
Synthesis and Reactivity of Novel Boranes Derived from Bulky Salicylaldimines: The Molecular Structure of a Maltolato Compound

Abstract
:
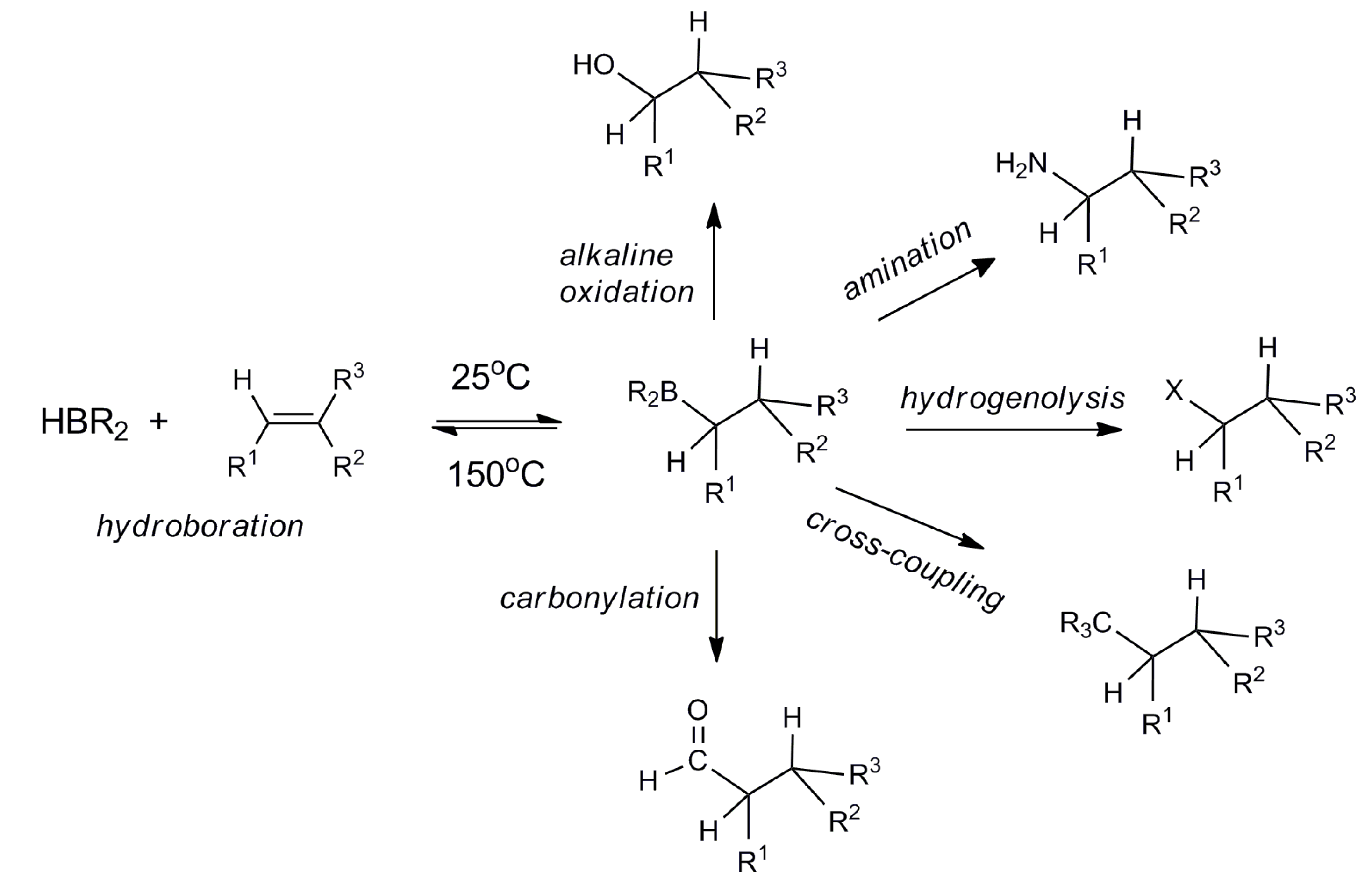
1. Introduction

2. Results and Discussion
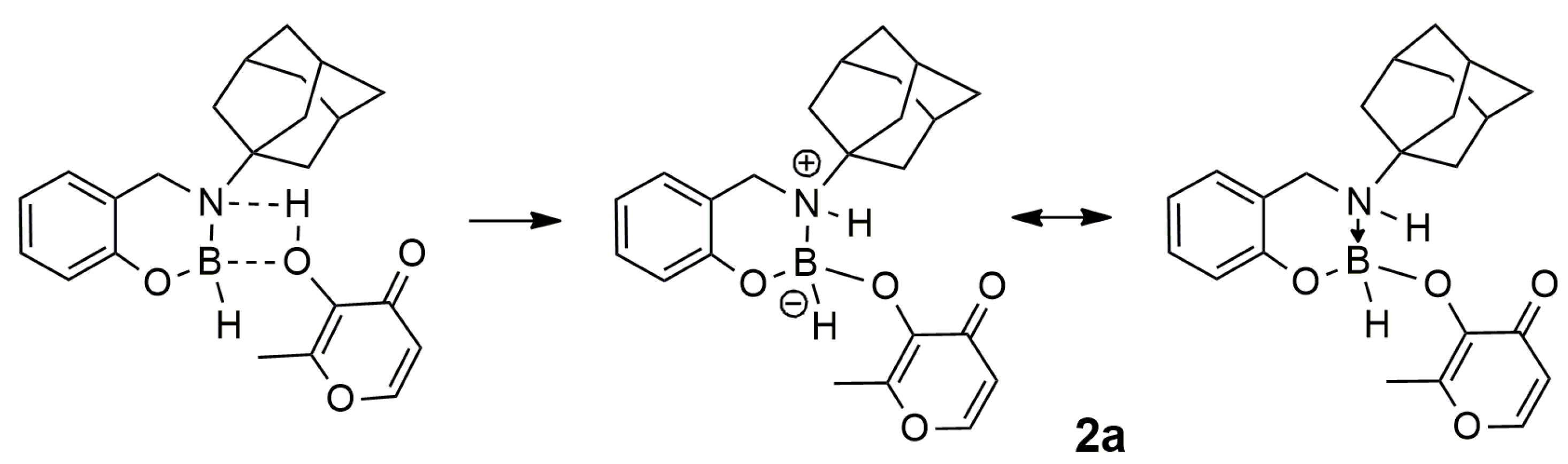
2.1. Synthesis and Reactivity

2.2. Molecular Structures


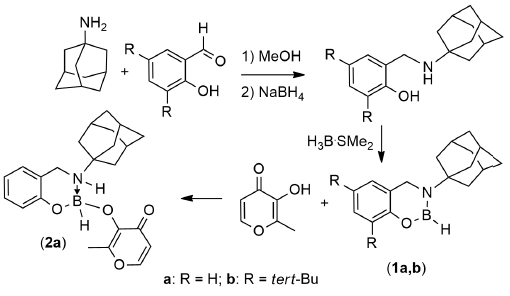{kind=link}
{kind=link}
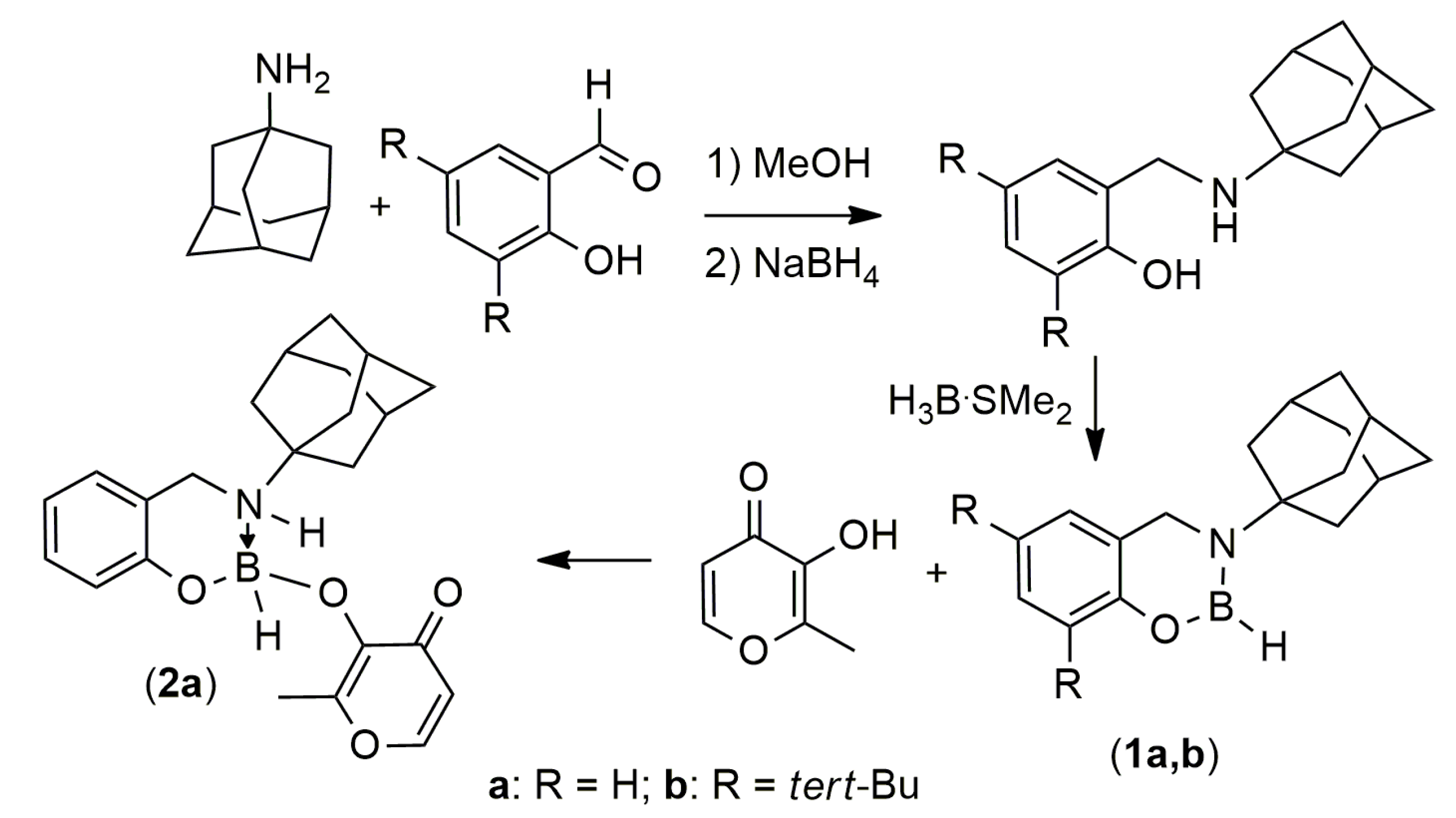{kind=link}
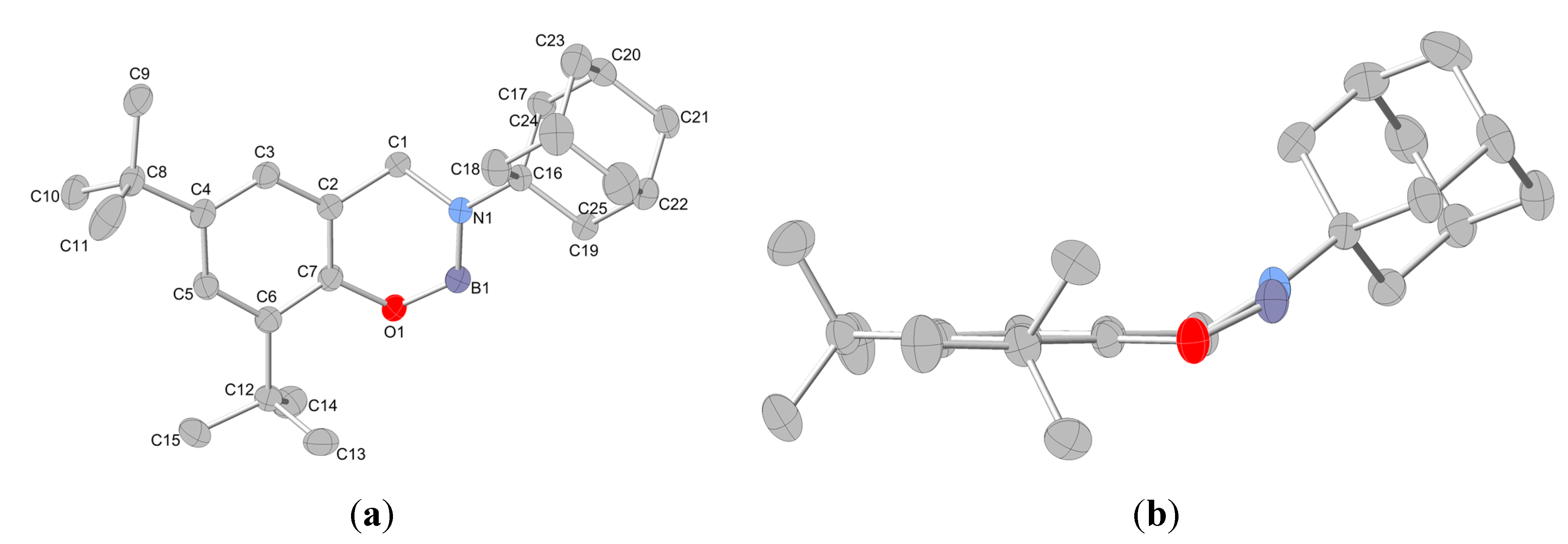{kind=link}
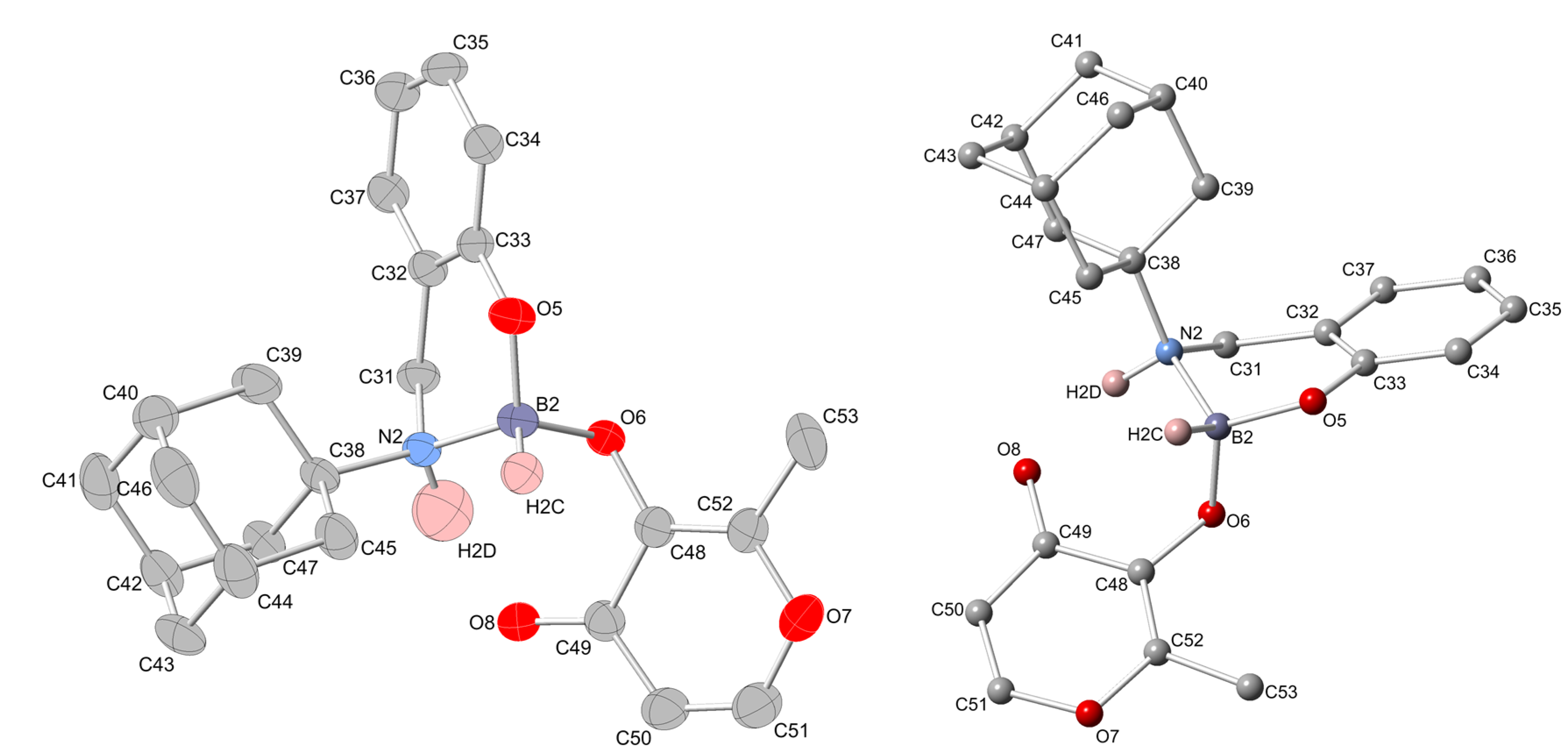{kind=link}
{kind=link}
| Complex | 1b | 2a |
|---|---|---|
| Formula | C25H38BNO | C23H28BNO4 |
| Molecular weight | 379.37 | 393.27 |
| Crystal system | Triclinic | Orthorhombic |
| Space group | Pī | Pna2(1) |
| a/Å | 9.1267(19) | 18.519(6) |
| b/Å | 11.676(2) | 17.315(5) |
| c/Å | 12.240(3) | 12.680(4) |
| α/° | 66.840(3) | 90 |
| β/° | 78.529(3) | 90 |
| γ/° | 67.354(3) | 90 |
| V/Å3 | 1105.0(4) | 4066(2) |
| Z | 2 | 8 |
| ρcalc./Mg·m−3 | 1.140 | 1.285 |
| Crystal size/mm3 | 0.60 × 0.45 × 0.20 | 0.45 × 0.20 × 0.10 |
| Temp/K | 198(1) | 188(1) |
| Radiation | Mo-Kα (λ = 0.71073 Å) | Mo-Kα (λ = 0.71073 Å) |
| μ/mm−1 | 0.067 | 0.086 |
| Total reflections | 7700 | 27270 |
| Total unique reflections | 4799 | 4981 |
| No. of variables | 405 | 541 |
| θ Range/° | 1.81 to 27.50 | 1.61 to 28.48 |
| Largest difference peak/hole/e Å−3 | 0.359 and −0.160 | 0.350 and −0.259 |
| S (GoF) on F2 | 1.042 | 1.082 |
| R1a (I > 2σ(I)) | 0.0427 | 0.0618 |
| wR2b (all data) | 0.1256 | 0.1780 |


3. Experimental Section
3.1. General
3.2. Synthesis of 3-(adamantan-1-yl)-3,4-dihydro-2H-benzo[e][1,3,2]oxazaborinane (1a)
3.3. Synthesis of 3-(adamantan-1-yl)-6,8-di-tert-butyl-3,4-dihydro-2H-enzo[e][1,3,2]oxazaborinane (1b)
3.4. Synthesis of 2a
3.5. X-ray Crystallography
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clay, J.M.; Vedejs, E. Hydroboration with pyridine borane at room temperature. J. Am. Chem. Soc. 2005, 127, 5766–5767. [Google Scholar] [CrossRef] [PubMed]
- Atkins, W.J., Jr.; Burkhardt, E.R.; Matos, K. Safe handling of boranes at scale. Org. Process Res. Dev. 2006, 10, 1292–1295. [Google Scholar] [CrossRef]
- Kanth, J.V.B. Borane-amine complexes for hydroboration. Aldrichim. Acta 2002, 35, 57–66. [Google Scholar]
- Fritschi, C.B.; Wernitz, S.B.; Vogels, C.M.; Shaver, M.P.; Decken, A.; Bell, A.; Westcott, S.A. 4,4,5,5-Tetraphenyl-1,3,2-dioxaborolane: A bulky borane for the transition metal catalysed hydroboration of alkenes. Eur. J. Inorg. Chem. 2008, 2008, 779–785. [Google Scholar] [CrossRef]
- Hunter, N.M.; Vogels, C.M.; Decken, A.; Bell, A.; Westcott, S.A. [Cp*IrCl2]2 catalyzed hydroborations of alkenes using a bulky dioxaborocine. Inorg. Chim. Acta 2011, 365, 408–413. [Google Scholar] [CrossRef]
- Cho, B.T. Recent advances in the synthetic applications of the oxazaborolidine-mediated asymmetric reduction. Tetrahedron 2006, 62, 7621–7643. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Tao, R. 2-[(Adamantan-1-ylamino)methyl]phenol. Acta Cryst. 2012, E68. [Google Scholar] [CrossRef]
- Wang, J.; Ma, C.; Wang, J.; Jo, H.; Canturk, B.; Fiorin, G.; Pinto, L.H.; Lamb, R.A.; Klein, M.L.; DeGrado, W.F. Discovery of novel dual inhibitors of the wild-type and the most prevalent drug-resistant mutant, S31N, of the M2 proton channel from influenza A virus. J. Med. Chem. 2013, 56, 2804–2812. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.N.; Srebnik, M.; Brown, H.C. Chiral oxazaborolidines as catalysts for the enantioselective addition of diethylzinc to aldehydes. Tetrahedron Lett. 1989, 30, 5551–5554. [Google Scholar] [CrossRef]
- Vogels, C.M.; Westcott, S.A. Recent advances in organic synthesis using transition metal-catalyzed hydroborations. Curr. Org. Chem. 2005, 9, 687–699. [Google Scholar] [CrossRef]
- Corey, E.J.; Azimioara, M.; Sarshar, S. X-Ray crystal structure of a chiral oxazaborolidine catalyst for enantioselective carbonyl reduction. Tetrahedron Lett. 1992, 33, 3429–3430. [Google Scholar] [CrossRef]
- Shan, N.; Zaworotko, M.J. The role of cocrystals in pharmaceutical science. Drug Discov. Today 2008, 13, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; DePue, L.J.; Struss, J.E.; Patel, B.P.; Parkin, S.; Atwood, D.A. Mononuclear Schiff base boron halides: Synthesis, characterization, and dealkylation of trimethyl phosphate. Inorg. Chem. 2006, 45, 9213–9224. [Google Scholar] [CrossRef] [PubMed]
- SAINT 7.23A; Bruker AXS, Inc.: Madison, WI, USA, 2006.
- SADABS 2008, George Sheldrick; Bruker AXS, Inc.: Madison, WI, USA, 2008.
- Sheldrick, G.M. SHELXTL. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourque, J.L.; Geier, S.J.; Vogels, C.M.; Decken, A.; Westcott, S.A. Synthesis and Reactivity of Novel Boranes Derived from Bulky Salicylaldimines: The Molecular Structure of a Maltolato Compound. Crystals 2015, 5, 91-99. https://doi.org/10.3390/cryst5010091
Bourque JL, Geier SJ, Vogels CM, Decken A, Westcott SA. Synthesis and Reactivity of Novel Boranes Derived from Bulky Salicylaldimines: The Molecular Structure of a Maltolato Compound. Crystals. 2015; 5(1):91-99. https://doi.org/10.3390/cryst5010091
Chicago/Turabian StyleBourque, Jeremy L., Stephen J. Geier, Christopher M. Vogels, Andreas Decken, and Stephen A. Westcott. 2015. "Synthesis and Reactivity of Novel Boranes Derived from Bulky Salicylaldimines: The Molecular Structure of a Maltolato Compound" Crystals 5, no. 1: 91-99. https://doi.org/10.3390/cryst5010091