
Effect of Intra- and Intermolecular Interactions on the Properties of para-Substituted Nitrobenzene Derivatives


Abstract
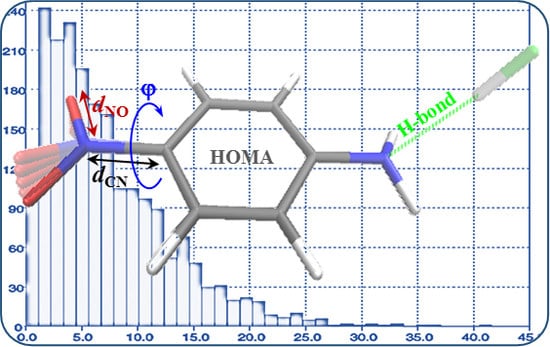
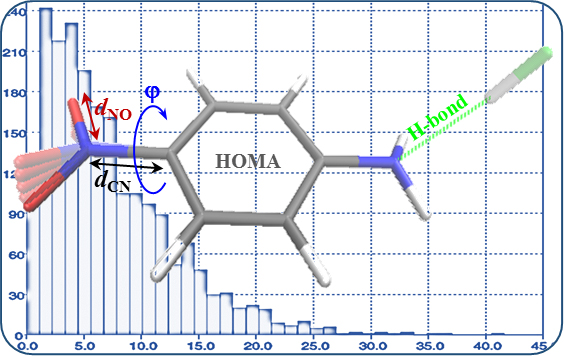
:
1. Introduction
2. Experimental and Computational Section
- (1)
- The search was performed for organic compounds containing para-substituted nitrobenzene fragment(s).
- (2)
- The search was restricted to structure measurements with the reported mean estimated standard deviation (esd) for the CC bond ≤ 0.005 Å, with 3D coordinates determined, not disordered, without errors, polymeric, ions and powder structures, and with R ≤ 0.05. Sometimes solvent molecules were also present in the crystal latice.
3. Results and Discussion
3.1. Analyses of Molecular Geometry Deformations Based on the Cambridge Structural Database
3.2. Quantum Chemistry Modeling
3.2.1. Consequences of the Nitro Group Rotation in p-Nitroaniline
3.2.2. Consequences of the H-Bond Formation at the Amino Group in p-Nitroaniline
4. Conclusions
- (1)
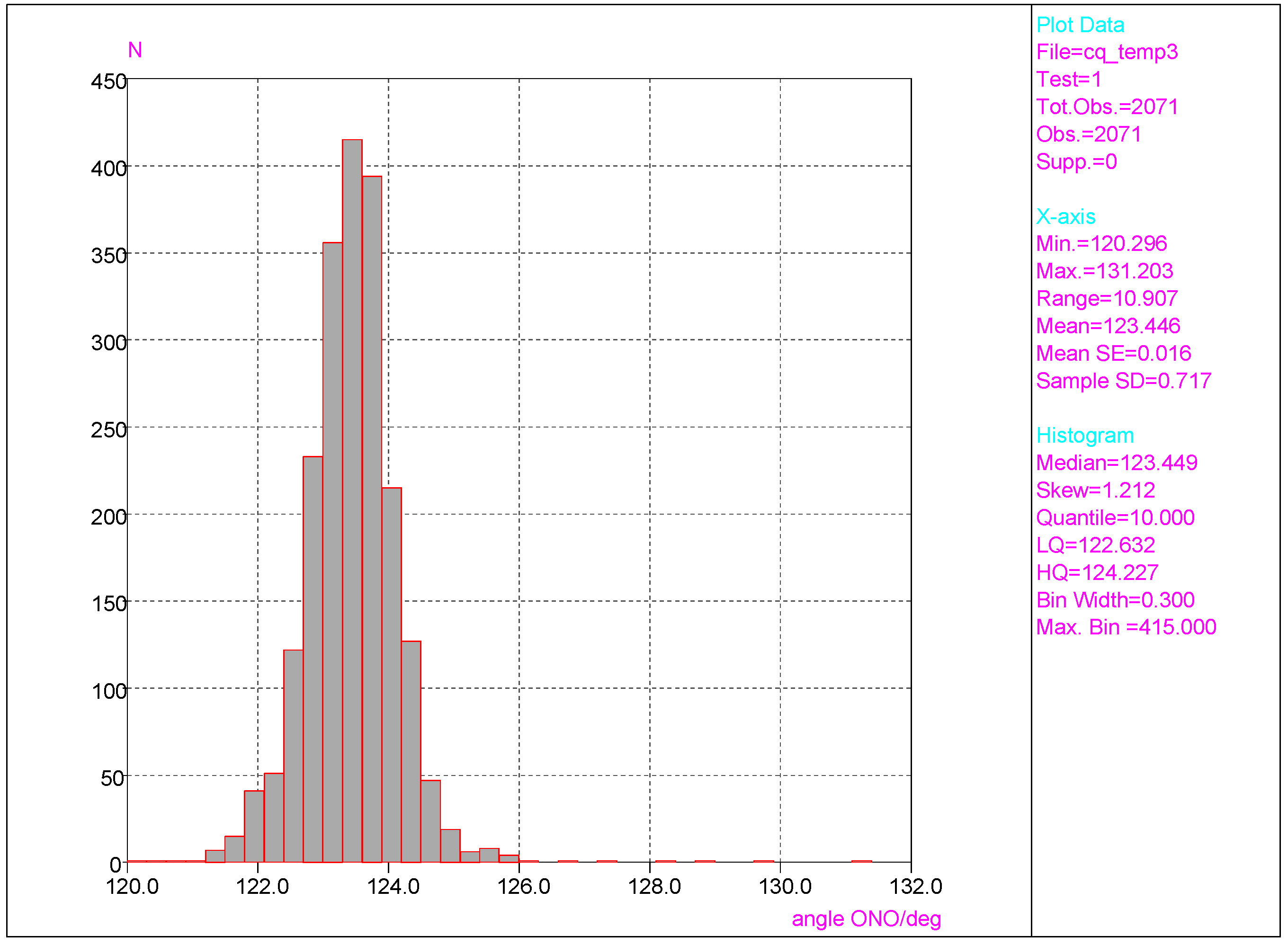
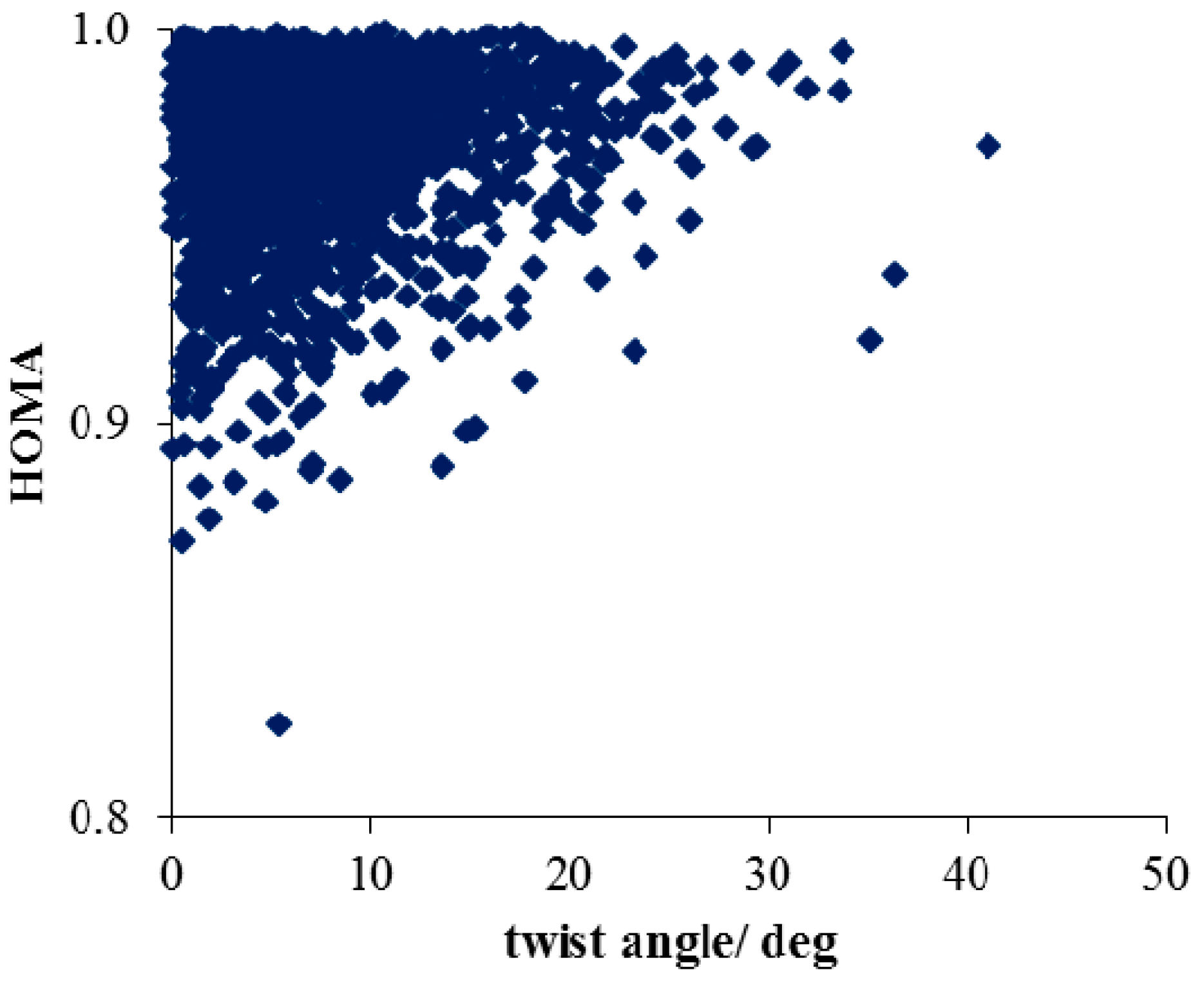
- Considering CSD data, deformations of the ONO angle are small, with the mean value (123.4 deg) close to the value for nitrobenzene in the crystal (123.2(1) deg) with a small value of esd (ca. 0.7). This indicates a rather small effect of the chaotic action of the neighboring molecules in the crystal lattice on this angle of the NO2 group. Similar results are found for the NO bond lengths.
- (2)
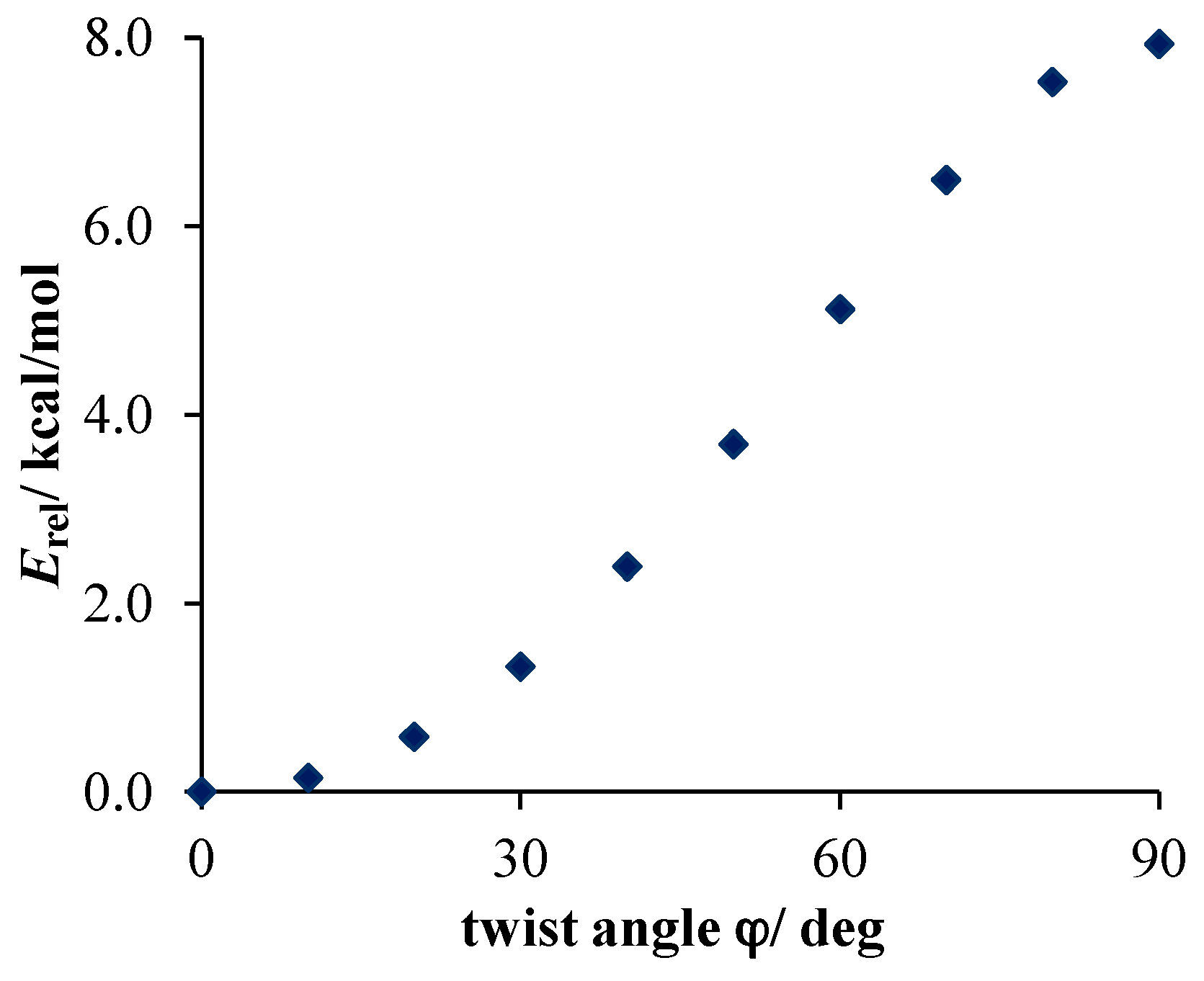
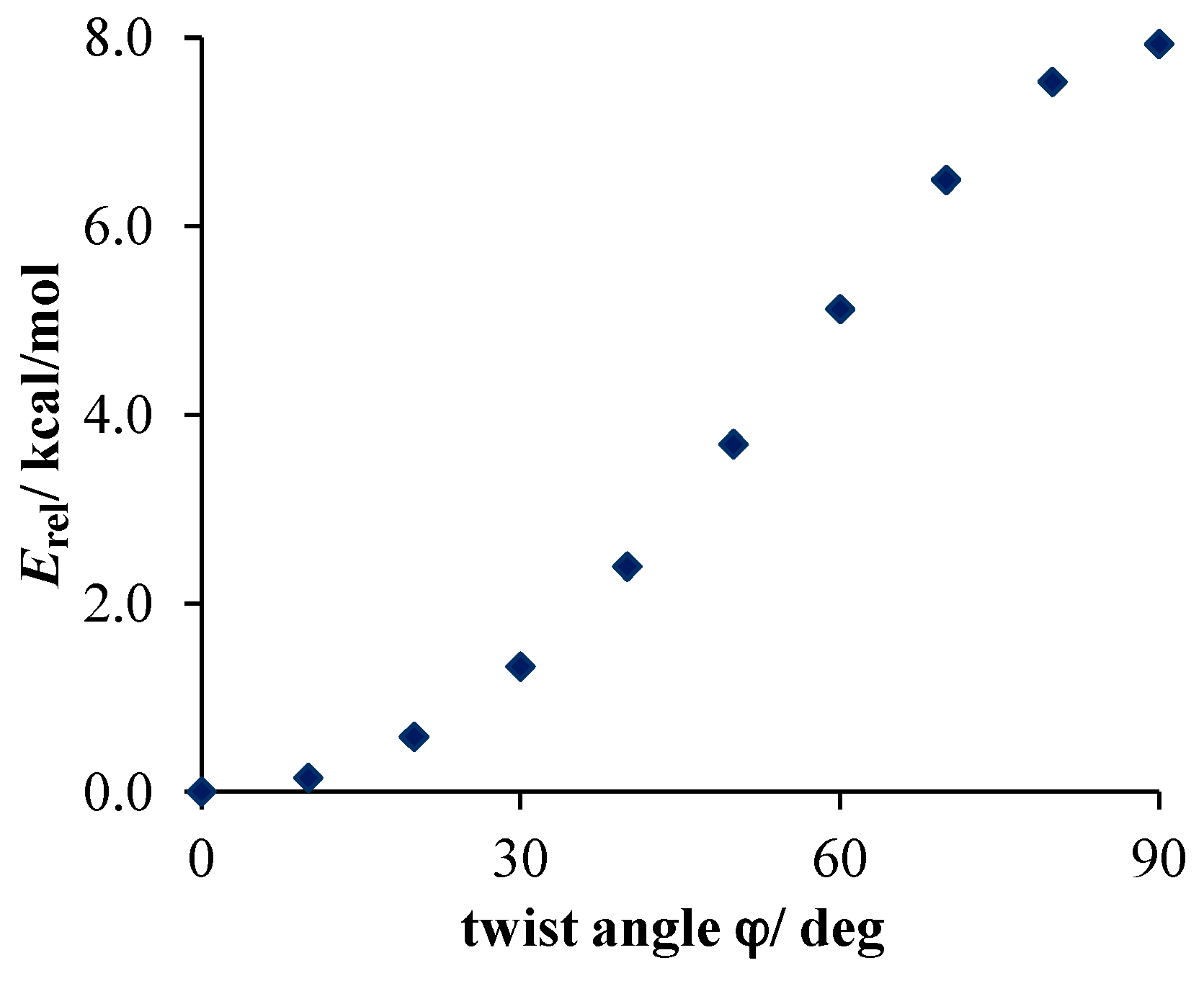
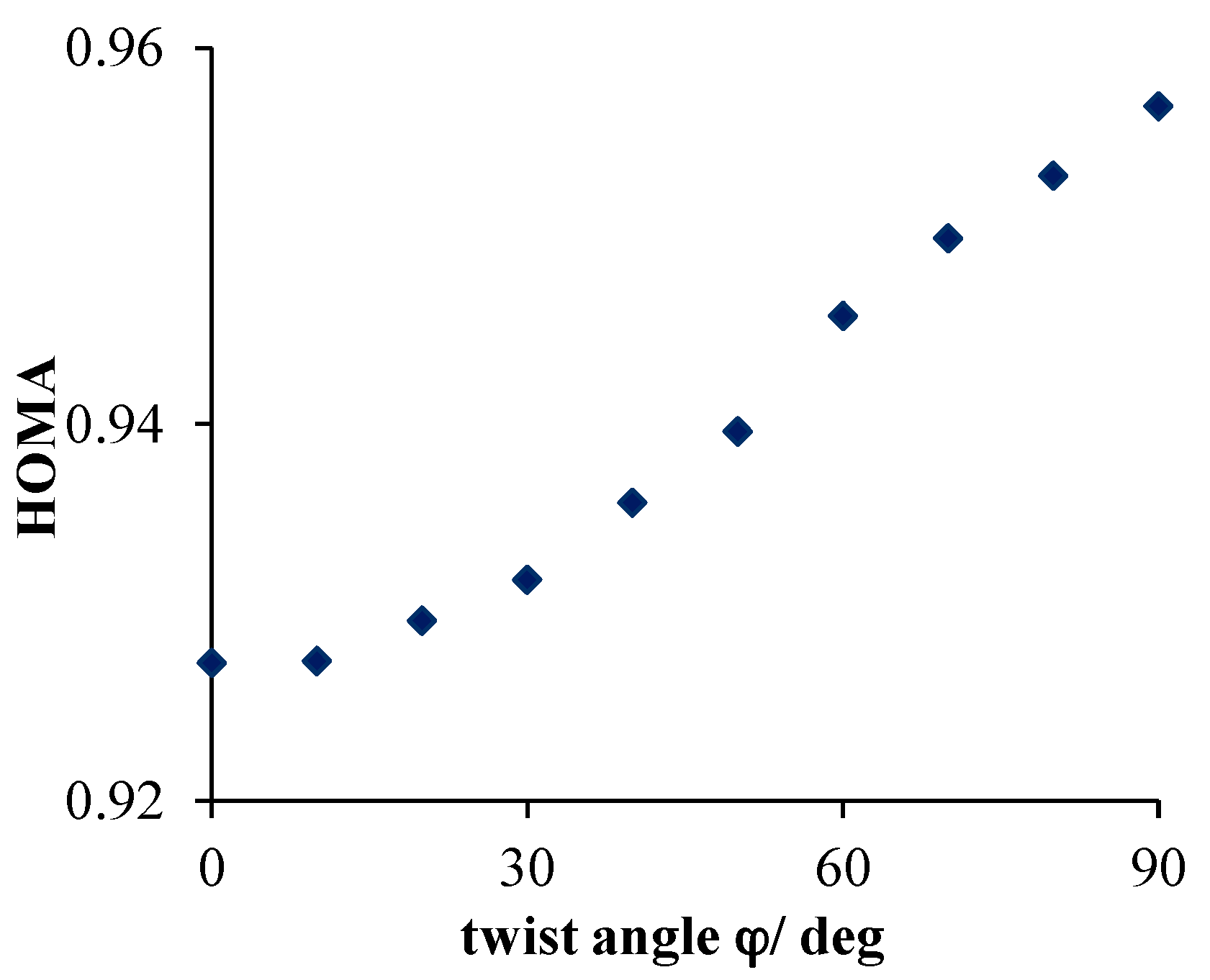
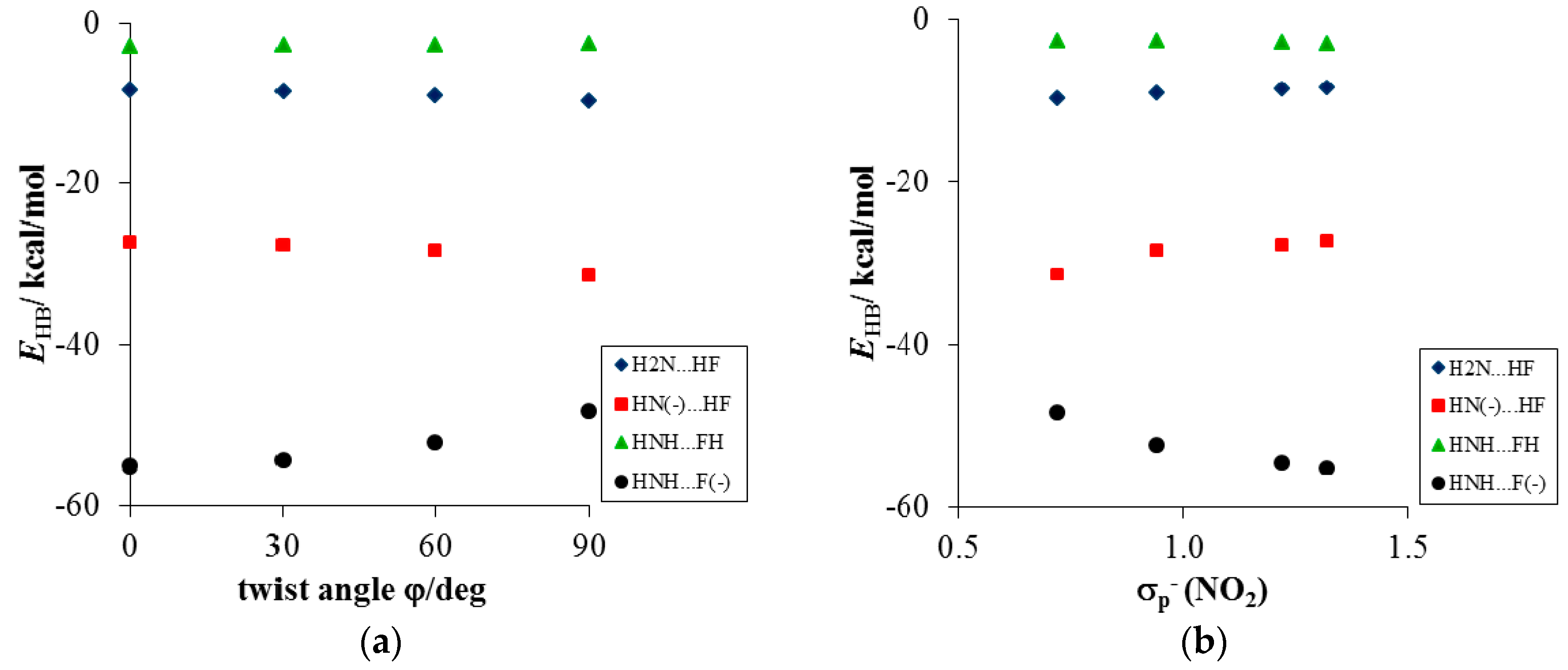
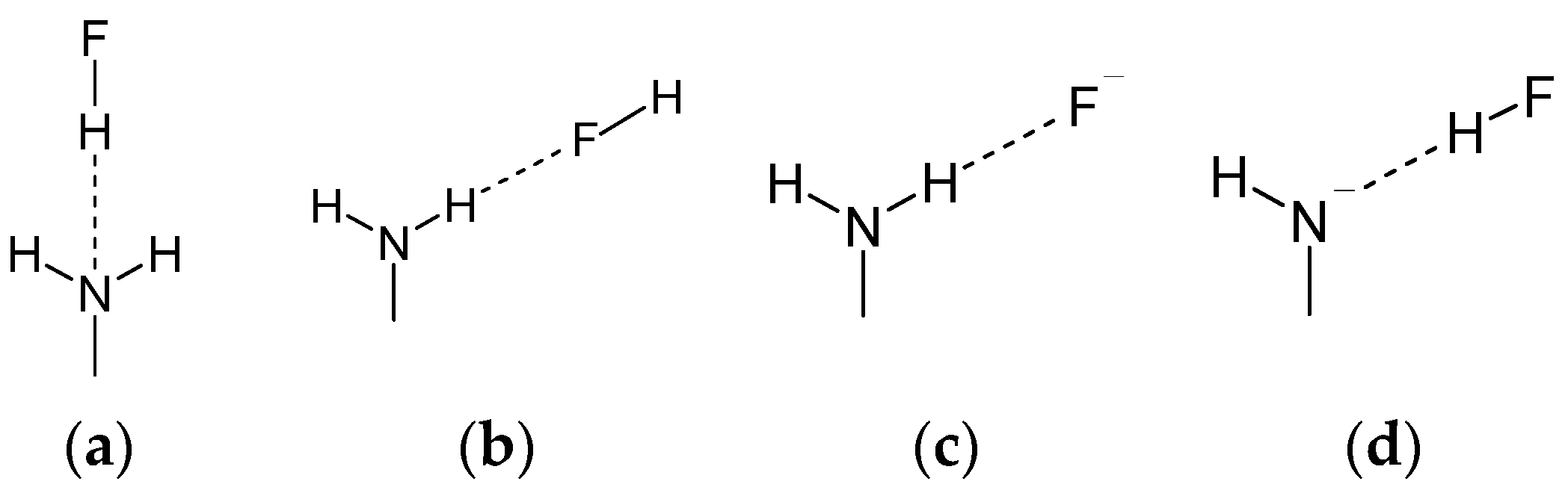
- Much stronger deformations are observed for the twist angle, with a maximum value of ~40 deg, but a mean value of 7.3 deg with a rather large esd (5.6 deg). This is in line with QM results for PNA showing that rotation of the NO2 group with respect to the benzene ring up to 30 degrees is associated with very small changes in energy of the system, namely less than 2.0 kcal/mol, whereas to achieve a perpendicular orientation ca. 8.0 kcal/mol is required.
- (3)
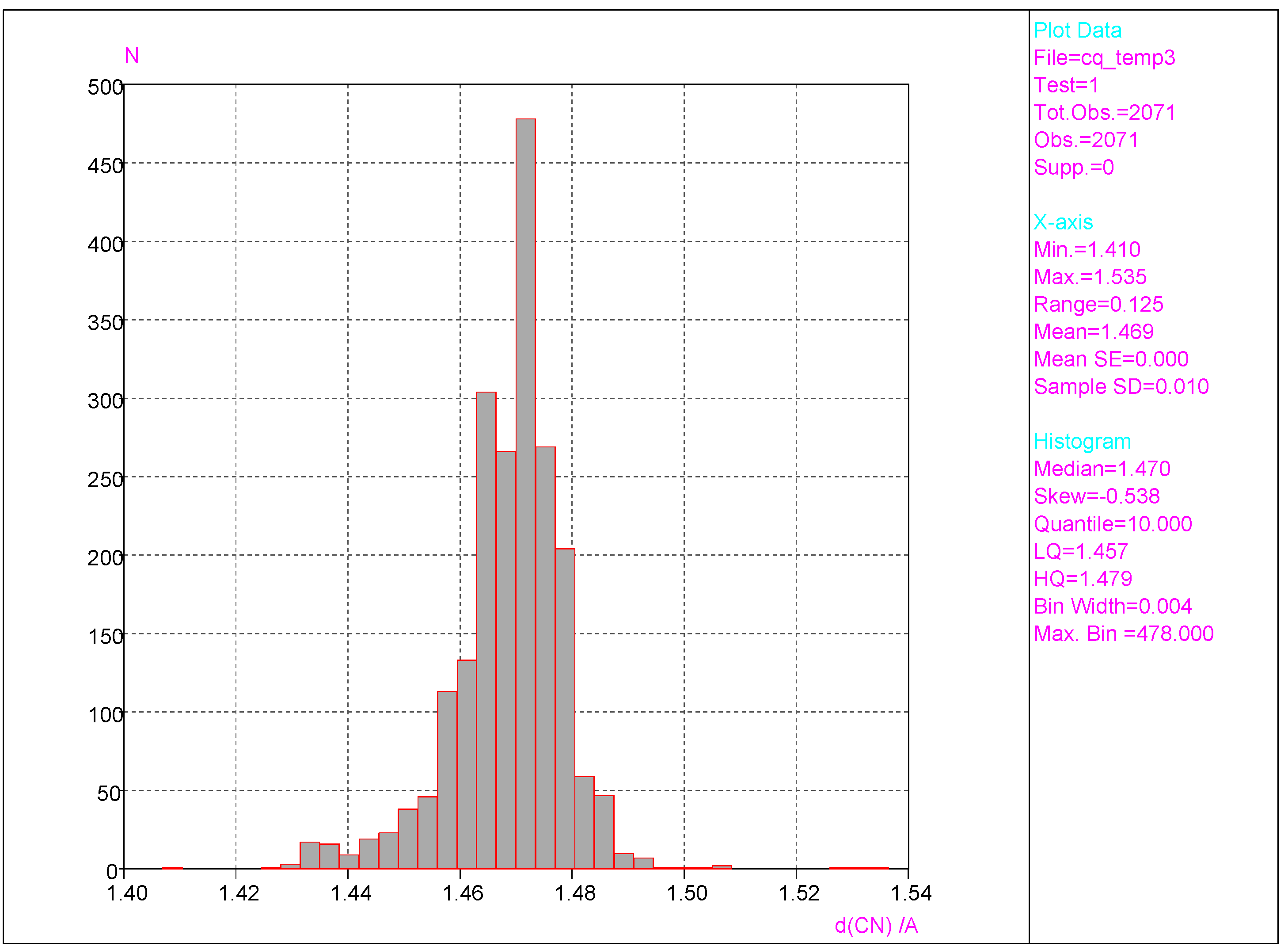
- Substantial range of the CN bond length is observed in the crystals of nitrobenzene derivatives (from 1.410 to 1.535 Å). Shortening of the CN bond length indicates an increased contribution of the quinoid structure and as a consequence a lower aromaticity of the ring. Such changes can be caused either by distortion from coplanar orientation between the NO2 group and the phenyl ring or by the effect of a substituent located in the para-position, as was confirmed by QM results for PNA and its H-bonded complexes.
- (4)
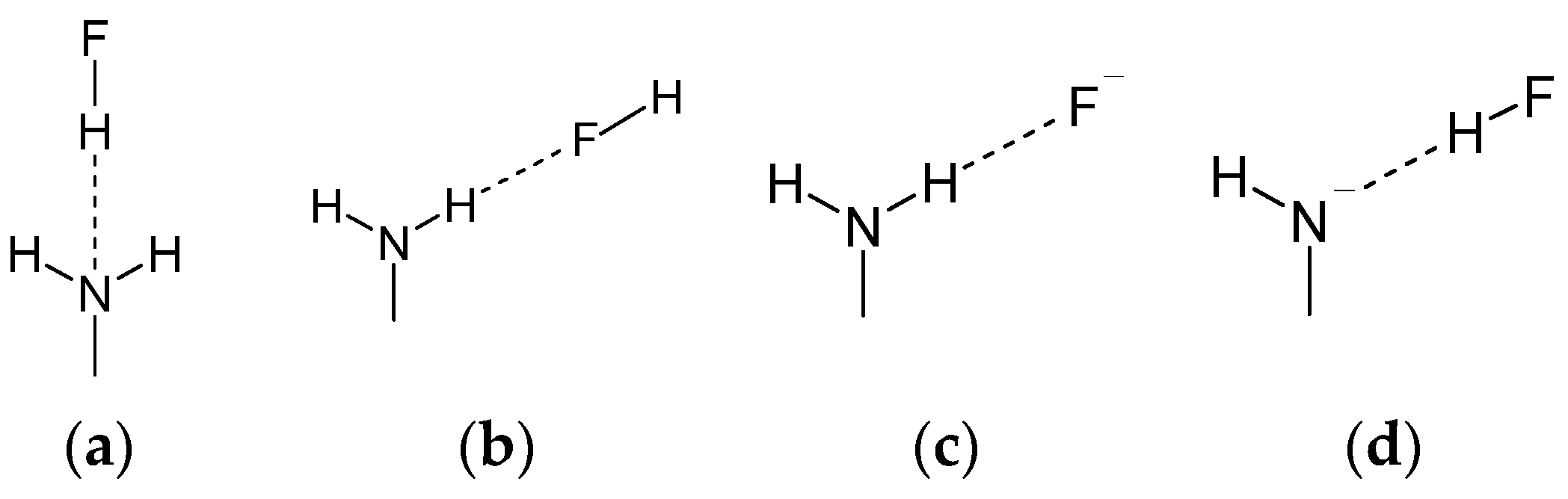
- Energy decomposition analysis of the H-bonds in the modeled complexes of aniline and PNA revealed that the amino group can form hydrogen bonds in a wide range of strengths (from −2.0 to −55.0 kcal/mol). The results support the view that these interactions are mostly provided by electrostatic attraction, however a significant covalent contribution has to be considered.
- (5)
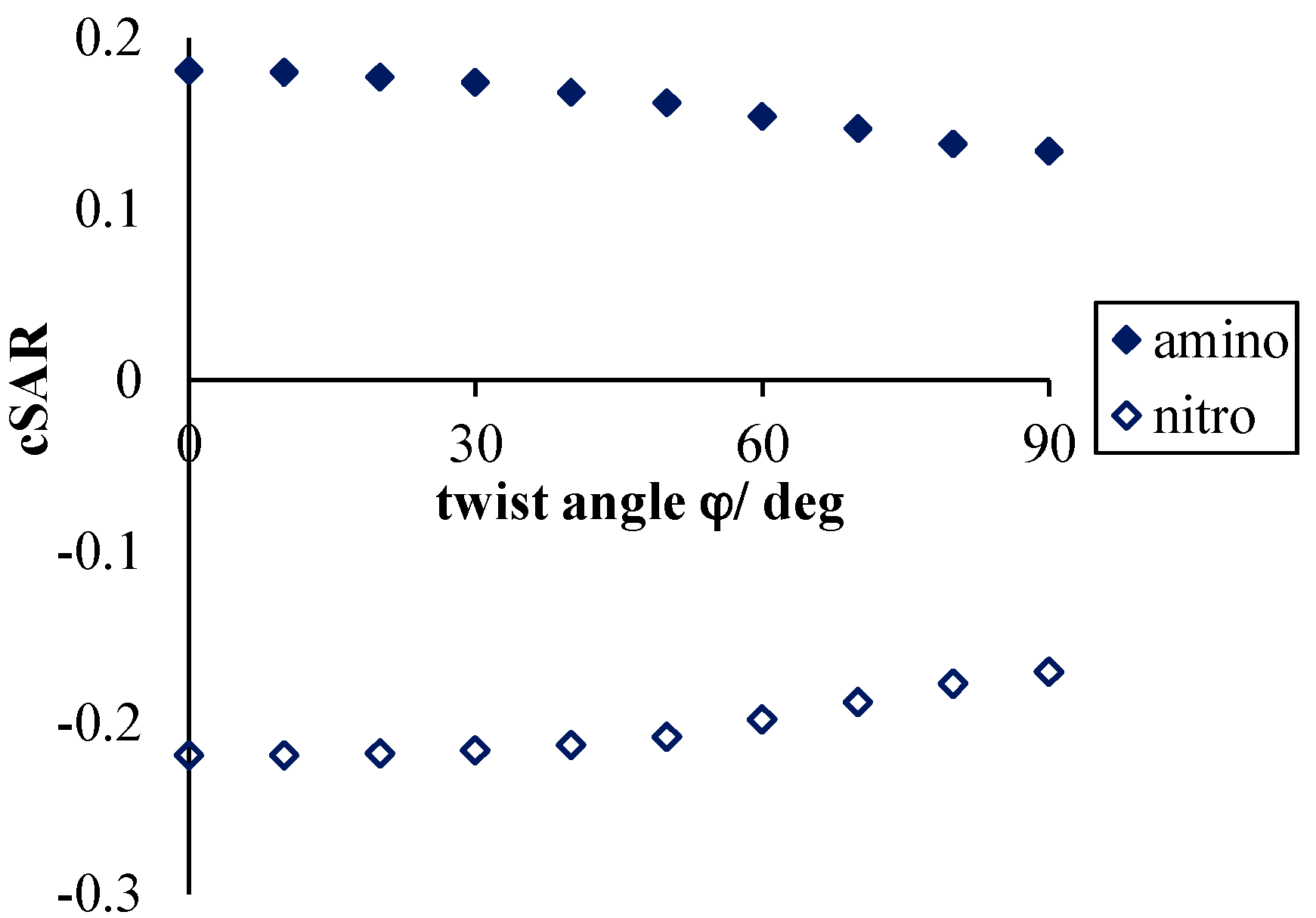
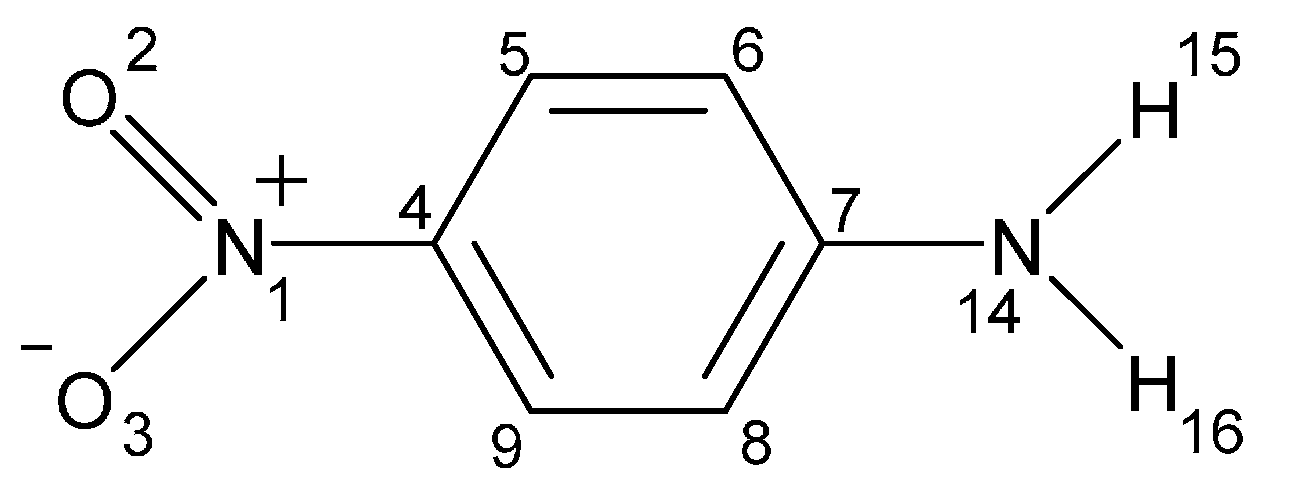
- Application of the VDD atomic charges and the cSAR concept allows an estimation of the magnitude of the intramolecular charge transfer. For all of the studied systems, a mutually dependent change in characteristics of both groups (NO2 and NH2) was observed. It was shown that the electron-attracting ability of the nitro group dramatically depends on the moiety to which it is attached (see Table 1) as well as on intermolecular interactions occurring even at distant parts of the studied systems.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, M.B. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th ed.; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Stasyuk, O.A.; Szatylowicz, H.; Krygowski, T.M.; Fonseca Guerra, C. How amino and nitro substituents direct electrophilic aromatic substitution in benzene: An explanation with Kohn–Sham molecular orbital theory and Voronoi deformation density analysis. Phys. Chem. Chem. Phys. 2016. [Google Scholar] [CrossRef] [PubMed]
- Campanelli, A.R.; Domenicano, A.; Ramondo, F.; Hargittai, I. Group electronegativities from benzene ring deformations: A quantum chemical study. J. Phys. Chem. A 2004, 108, 4940–4948. [Google Scholar] [CrossRef]
- Exner, O. A critical compilation of substituent constants. Chapter 10. In Correlation Analysis in Chemistry: Recent Advances; Chapman, N.B., Shorter, J., Eds.; Plenum Press: New York, NY, USA, 1978; pp. 439–540. [Google Scholar]
- Dobrowolski, M.A.; Krygowski, T.M.; Cyrański, M.K. Substituent constants (σp−) of the rotated nitro group. The interplay between the substituent effect of a rotated −NO2 group and H-bonds affecting π-electron delocalization in 4-nitrophenol and 4-nitrophenolate complexes: A B3LYP/6–311+G** study. Croat. Chim. Acta 2009, 82, 139–147. [Google Scholar]
- Xu, J. Synthesis of β-lactams with π electron-withdrawing substituents. Tetrahedron 2012, 68, 10696–10747. [Google Scholar] [CrossRef]
- Zhang, C. Review of the establishment of nitro group charge method and its applications. J. Hazard. Mater. 2009, 161, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Badgujar, D.M.; Talawar, M.B.; Asthana, S.N.; Mahulikar, P.P. Advances in science and technology of modern energetic materials: An overview. J. Hazard. Mater. 2008, 151, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Rouchaud, J.; Neus, O.; Cools, K.; Bulcke, R. Dissipation of the triketone mesotrione herbicide in the soil of corn crops grown on different soil types. Toxicol. Environ. Chem. 2000, 77, 31–40. [Google Scholar] [CrossRef]
- Irle, S.; Krygowski, T.M.; Niu, J.E.; Schwarz, W.H.E. Substituent effects of −NO and −NO2 groups in aromatic systems. J. Org. Chem. 1995, 60, 6744–6755. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Data Centre (CCDC). Vista—A Program for the Analysis and Display of Data Retrieved from the CSD; Cambridge Crystallographic Data Centre: Cambridge, UK, 1994. [Google Scholar]
- Te Velde, G.; Bickelhaupt, F.M.; van Gisbergen, S.J.A.; Fonseca Guerra, C.; Baerends, E.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF 2012; SCM, Theoretical Chemistry, Vrije Universiteit: Amsterdam, The Netherlands, 2012.
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef] [PubMed]
- Snijders, J.G.; Baerends, E.J.; Vernooijs, P. Roothaan-Hartree-Fock-Slater atomic wave functions. Single-zeta, double-zeta, and extended Slater-type basis sets for 87Fr-103Lr. Atom. Data Nucl. Data Tables 1981, 26, 483–509. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method: I. The transition state method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. A theoretical study of the ethylene-metal bond in complexes between Cu+, Ag+, Au+, Pt0, or Pt2+ and ethylene, based on the Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1558–1565. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. CO, CS, N2, PF3, and CNCH3 as σ donors and π acceptors. A theoretical study by the Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Grabowski, S.J. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Bickelhaupt, F.M.; van Eikema Hommes, N.J.R.; Fonseca Guerra, C.; Baerends, E.J. The carbon-lithium electron pair bond in (CH3Li)n (n = 1, 2, 4). Organometallics 1996, 15, 2923–2931. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Handgraaf, J.-W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD Methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Sadlej-Sosnowska, N. On the way to physical interpretation of Hammett constants: How substituent active space impacts on acidity and electron distribution in p-substituted benzoic acid molecules. Pol. J. Chem. 2007, 81, 1123–1134. [Google Scholar]
- Sadlej-Sosnowska, N. Substituent active region—A gate for communication of substituent charge with the rest of a molecule: Monosubstituted benzenes. Chem. Phys. Lett. 2007, 447, 192–196. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Sadlej-Sosnowska, N. Towards physical interpretation of Hammett constants: Charge transferred between active regions of substituents and a functional group. Struct. Chem. 2011, 22, 17–22. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Szatylowicz, H.; Stasyuk, O.A.; Dominikowska, J.; Palusiak, M. Aromaticity from the Viewpoint of Molecular Geometry: Application to Planar Systems. Chem. Rev. 2014, 114, 6383–6422. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, J.; Krygowski, T.M. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 3839–3842. [Google Scholar] [CrossRef]
- Krygowski, T.M. Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of π-electron systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. [Google Scholar] [CrossRef]
- Boese, R.; Blaser, D.; Nussbaummer, M.; Krygowski, T.M. Low temperature crystal and molecular structure of nitrobenzene. Struct. Chem. 1992, 3, 363–368. [Google Scholar] [CrossRef]
- Domenicano, A.; Schultz, G.; Hargittai, I.; Colapietro, M.; Portalone, G.; George, P.; Bock, C.W. Molecular structure of nitrobenzene in the planar and orthogonal conformations—A concerted study by electron diffraction, X-ray crystallography, and molecular orbital calculations. Struct. Chem. 1989, 1, 107–122. [Google Scholar] [CrossRef]
- Di Rienzo, F.; Domenicano, A.; Riva di Sanseverino, L. Structural studies of benzene-derivatives. 8. Refinement of the crystal-structure of para-dinitrobenzene. Acta Cryst. Sect. B 1980, B36, 586–591. [Google Scholar] [CrossRef]
- Tonogaki, M.; Kawata, T.; Ohba, S.; Iwata, Y.; Shibuya, I. Electron-density distribution in crystals of p-nitrobenzene derivatives. Acta Crystallogr. Sect. B-Struct. Sci. 1993, 49, 1031–1039. [Google Scholar] [CrossRef]
- Cantillo, D.; Avalos, M.A.; Babiano, R.; Cintas, P.; Jimenez, J.L.; Light, M.E.; Palacios, J.C.; Rodriguez, V. Push-pull 1,3-thiazolium-5-thiolates. Formation via concerted and stepwise pathways, and theoretical evaluation of NLO properties. Org. Biomol. Chem. 2010, 8, 5367–5374. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Wisiorowski, M.; Nakata, K.; Fujio, M.; Tsuno, Y. Changes of the aromatic character of the ring in exocyclically substituted derivatives of benzylic cation as a result of varying charge at the exo-carbon atom. Bull. Chem. Soc. Jpn. 1996, 69, 2275–2279. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Ejsmont, K.; Stepien, M.K.; Poater, J.; Sola, M. Relation between the substituent effect and aromaticity. J. Org. Chem. 2004, 69, 6634–6640. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Stepien, B.T.; Cyranski, M.K.; Ejsmont, K. Relation between resonance energy and substituent resonance effect in P-phenols. J. Phys. Org. Chem. 2005, 18, 886–891. [Google Scholar] [CrossRef]
- Stasyuk, O.A.; Szatylowicz, H.; Fonseca Guerra, C.; Krygowski, T.M. Theoretical study of electron-attracting ability of the nitro group: Classical and reverse substituent effects. Struct. Chem. 2015, 26, 905–913. [Google Scholar] [CrossRef]
- Szatylowicz, H.; Siodla, T.; Stasyuk, O.A.; Krygowski, T.M. Towards physical interpretation of substituent effects: The case of meta- and para-substituted anilines. Phys. Chem. Chem. Phys. 2016. [Google Scholar] [CrossRef] [PubMed]
- Szatylowicz, H.; Krygowski, T.M.; Hobza, P. How the shape of the NH2 group depends on the substituent effect and H-bond formation in derivatives of aniline. J. Phys. Chem. A 2007, 111, 170–175. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | cSAR(H) (ortho-, meta- and para-Positions) | cSAR(NO2) | cSAR(NH2) |
|---|---|---|---|
| benzene | 0.0 | - | - |
| nitrobenzene | 0.033, 0.022, 0.036 | −0.146 | - |
| aniline | −0.044, −0.001, −0.039 | - | 0.129 |
| p-nitroaniline 0 | −0.017, 0.032, - | −0.219 | 0.198 |
| p-nitroaniline 90 | −0.019, 0.029, - | −0.170 | 0.156 |
| p-nitroanilide anion | −0.063/−0.085, 2 −0.026, - | −0.509 | - |
| p-nitroanilide anion 90 | −0.080/−0.104, 2 −0.045, - | −0.348 | - |
| Complex | Angle φ/deg | dX-H/Å | ΔEdef | ΔEint | ΔEPauli | ΔVelstat | ΔEdisp | ΔEoi | ΔEoiσ | ΔEoiπ | ΔEHB |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2N···HF | |||||||||||
| aniline | 0.971 | 1.37 | −12.72 | 21.14 | −17.91 | −1.55 | −14.40 | −11.35 | |||
| PNA | 0 | 0.967 | 1.42 | −9.67 | 17.49 | −13.42 | −1.58 | −12.16 | −8.25 | ||
| 30 | 0.967 | 1.41 | −9.87 | 17.78 | −13.76 | −1.58 | −12.31 | −8.46 | |||
| 60 | 0.968 | 1.36 | −10.43 | 18.53 | −14.66 | −1.58 | −12.71 | −9.07 | |||
| PNA90 | 90 | 0.970 | 1.29 | −10.97 | 19.20 | −15.52 | −1.58 | −13.06 | −9.68 | ||
| HN−···HF | |||||||||||
| aniline | 1.069 | 8.70 | −40.69 | 31.63 | −41.29 | −1.48 | −29.54 | −27.48 | −2.07 | −31.99 | |
| PNA | 0 | 1.065 | 8.43 | −35.61 | 50.65 | −45.59 | −1.54 | −39.12 | −36.02 | −3.10 | −27.18 |
| 30 | 1.067 | 8.50 | −36.07 | 51.26 | −46.05 | −1.54 | −39.74 | −27.57 | |||
| 60 | 1.075 | 9.42 | −37.65 | 53.46 | −47.61 | −1.53 | −41.98 | −28.23 | |||
| PNA90 | 90 | 1.175 | 22.26 1 | −53.60 | 78.08 | −61.37 | −1.47 | −68.86 | −63.72 | −5.14 | −31.34 |
| HNH···F− | |||||||||||
| aniline | 1.174 | 12.24 | −54.36 | 64.88 | −58.37 | −0.97 | −59.90 | −51.22 | −8.67 | −42.12 | |
| PNA | 0 | 1.180 | 12.82 | −67.83 | 34.89 | −54.94 | −0.98 | −46.80 | −37.55 | −9.25 | −55.01 |
| 30 | 1.174 | 12.32 | −66.68 | 34.36 | −54.08 | −0.98 | −45.97 | −54.36 | |||
| 60 | 1.161 | 11.30 | −63.51 | 33.19 | −51.71 | −0.98 | −44.01 | −52.21 | |||
| PNA90 | 90 | 1.133 | 8.08 | −56.37 | 30.90 | −46.72 | −0.99 | −39.56 | −32.61 | −6.95 | −48.29 |
| HNH···FH | |||||||||||
| aniline | 1.007 | 0.01 | −1.94 | 1.78 | −2.22 | −0.64 | −0.86 | −0.79 | −0.07 | −1.93 | |
| PNA | 0 | 1.009 | 0.01 | −2.91 | 2.11 | −3.20 | −0.69 | −1.12 | −1.02 | −0.10 | −2.90 |
| 30 | 1.012 | 0.00 | −2.78 | 2.09 | −3.09 | −0.71 | −1.08 | −2.78 | |||
| 60 | 1.013 | 0.00 | −2.63 | 2.07 | −2.96 | −0.71 | −1.04 | −2.63 | |||
| PNA90 | 90 | 1.008 | 0.01 | −2.54 | 1.95 | −2.82 | −0.67 | −1.01 | −0.92 | −0.09 | −2.53 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szatylowicz, H.; Stasyuk, O.A.; Fonseca Guerra, C.; Krygowski, T.M. Effect of Intra- and Intermolecular Interactions on the Properties of para-Substituted Nitrobenzene Derivatives. Crystals 2016, 6, 29. https://doi.org/10.3390/cryst6030029
Szatylowicz H, Stasyuk OA, Fonseca Guerra C, Krygowski TM. Effect of Intra- and Intermolecular Interactions on the Properties of para-Substituted Nitrobenzene Derivatives. Crystals. 2016; 6(3):29. https://doi.org/10.3390/cryst6030029
Chicago/Turabian StyleSzatylowicz, Halina, Olga A. Stasyuk, Célia Fonseca Guerra, and Tadeusz M. Krygowski. 2016. "Effect of Intra- and Intermolecular Interactions on the Properties of para-Substituted Nitrobenzene Derivatives" Crystals 6, no. 3: 29. https://doi.org/10.3390/cryst6030029
APA StyleSzatylowicz, H., Stasyuk, O. A., Fonseca Guerra, C., & Krygowski, T. M. (2016). Effect of Intra- and Intermolecular Interactions on the Properties of para-Substituted Nitrobenzene Derivatives. Crystals, 6(3), 29. https://doi.org/10.3390/cryst6030029