melt Interfaces" Crystals 14, no. 4: 292. https://doi.org/10.3390/cryst14040292