High-Pressure Reactivity of Kr and F2—Stabilization of Krypton in the +4 Oxidation State
by
 ,
,
Dominik Kurzydłowski
1,2,* ,
,
Magdalena Sołtysiak
2,
Aleksandra Dżoleva
2 and
Patryk Zaleski-Ejgierd
3,* 1
Centre of New Technologies, University of Warsaw, Warsaw 02-097 , Poland
2
Faculty of Mathematics and Natural Sciences, Cardinal Stefan Wyszyński University, Warsaw 01-038 , Poland
3
Faculty of Physics, IFT, University of Warsaw, Warsaw 02-093, Poland
*
Authors to whom correspondence should be addressed.
Crystals 2017, 7(11), 329; https://doi.org/10.3390/cryst7110329
Submission received: 29 September 2017
/
Revised: 24 October 2017
/
Accepted: 25 October 2017
/
Published: 28 October 2017
(This article belongs to the Special Issue Structure and Properties of Fluoride-based Materials)
Abstract
:Since the synthesis of the first krypton compound, several other Kr-bearing connections have been obtained. However, in all of them krypton adopts the +2 oxidation state, in contrast to xenon which forms numerous compounds with an oxidation state as high as +8. Motivated by the possibility of thermodynamic stabilization of exotic compounds with the use of high pressure (exceeding 1 GPa = 10 kbar), we present here theoretical investigations into the chemistry of krypton and fluorine at such large compression. In particular we focus on krypton tetrafluoride, KrF4, a molecular crystal in which krypton forms short covalent bonds with neighboring fluorine atoms thus adopting the +4 oxidation state. We find that this hitherto unknown compound can be stabilized at pressures below 50 GPa. Our results indicate also that, at larger compressions, a multitude of other KrmFn fluorides should be stable, among them KrF which exhibits covalent Kr–Kr bonds. Our results set the stage for future high-pressure synthesis of novel krypton compounds.
1. Introduction
Among the non-radioactive group 18 elements (He, Ne, Ar, Kr, Xe) only the two heaviest, krypton and xenon, form compounds isolable in macroscopic quantities. With its lowest first ionization potential (12.13 eV) xenon forms numerous compounds in which it adopts oxidation states +1/2, +2, +4, +6 and +8 [1,2,3,4,5,6]. The relatively large thermodynamic and/or kinetic stability of these species enabled characterization of over 150 crystal structures for Xe-bearing systems.
Due to its higher ionization potential (14.0 eV), krypton is much less reactive and forms compounds only in the +2 oxidation state [7,8,9]. All of these connections are derived from krypton difluoride (KrF2), a compound discovered in 1963 [10,11,12,13], only a year after Bartlett’s landmark synthesis of the first xenon compound [14,15]. In contrast to xenon difluoride (XeF2), KrF2 is thermodynamically unstable (ΔHf = 60.2 kJ mol−1, at 93 °C [16]) and thus hard to synthesize and handle. Higher fluorides of krypton (KrF4, KrF6) are not known (initial reports of the synthesis of KrF4 [10,11] have never been confirmed), again in contrast to xenon which does form both XeF4 and XeF6. Despite the mediocre reactivity of krypton a substantial number of compounds and complexes containing this element were speculated to exist in the gas phase [17,18,19,20,21,22,23].
The next group 18 element, argon, exhibits an even higher first ionization potential (15.8 eV), and consequently forms transient species which can be stabilized only in low-temperature matrices or in the gas-phase [24,25,26,27,28,29]. The decreased reactivity of argon is also manifested by the fact that at ambient pressure it does not form any fluoride, even at low temperature. However, our recent computational investigation indicated that argon difluoride (ArF2) can be thermodynamically stabilized (i.e., possess a negative enthalpy of formation) at pressures above 60 GPa (=600 kbar) [30].
While 60 GPa may seem high on the pressure scale, such conditions are now routinely achieved in experiments conducted with the use of diamond anvil cells (DACs). In recent years, there is a growing interest in the high-pressure chemistry of noble gases concerning both experiment [31,32,33,34,35], and theory [36,37,38,39,40]. In particular, much attention has been paid to the high-pressure phase transformations of XeF2 [41,42,43,44]. At ambient pressure this compound forms a molecular crystal composed of weakly interacting XeF2 molecules (in the form of linear and symmetric F–Xe–F units). Early high-pressure investigations indicated that above 50 GPa XeF2 adopts a graphite-like hexagonal structure, and upon further compression to 70 GPa transforms to a metallic three-dimensional fluorite-like structure [41]. These findings were put into doubt by theory [42], and subsequent experiments indicated that XeF2 remains molecular and insulating up to 80 GPa [43]. Furthermore, a recent theoretical investigation on the high-pressure thermodynamic stability of XemFn phases indicated that XeF2 should decompose into XeF4 and Xe2F above 81 GPa [44]. Interestingly Xe2F, which is not known at ambient conditions, was predicted to exhibit direct Xe–Xe bonds [44].
Noble-gas compounds are text-book examples of systems exhibiting hypervalency, multi-center and charge-shift bonding [45]. Motivated by the peculiar chemistry of Xe/F mixtures at large compression, we performed a systematic theoretical study on the high-pressure thermodynamic stability of various KrmFn crystalline compounds. Our aim was to verify whether novel krypton fluorides might be stabilized at large compression. Employing the Density Functional Theory (DFT) we analyzed 11 possible stoichiometries including both species that are krypton rich (Kr6F, Kr4F, Kr3F, Kr2F, Kr3F2), fluorine rich (Kr2F3, KrF2, KrF3, KrF4, KrF6), as well as KrF. Our study models the 0–200 GPa pressure range which is accessible to modern experiments.
We found that novel connections between krypton and fluorine should indeed stabilize at large compression. In particular, our results indicate that fluorine should oxidize krypton difluoride at high pressure to yield KrF4—a first example of a compound containing krypton in the +4 oxidation state. We also predict formation of KrF at 100 GPa, which is a compound containing direct Kr–Kr bonds. By comparing our results with previous investigations, we propose general trends in the high-pressure reactivity of noble-gas difluorides.
2. Results
For each of the studied KrmFn compounds we propose crystal structures and perform geometry optimization at high pressure utilizing the DFT method in its periodic approach. We also optimize the structures of solid Kr and F2. Due to the large complexity of solid-state structures, proposing good high-pressure candidate crystal structures is challenging. In recent years, considerable effort was put into developing algorithms which enable effective structure search for a solid compound of given composition [46,47,48,49]. In this study, we performed crystal structure searches employing the USPEX (Universal Structure Predictor: Evolutionary Xtallography) evolutionary algorithm [50,51]—in all of them the only input information was the stoichiometry of the compound and the pressure at which the search was conducted (for more computational details see Materials and Methods).
Our calculations do not include temperature effects, and thus give the thermodynamic stability at the 0 K temperature limit. Due to the large computational burden associated with calculating vibrational properties of solids, neglecting finite-temperature corrections is a standard approach in high-pressure computational chemistry. At the T = 0 K limit, the Gibbs free energy (G) can be equated to the enthalpy (H), and hence at a given pressure the thermodynamic stability of a compound will be governed by its enthalpy.
In this work we define the enthalpy of formation, ΔHf(KrmFn) at a given pressure as:
where H(KrmFn), H(Kr), and H(F2) are the calculated enthalpies of KrmFn, Kr, and F2 at each pressure. Equation (1) gives ΔHf(KrmFn) values per atom, and ensures that ΔHf(Kr) = ΔHf(F2) = 0. In order to assess the relative thermodynamic stability of various KrmFn phases, as well as their decomposition into Kr and F2, we use the enthalpies of formation defined above to construct the so-called tie-line plot [52]. In this representation, ΔHf(KrmFn) values are plotted against the mole fraction of F atoms. It can be shown that a phase is thermodynamically stable if no tie-line passes below it [52].
ΔHf(KrmFn) = [H(KrmFn) − m·H(Kr) − n/2·H(F2)]/(m + n),
2.1. Pressures Up to 50 GPa
We start our discussion with the results obtained in the pressure regime up to 50 GPa. We find that the thermodynamic stability of various phases differs qualitatively in this pressure range compared to results obtained at higher compression (see Section 2.2). In Figure 1a we display a tie-line plot of the Kr/F system for pressures of 0 GPa (1 bar = 10−4 GPa; ambient pressure thus equals effectively to 0 GPa), 15 GPa and 50 GPa.
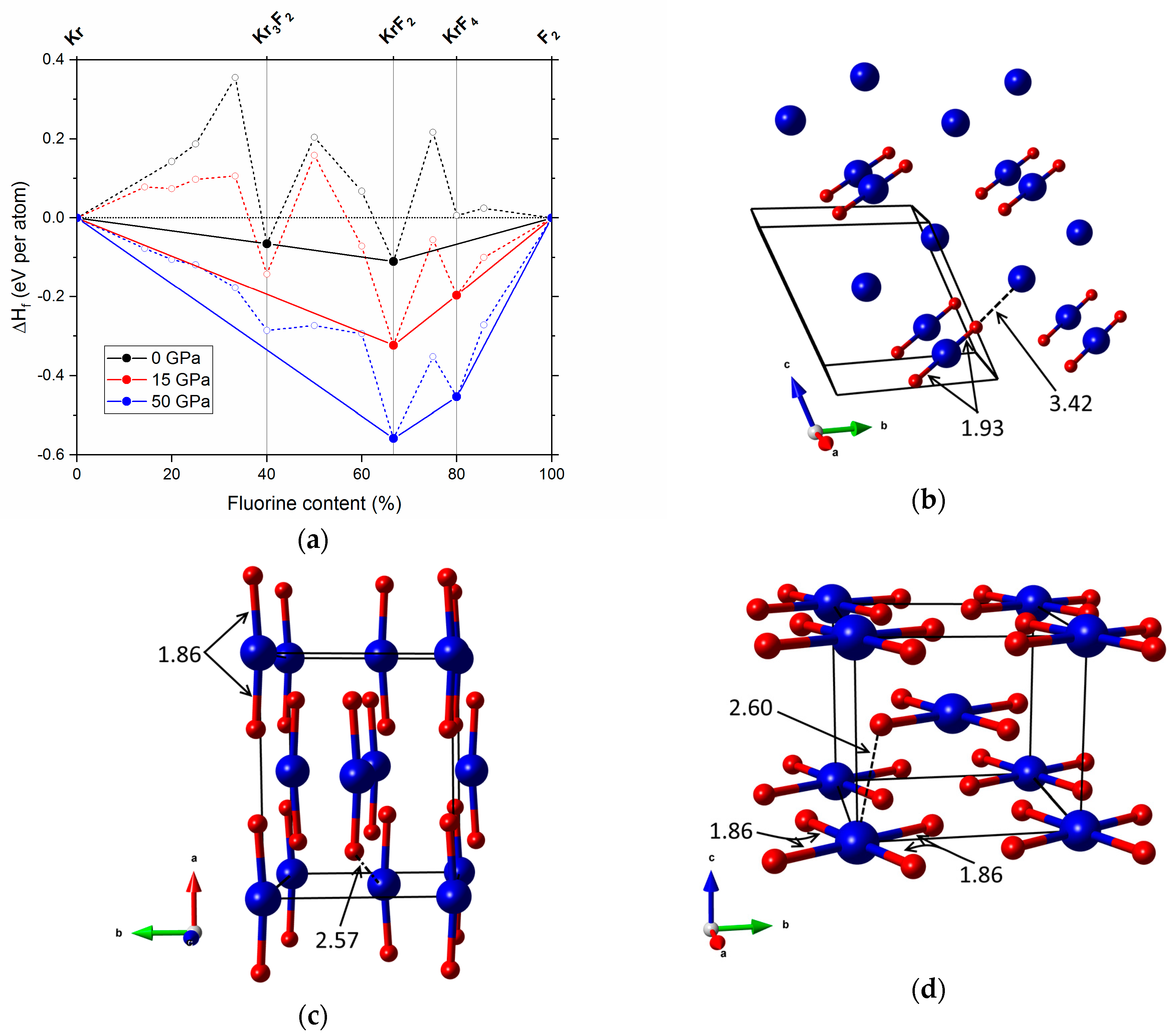
At ambient pressure two compositions are located on the convex hull (i.e., there are no tie-lines passing below them, and hence they are thermodynamically stable). These are Kr3F2 and KrF2. Inspection of the structure of the former compound (Figure 1b), indicates that it is composed of linear F–Kr–F molecules, characterized by a Kr–F bond length of 1.93 Å, and unbound Kr atoms (nearest Kr–F contact at 3.42 Å). Therefore, a more realistic formulation of Kr3F2 would be 2Kr·KrF2—that is a co-crystal of Kr and KrF2. This phase quickly destabilizes at high pressures, and is predicted to decompose into Kr and KrF2 already at 1 GPa.
The other stable compound at ambient conditions is KrF2. At 0 GPa, our evolutionary searches identify two structures of I4/mmm and P42/mnm symmetry which are nearly degenerate in terms of enthalpy. These correspond respectively to the experimentally known ambient-pressure α and β polymorphs of KrF2 [53,54]. Both of them contain linear F–Kr–F units with a Kr–F bond length of 1.93 Å. Application of pressures up to 6 GPa leads to thermodynamic stabilization of the I4/mmm structure over P42/mnm. Above 6 GPa we predict a phase transition from I4/mmm to a structure of Cmcm symmetry composed of slightly bent F–Kr–F molecules (173° at 50 GPa, see Figure 1c). This polymorph of KrF2, which is isostructural to the predicted high-pressure phase of ArF2 [30], remains the most stable structure up to 200 GPa. Even at that pressure it retains its molecular character with Kr–F bonds (1.78 Å) still much shorter than the Kr–F intermolecular contacts (2.19 Å).
We predict that at 0 GPa KrF2 is marginally stable with respect to decomposition into Kr and F2 (ΔHf = −0.08 eV/atom). In contrast, the experimental enthalpy of formation at ambient pressure is slightly positive (0.21 eV/atom [16]). This value was well reproduced with rigorous coupled-cluster calculations of Dixon et al. [55]. In our previous study, we found that the Perdew–Burke–Ernzerhof (PBE) functional used here tends to overestimate the thermodynamic stability of noble-gas compounds, and that more accurate values can be generally achieved with the application of the hybrid HSE06 functional [30]. Indeed, calculation performed with the use of this functional gives ΔHf(KrF2) = 0.06 eV/atom, much closer to the theoretical value of Dixon et al. (0.21 eV/atom, [55]). However, the high computational cost of the HSE06 functional severely restricts the number of compounds that can be studied. Therefore, here we opted to work with the less accurate PBE functional, which however enabled us to screen a much larger array of possible stoichiometries. We emphasize that the pressure dependence of ΔHf is the same at both the PBE and HSE06 level of theory (see Figure S2 in the Supplementary Material, SM). This gives us confidence that the thermodynamic stabilization of krypton fluorides that we observe at high pressure is not a computational artifact, although the pressure at which a given compound becomes stable might be underestimated (vide infra the case of KrF4).
The first novel krypton fluoride that becomes thermodynamically stable at elevated pressure is KrF4; it becomes part of the convex hull already at 15 GPa (Figure 1a). This indicates that KrF4 should be accessible at high pressures through a reaction of KrF2 with F2 or by reacting Kr and F2 in a 1:2 or greater ratio. Considering the abovementioned over-binding of KrmFn compounds at the applied level of theory (DFT/PBE) the experimental pressure of synthesis might be higher by about 25 GPa, i.e., reach 40 GPa. Indeed, HSE06 calculations indicate that KrF4 should be stable above that pressure (Figure S2 in SM). We note that such pressure is still in the range of modern experimental techniques.
The most stable structure of KrF4 in the entire considered pressure range—up to 200 GPa—is a molecular crystal of I4/m symmetry composed of weakly interacting square-planar KrF4 units (Figure 1d). Even at 200 GPa, the Kr–F bond within these molecules (1.77 Å) is more than 20% shorter than the closest Kr–F intermolecular contact (2.26 Å). In fact, the crystal structure of KrF4 is analogous to the predicted high-pressure phase of XeF4, which is also molecular even at large compression [44].
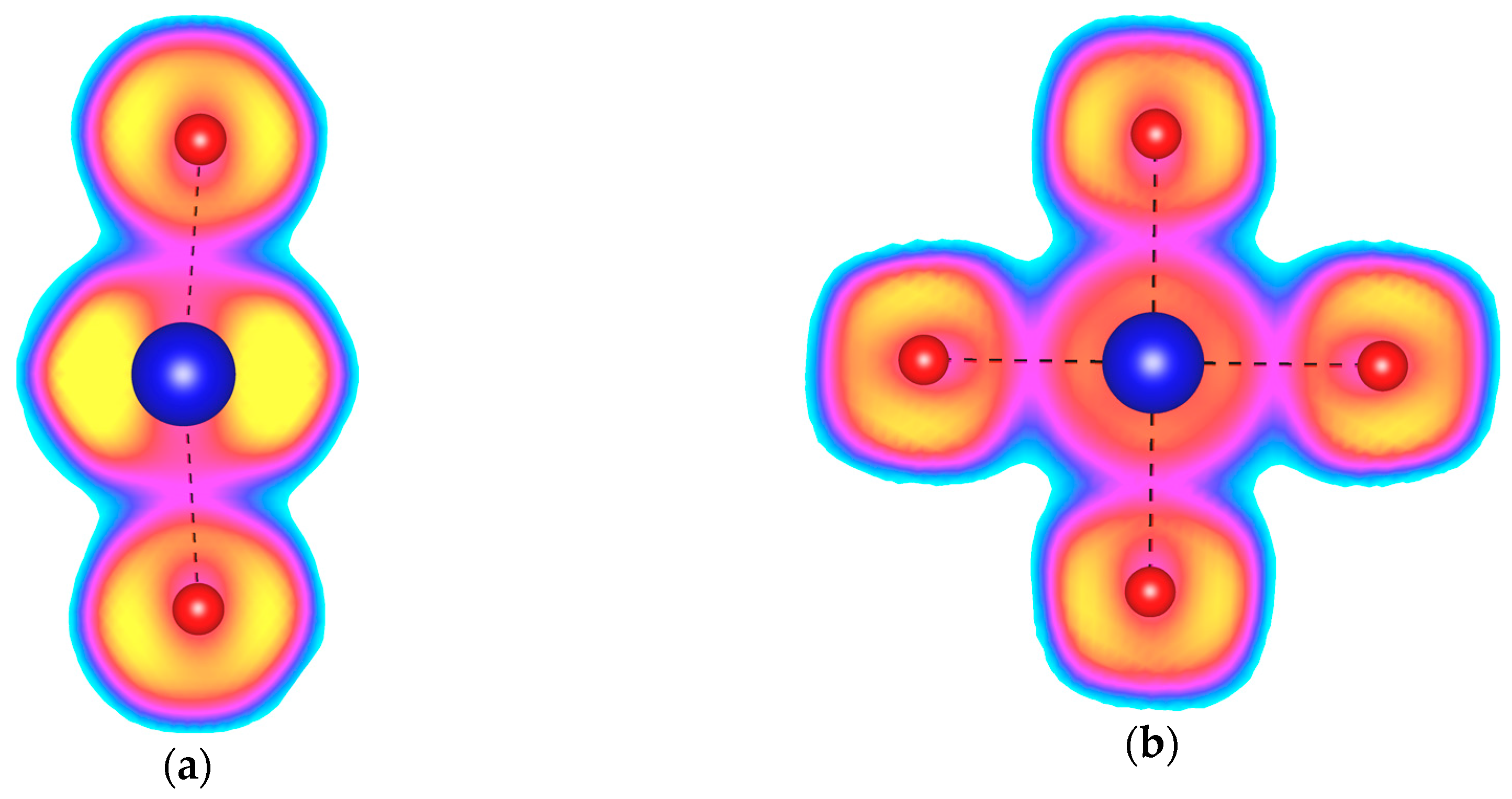
To confirm the presence of four covalent Kr–F bonds in KrF4, we perform an analysis of the Electron Localization Function (ELF) [56]. This dimensionless function, defined for every point in the crystal structure, spans values from 0 to 1. Values close to 1 are generally interpreted as indicating paired electrons (i.e., lone pairs, bonds, and core electrons). Typical covalent bonds are characterized by ELF values of exceeding 0.7; however, previous studies indicated that the maximum ELF value for the Xe–F bond in XeF2 is only 0.5 [42,44]. This can be rationalized by taking into account that this bond arises from a three-center two-electron interaction, and is characterized by a bond order of 1/2. By analogy, the same applies to the Kr–F bond in KrF2, for which we obtain a maximum ELF value of 0.4 (Figure 2a). This is slightly lower than the value found for XeF2 in accordance with the fact that Kr–F is a weaker bond. More importantly, we find the same ELF values of 0.4 for Kr–F bonds in the KrF4 molecules present in the I4/m structure (Figure 2b), which indicates that in this particular phase krypton forms four covalent connections to fluorine atoms; therefore, it can be assigned an oxidation state of +4.
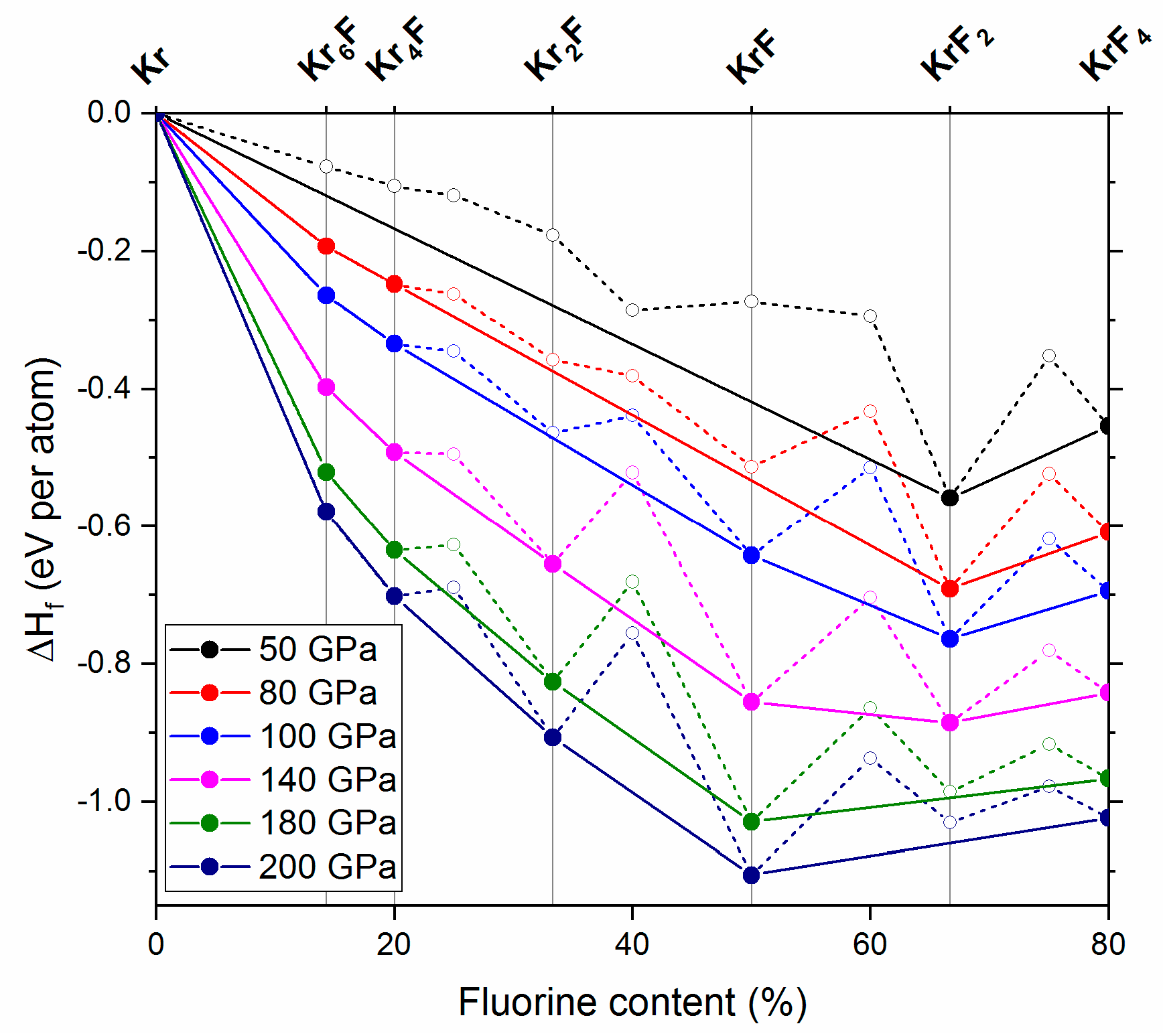
2.2. The 50–200 GPa Pressure Range
Our calculations indicate that KrF4 remains thermodynamically stable up to the highest pressure considered in this study (200 GPa). We also do not find any pressure range at which KrF6—the only KrmFn compound with greater fluorine content than KrF4—would exhibit thermodynamic stability. However, we do find that some less-fluorine rich KrmFn phases stabilize at pressures larger than 50 GPa. In particular, on the krypton-rich side of the phase diagram we find Kr6F and Kr4F becoming part of the convex hull at approx. 80 GPa (Figure 3), and remaining thermodynamically stable up to 200 GPa. Therefore, one should expect formation of these compounds in krypton-rich Kr/F mixtures upon compression.
At higher pressures we predict stabilization of KrF (at 100 GPa), and Kr2F (at 140 GPa). Both of them remain on the convex hull up to 200 GPa. Interestingly, we find that KrF2 becomes thermodynamically unstable above 180 GPa—above that pressure it should spontaneously decompose into the hitherto unknown KrF and KrF4.
Within their pressure stability range, Kr6F and Kr4F do not exhibit any phase transitions. The lowest-enthalpy structures of these krypton-rich phases are formed by closely packed Kr atoms decorated with isolated F atoms (see Figure S3 in the SM). As shown in Table 1, the Kr–F distances found in these compounds are considerably longer than the covalent Kr–F bonds in KrF2 and KrF4. On the other hand, Kr–Kr contacts are comparable to those found in solid Kr at the same pressure. These characteristic features indicate that the building blocks of both Kr6F and Kr4F consist of atoms rather than molecules, and that these compounds lack covalent bonding. The latter point is confirmed by the analysis of the ELF function for these compounds, which indicates that ELF values calculated between all of the atoms do not exceed 0.1. This number is comparable to the value found between Kr atoms in solid krypton at the same pressure (Figure S4 in SM).
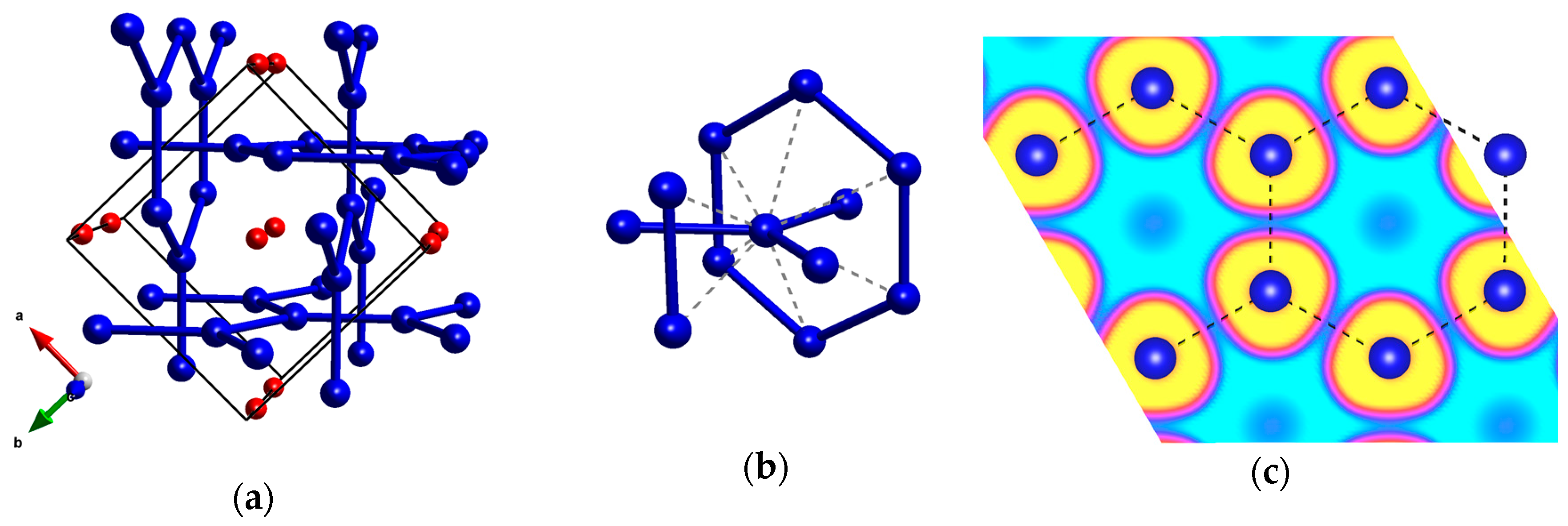
For Kr2F, our structure investigations identified a phase with I4/mcm symmetry, one that remains the lowest-enthalpy structure in the whole pressure range studied. This polymorph is isostructural with a previously proposed high-pressure phase of Xe2F [44]. Similarly to that structure, it contains an interpenetrating network of graphitic Kr layers (Figure 4a) with a Kr–Kr bond of about 2.5 Å at 150 GPa, which is ca 8% shorter than the Kr–Kr separation in solid krypton at the same pressure (Table 1). The second-nearest Kr–Kr contacts in Kr2F are much longer (2.83–2.84 Å, Figure 4b), and the length of Kr–F contacts hints at no covalent bonding between krypton and fluorine (Table 1). In case of Xe2F, Peng et al. indicated formation of covalent Xe–Xe bonds [44]. For Kr2F we find, however, that ELF values between nearest-neighbor Kr atoms do not exceed 0.15 (Figure 4c), which hints at lack of Kr–Kr covalent bonding within the graphitic layers. Therefore, although the structure of Kr2F features more directional contacts than Kr6F and Kr4F, it seems that it also can be described as built of relatively weekly bonded Kr and F atoms.
Similarly to Kr2F, we find that KrF exhibits only one polymorphic form in the whole pressure range studied. This structure of P2/m symmetry contains chains of Kr atoms (Figure 5a) with an extremely short Kr–Kr bond of 2.29 Å at 150 GPa (which is about 15% shorter than the Kr–Kr contact in solid krypton; see Table 1). At the same time, Kr–F and F–F shortest distances are considerably longer than those found in KrF2 and F2, respectively, while secondary Kr–Kr contacts are slightly longer to those found in solid Kr (Table 1, Figure 5b). These structural features indicate formation of Kr–Kr covalent bonds within the Kr chains. Indeed, inspection of the ELF function shows that the maximum ELF value along the shortest Kr–Kr distance is 0.4, comparable to what is found for KrF2 and KrF4 (Figure 5c). We note that Peng et al. found XeF to be thermodynamically stable at 100 GPa and 140 K [44]. However, their lowest enthalpy structure contained Xe–Xe dimers, instead of Kr chains found by us for KrF.
3. Discussion
Previous theoretical studies on the reactivity of Xe/F [44] and Ar/F [30] systems indicated that application of high pressure leads to stabilization of compounds with high fluorine content (XeF2, XeF4, XeF6, ArF2) with respect to pure elements. We have found the same trend for the Kr/F system studied here—in particular we find that large compression should lead to thermodynamic stabilization of a crystal containing KrF4 molecules. If obtained, this compound would constitute the first example of a chemical connection containing krypton in the +4 oxidation state.
The increased reactivity of krypton compared to argon is evident from this study. We find that the stabilization pressure of KrF4 is 15 GPa (more adequately approx. 40 GPa after taking into account overbinding due to the employed functional), while a compound containing ArF4 molecules is predicted to form only above 250 GPa [30]—see Table 2. On the other hand, krypton remains less reactive than xenon at high pressure—our results indicate that KrF6 is not stable at least up to 200 GPa, while XeF6 can be obtained at ambient conditions.
Pressures exceeding 50 GPa should lead to stabilization of krypton-rich fluorides (Kr6F and Kr4F at 80 GPa, Kr2F at 140 GPa). This is in analogy with previous results for the Xe/F system, where stabilization of Xe2F was predicted to occur above 60 GPa [44] (we note that in that study Xe6F and Xe4F compositions were not taken into account). We do not observe formation of direct bonds between noble-gas atoms in KrmF (m = 2, 4, 6) compounds, in contrast to Xe2F, which does exhibit Xe–Xe bonds. We do however find Kr–Kr bond for KrF, which becomes stable at 100 GPa.
The enhanced thermodynamic stability of KrF at large compression influences the stability of KrF2, which is predicted to decompose into KrF and KrF4 at 180 GPa. Still, KrF2 is more stable at large compression than XeF2, which in turn is predicted to disproportionate into Xe2F and XeF4 above 81 GPa. Generally, it seems that the thermodynamic stability of noble-gas difluorides at high pressure increases, when moving up group 18 with ArF2 predicted to dominate the convex hull of the Ar/F system up to 200 GPa [30].
We find that F–Kr–F molecules in KrF2 undergo slight bending in response to compression, in analogy to ArF2, but we do not encounter any signs of auto-ionization, as predicted for XeF2 [42]. Interestingly, our structure searches do not identify any phases containing KrF+ and Kr2F3+ cations, which are known to form upon reaction of KrF2 with fluoride acceptors [7]. This might hint at the fact that the fluoride-donor properties of KrF2 are suppressed at large compression. In general, it seems that in the case of the Kr/F system, ionic bonding is not a major driving force in stabilization of high-pressure phases, in contrast to what is found for the N/F system [57].
We hope that the insight into the high-pressure chemistry of krypton offered by this study will motivate future investigation, in particular, experimental attempts to obtain KrF4 and KrF. As already noted, the pressures necessary for the synthesis reported here are expected to be underestimated by about 25 GPa; nonetheless, even with this correction they fall well within the experimentally accessible pressure range achievable even with the use of commercially available diamond anvil cells. Most importantly, we note that both compounds should be accessible by a convenient pathway involving the high-pressure decomposition of KrF2; such synthesis scheme should be preferred over the direct synthesis from Kr and F2, in particular due to the large reactivity and toxicity of fluorine.
Finally, we note that a detailed study of the electronic properties of the KrmFn compounds reported here would also be of interest. Analyzing the bands structure of these connections is beyond the scope of this study, we only note that our calculations indicate that Kr, F2, KrF2 and KrF4 are semiconductors at 150 GPa, while Kr6F, Kr4F, Kr2F and KrF are metallic at the same pressure (see Table S3 in SM).
4. Materials and Methods
Our study is based on periodic DFT calculations which were performed with the VASP 5.2 program [58,59,60]. We utilized the generalized gradient approximation (GGA) in the form of the Perdew–Burke–Ernzerhof (PBE) functional [61], which was successfully applied in previous high-pressure studies on xenon fluorides [42,44], as well as a recent study on the ambient-pressure structures of KrF2 [62]. The projector augmented-wave (PAW) method was used [63], as implemented in the VASP 5.2 code. Our calculations do not included van der Waals corrections the DFT functional. We have found, however, that inclusion of such dispersion corrections (in the form of the D3 correction [64]) lead to negligible differences in the high-pressure thermodynamic stability of KrF2 and KrF4 (see Figures S1 and S2 in SM), in accordance with previous results for the Ar/F system [30].
The cut-off energy of the plane waves was set to 600 eV with a self-consistent-field convergence criterion of 1 × 10−5 eV. Valence electrons (Kr: 4s, 4p; F: 2s, 2p) were treated explicitly, while VASP pseudopotentials, accounting for scalar relativistic effects, were used for the description of core and semi-core electrons. The spacing of the k-point mesh was set at 2π × 0.06 Å−1. All structures were optimized in 10 GPa steps (5 GPa below 20 GPa) using a conjugate gradient algorithm. Structure visualization was performed with the VESTA software [65], while symmetry recognition with the online FINDSYM program [66].
For each pressure, we optimize the structure of the studied KrmFn phases, as well as that of elemental Kr and F2. From the resulting enthalpies we derive the enthalpies of formation of KrmFn phases at each pressure. At ambient pressure, krypton adopts the face-centered cubic (fcc) structure [67]. In our calculations, already at slightly elevated pressure (approx. 10 GPa) krypton adopts a hexagonal close-packed (hcp) structure and maintains it in the whole considered pressure range (0–200 GPa) We also note that, above 50 GPa, the ambient pressure structure of F2 (C2/c space group [68]) symmetrizes spontaneously to a Cmca structure which is analogous to the high-pressure molecular polymorph of Cl2 [69]. For a comparison of the experimental and calculated crystal structures of α-F2 and Kr at ambient pressure see Table S4 in the SM.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/7/11/329/s1, Table S1: Structure parameters of the most stable polymorphs of Kr3F2, KrF2 and KrF4, Table S2: Structure parameters of the most stable polymorphs of K6F, Kr4F, Kr2F and KrF, Table S3: Calculated band gaps for the lowest enthalpy structures of KrmFn compounds at 150 GPa, Table S4: Comparison of the experimental and calculated (PBE method) crystal structures of α-F2 and Kr at ambient pressure, Figure S1: Comparison of the relative enthalpy of the ambient pressure P42/mnm structure of KrF2 with respect to the high-pressure Cmcm structure obtained with three different computational methods (PBE, PBE+D3, HSE06), Figure S2: Comparison of the enthalpy change of the reaction KrF2 + F2 -> KrF3 obtained with three different computational methods (PBE, PBE+D3, HSE06). The ambient pressure CCSD(T) value calculated with the use of data from Dixon et al. (ref. [55]) is marked with a star. Figure S3: (a) The P–1 structure of Kr6F obtained at 150 GPa; (b) the C2/m structure of Kr4F at 150 GPa, Figure S4: The ELF function at 150 GPa for: (a) the P–1 structure of Kr6F; (b) the C2/m structure of Kr4F; (c) the hcp structure of Kr.
Acknowledgments
Dominik Kurzydłowski acknowledges the support from the Polish National Science Centre (NCN) within grant No. UMO-2014/13/D/ST5/02764. This research was carried out with the support of the Interdisciplinary Centre for Mathematical and Computational Modelling (ICM) University of Warsaw under grant No. GA67–13.
Author Contributions
Dominik Kurzydłowski and Patryk Zaleski-Ejgierd conceived and designed the research; Magdalena Sołtysiak and Aleksandra Dżoleva performed the calculations; Dominik Kurzydłowski Magdalena Sołtysiak, Aleksandra Dżoleva and Patryk Zaleski-Ejgierd analyzed the data; Dominik Kurzydłowski wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Tramšek, M.; Žemva, B. Synthesis, properties and chemistry of xenon(II) fluoride. Acta Chim. Slov. 2006, 105–116. [Google Scholar] [CrossRef]
- Grochala, W. Atypical compounds of gases, which have been called “noble”. Chem. Soc. Rev. 2007, 36, 1632–1655. [Google Scholar] [CrossRef] [PubMed]
- Nabiev, S.S.; Sokolov, V.B.; Chaivanov, B.B. Structure of simple and complex noble gas fluorides. Crystallogr. Rep. 2011, 56, 774–791. [Google Scholar] [CrossRef]
- Brock, D.S.; Schrobilgen, G.J.; Žemva, B. Noble-Gas Chemistry. In Comprehensive Inorganic Chemistry II; Elsevier: Cambridge, MA, USA, 2013; pp. 755–822. [Google Scholar]
- Nabiev, S.S.; Sokolov, V.B.; Chaivanov, B.B. Molecular and crystal structures of noble gas compounds. Russ. Chem. Rev. 2014, 83, 1135–1180. [Google Scholar] [CrossRef]
- Haner, J.; Schrobilgen, G.J. The chemistry of Xenon(IV). Chem. Rev. 2015, 115, 1255–1295. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.F.; Mercier, H.P.A.; Schrobilgen, G.J. The chemistry of krypton. Coord. Chem. Rev. 2002, 233–234, 1–39. [Google Scholar] [CrossRef]
- Lozinšek, M.; Schrobilgen, G.J. The world of krypton revisited. Nat. Chem. 2016, 8, 732. [Google Scholar] [CrossRef] [PubMed]
- Lozinšek, M.; Mercier, H.P.A.; Brock, D.S.; Žemva, B.; Schrobilgen, G.J. Coordination of KrF2 to a Naked Metal Cation, Mg2+. Angew. Chem. Int. Ed. 2017, 56, 6251–6254. [Google Scholar] [CrossRef] [PubMed]
- Grosse, A.V.; Kirshenbaum, A.D.; Streng, A.G.; Streng, L.V. Krypton Tetrafluoride: Preparation and Some Properties. Science 1963, 139, 1047–1048. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.H.; Verdier, P.H. Krypton Tetrafluoride: 19F High-Resolution Magnetic Resonance Spectrum. J. Chem. Phys. 1964, 40, 2057. [Google Scholar] [CrossRef]
- Turner, J.J.; Pimentel, G.C. Krypton Fluoride: Preparation by the Matrix Isolation Technique. Science 1963, 140, 974–975. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, F.; Malm, J.G.; Hindman, J.C. The Preparation and Nuclear Magnetic Resonance of Krypton Difluoride. J. Am. Chem. Soc. 1965, 87, 25–28. [Google Scholar] [CrossRef]
- Bartlett, N. Xenon hexafluoroplatinate(V) Xe+[PtF6]−. Proc. Chem. Soc. 1962, 6, 218. [Google Scholar]
- Hargittai, I. Neil Bartlett and the first noble-gas compound. Struct. Chem. 2009, 20, 953–959. [Google Scholar] [CrossRef]
- Gunn, S.R. Heat of formation of krypton difluoride. J. Phys. Chem. 1967, 71, 2934–2937. [Google Scholar] [CrossRef]
- Chen, J.L.; Yang, C.Y.; Lin, H.J.; Hu, W.P. Theoretical prediction of new noble-gas molecules FNgBNR (Ng = Ar, Kr, and Xe; R = H, CH3, CCH, CHCH2, F, and OH). Phys. Chem. Chem. Phys. 2013, 15, 9701. [Google Scholar] [CrossRef] [PubMed]
- Gronowski, M.; Turowski, M.; Kołos, R. Quantum Chemical Study on HKrC5N, HXeC5N, and Related Rare Gas Compounds. J. Phys. Chem. A 2015, 119, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Manna, D.; Ghanty, T.K. Theoretical Prediction of Noble Gas Inserted Thioformyl Cations: HNgCS+ (Ng = He, Ne, Ar, Kr, and Xe). J. Phys. Chem. A 2015, 119, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.A.; McDowell, S.A.C. Comparative computational study of model halogen-bonded complexes of FKrCl. J. Phys. Chem. A 2015, 119, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, M.; Zhou, M.; Andrada, D.M.; Frenking, G. Experimental and theoretical studies of the infrared spectra and bonding properties of NgBeCO3 and a comparison with NgBeO (Ng = He, Ne, Ar, Kr, Xe). J. Phys. Chem. A 2015, 119, 2543–2552. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.; Pan, S.; Mandal, S.; Orozco, M.; Merino, G.; Chattaraj, P.K. Noble gas supported B3+ cluster: Formation of strong covalent noble gas–boron bonds. RSC Adv. 2016, 6, 78611–78620. [Google Scholar] [CrossRef]
- Chen, W.; Chen, G.H.; Wu, D.; Wang, Q. BNg3F3 : The first three noble gas atoms inserted into mono-centric neutral compounds—A theoretical study. Phys. Chem. Chem. Phys. 2016, 18, 17534–17545. [Google Scholar] [CrossRef] [PubMed]
- Khriachtchev, L.; Pettersson, M.; Runeberg, N.; Lundell, J.; Räsänen, M. A stable argon compound. Nature 2000, 406, 874–876. [Google Scholar] [PubMed]
- Evans, C.J.; Gerry, M.C.L. The microwave spectra and structures of Ar–AgX (X = F, Cl, Br). J. Chem. Phys. 2000, 112, 1321–1329. [Google Scholar] [CrossRef]
- Grochala, W.; Khriachtchev, L.; Räsänen, M. Noble-gas chemistry. In Physics & Chemistry at Low Temperatures; Khriachtchev, L., Ed.; Pan Stanford Publishing: Boca Raton, FL, USA, 2011; pp. 419–446. [Google Scholar]
- Grandinetti, F. Review: Gas-phase ion chemistry of the noble gases: Recent advances and future perspectives. Eur. J. Mass Spectrom. 2011, 17, 423–463. [Google Scholar] [CrossRef] [PubMed]
- Young, N.A. Main group coordination chemistry at low temperatures: A review of matrix isolated Group 12 to Group 18 complexes. Coord. Chem. Rev. 2013, 257, 956–1010. [Google Scholar] [CrossRef]
- Wang, X.; Andrews, L.; Brosi, F.; Riedel, S. Matrix Infrared Spectroscopy and Quantum-Chemical Calculations for the Coinage-Metal Fluorides: Comparisons of Ar-AuF, Ne-AuF, and Molecules MF2 and MF3. Chem. A Eur. J. 2013, 19, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Kurzydłowski, D.; Zaleski-Ejgierd, P. High-pressure stabilization of argon fluorides. Phys. Chem. Chem. Phys. 2016, 18, 2309–2313. [Google Scholar] [CrossRef] [PubMed]
- Somayazulu, M.; Dera, P.; Smith, J.; Hemley, R.J. Structure and stability of solid Xe(H2)n. J. Chem. Phys. 2015, 142, 104503. [Google Scholar] [CrossRef] [PubMed]
- Dewaele, A.; Worth, N.; Pickard, C.J.; Needs, R.J.; Pascarelli, S.; Mathon, O.; Mezouar, M.; Irifune, T. Synthesis and stability of xenon oxides Xe2O5 and Xe3O2 under pressure. Nat. Chem. 2016, 8, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Howie, R.T.; Turnbull, R.; Binns, J.; Frost, M.; Dalladay-Simpson, P.; Gregoryanz, E. Formation of Xenon-nitrogen compounds at high pressure. Sci. Rep. 2016, 6, 34896. [Google Scholar] [CrossRef] [PubMed]
- Dewaele, A.; Pépin, C.M.; Geneste, G.; Garbarino, G. Reaction between nickel or iron and xenon under high pressure. High Press. Res. 2017, 37, 137–146. [Google Scholar] [CrossRef]
- Dong, X.; Oganov, A.R.; Goncharov, A.F.; Stavrou, E.; Lobanov, S.; Saleh, G.; Qian, G.R.; Zhu, Q.; Gatti, C.; Deringer, V.L.; et al. A stable compound of helium and sodium at high pressure. Nat. Chem. 2017, 9, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Jung, D.Y.; Oganov, A.R.; Glass, C.W.; Gatti, C.; Lyakhov, A.O. Stability of xenon oxides at high pressures. Nat. Chem. 2013, 5, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Schwerdtfeger, P. Xenon Suboxides Stable under Pressure. J. Phys. Chem. Lett. 2014, 5, 4336–4342. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Chen, Y.; Xiang, S.; Kuang, X.; Bi, Y.; Chen, H. High-temperature- and high-pressure-induced formation of the Laves-phase compound XeS2. Phys. Rev. B 2016, 93, 214112. [Google Scholar] [CrossRef]
- Zaleski-Ejgierd, P.; Lata, P.M. Krypton oxides under pressure. Sci. Rep. 2016, 6, 18938. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Wang, X.; Botana, J.; Miao, M. Noble gas bond and the behaviour of XeO3 under pressure. Phys. Chem. Chem. Phys. 2017, 19, 27463–27467. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Debessai, M.; Yoo, C.S. Two- and three-dimensional extended solids and metallization of compressed XeF2. Nat. Chem. 2010, 2, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Kurzydłowski, D.; Zaleski-Ejgierd, P.; Grochala, W.; Hoffmann, R. Freezing in resonance structures for better packing: XeF2 becomes (XeF+)(F−) at large compression. Inorg. Chem. 2011, 50, 3832–3840. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Huang, X.; Huang, Y.; Pan, L.; Li, F.; Li, X.; Liu, M.; Liu, B.; Cui, T. Confirmation of the Structural Phase Transitions in XeF2 under High Pressure. J. Phys. Chem. C 2017, 121, 6264–6271. [Google Scholar] [CrossRef]
- Peng, F.; Botana, J.; Wang, Y.; Ma, Y.; Miao, M. Unexpected trend in stability of Xe–F compounds under pressure driven by Xe–Xe covalent bonds. J. Phys. Chem. Lett. 2016, 4562–4567. [Google Scholar] [CrossRef] [PubMed]
- Braïda, B.; Hiberty, P.C. The essential role of charge-shift bonding in hypervalent prototype XeF2. Nat. Chem. 2013, 5, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Oganov, A.R.; Lyakhov, A.O.; Valle, M. How evolutionary crystal structure prediction works—And why. Acc. Chem. Res. 2011, 44, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Zurek, E.; Grochala, W. Predicting crystal structures and properties of matter under extreme conditions via quantum mechanics: The pressure is on. Phys. Chem. Chem. Phys. 2015, 17, 2917–2934. [Google Scholar] [CrossRef] [PubMed]
- Needs, R.J.; Pickard, C.J. Perspective: Role of structure prediction in materials discovery and design. APL Mater. 2016, 4, 53210. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Lv, J.; Ma, Y. Materials discovery at high pressures. Nat. Rev. Mater. 2017, 2, 17005. [Google Scholar] [CrossRef]
- Glass, C.W.; Oganov, A.R.; Hansen, N. USPEX—Evolutionary crystal structure prediction. Comput. Phys. Commun. 2006, 175, 713–720. [Google Scholar] [CrossRef]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Hennig, R.G.; Ashcroft, N.W.; Hoffmann, R. Emergent reduction of electronic state dimensionality in dense ordered Li-Be alloys. Nature 2008, 451, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.F.; Dixon, D.A.; Schrobilgen, G.J. X-ray crystal structures of α-KrF2, [KrF][MF6] (M = As, Sb, Bi), [Kr2F3][SbF6]·KrF2, [Kr2F3]2[SbF6]2·KrF2, and [Kr2F3][AsF6]·[KrF][AsF6]; synthesis and characterization of [Kr2F3][PF6]·nKrF2 ; and theoretical studies of KrF2, KrF+, Kr2F3+, and the [KrF][MF6] (M = P, As, Sb, Bi) ion pairs. Inorg. Chem. 2001, 40, 3002–3017. [Google Scholar] [PubMed]
- Burbank, R.D.; Falconer, W.E.; Sunder, W.A. Crystal Structure of Krypton Difluoride at −80 °C. Science 1972, 178, 1285–1286. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.A.; Wang, T.H.; Grant, D.J.; Peterson, K.A.; Christe, K.O.; Schrobilgen, G.J. Heats of formation of krypton fluorides and stability predictions for KrF4 and KrF6 from high level electronic structure calculations. Inorg. Chem. 2007, 46, 10016–10021. [Google Scholar] [CrossRef] [PubMed]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The electron localization function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Kurzydłowski, D.; Zaleski-Ejgierd, P. Hexacoordinated nitrogen(V) stabilized by high pressure. Sci. Rep. 2016, 6, 36049. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Galy, J.; Matar, S.F. ns2np4 (n = 4, 5) lone pair triplets whirling in M*F2E3 (M* = Kr, Xe): Stereochemistry and ab initio analyses. Solid State Sci. 2017, 64, 114–124. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA : A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Stokes, H.T.; Hatch, D.M. FINDSYM : Program for identifying the space-group symmetry of a crystal. J. Appl. Crystallogr. 2005, 38, 237–238. [Google Scholar] [CrossRef]
- Figgins, B.F.; Smith, B.L. Density and expansivity of solid krypton. Philos. Mag. 1960, 5, 186–188. [Google Scholar] [CrossRef]
- Pauling, L.; Keaveny, I.; Robinson, A.B. The crystal structure of α-fluorine. J. Solid State Chem. 1970, 2, 225–227. [Google Scholar] [CrossRef]
- Johannsen, P.G.; Holzapfel, W.B. Effect of pressure on Raman spectra of solid chlorine. J. Phys. C Solid State Phys. 1983, 16, L1177–L1179. [Google Scholar] [CrossRef]
Figure 1.
(a) Pressure dependence of the enthalpy of formation per atom of various KrmFn phases at 0, 15 and 50 GPa. The compounds stable at 0 GPa (Kr3F2, KrF3), 15 GPa (KrF2, KrF4) and 50 GPa (KrF2, KrF4) are marked in the upper part of the graph; (b) The P1 structure of Kr3F2 (=2Kr·KrF2) at 0 GPa; (c) The Cmcm structure of KrF2 at 50 GPa; (d) the I4/m structure of KrF4 at 50 GPa; blue/red balls depict Kr/F atoms; Kr–F bonds (solid lines) and shortest intermolecular Kr–F contacts (dashed lines) are given in Å.
Figure 1.
(a) Pressure dependence of the enthalpy of formation per atom of various KrmFn phases at 0, 15 and 50 GPa. The compounds stable at 0 GPa (Kr3F2, KrF3), 15 GPa (KrF2, KrF4) and 50 GPa (KrF2, KrF4) are marked in the upper part of the graph; (b) The P1 structure of Kr3F2 (=2Kr·KrF2) at 0 GPa; (c) The Cmcm structure of KrF2 at 50 GPa; (d) the I4/m structure of KrF4 at 50 GPa; blue/red balls depict Kr/F atoms; Kr–F bonds (solid lines) and shortest intermolecular Kr–F contacts (dashed lines) are given in Å.

Figure 2.
The Electron Localization Function (ELF) plotted for (a) the KrF2 molecules found in the Cmcm phase of KrF2 at 150 GPa; (b) the KrF4 molecules found in the I4/m phase of KrF4 at 150 GPa. Yellow color depicts ELF values above 0.9, cyan—below 0.1, while magenta corresponds to values of 0.4. Kr–F covalent bonds are depicted with a black dashed line.
Figure 2.
The Electron Localization Function (ELF) plotted for (a) the KrF2 molecules found in the Cmcm phase of KrF2 at 150 GPa; (b) the KrF4 molecules found in the I4/m phase of KrF4 at 150 GPa. Yellow color depicts ELF values above 0.9, cyan—below 0.1, while magenta corresponds to values of 0.4. Kr–F covalent bonds are depicted with a black dashed line.
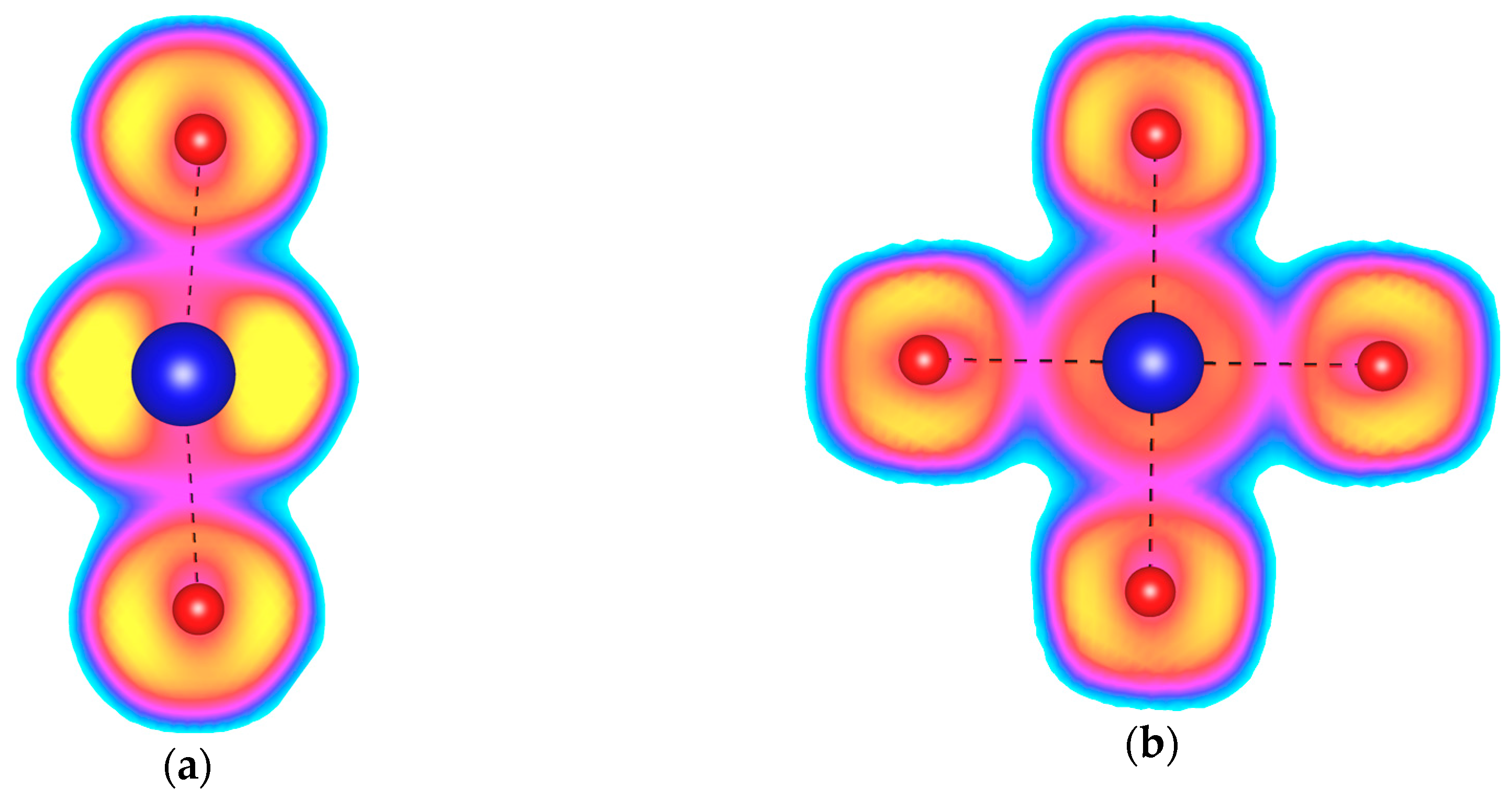
Figure 3.
Pressure dependence of the enthalpy of formation per atom of various KrmFn phases at 50, 80, 100, 140, 180 and 200 GPa. The compounds which stabilize at 80 GPa (Kr6F, Kr4F), 100 GPa (KrF) and 140 GPa (Kr2F) are marked together with KrF2 and KrF4 in the upper part of the graph; note that KrF2 is predicted to decompose above 180 GPa.
Figure 3.
Pressure dependence of the enthalpy of formation per atom of various KrmFn phases at 50, 80, 100, 140, 180 and 200 GPa. The compounds which stabilize at 80 GPa (Kr6F, Kr4F), 100 GPa (KrF) and 140 GPa (Kr2F) are marked together with KrF2 and KrF4 in the upper part of the graph; note that KrF2 is predicted to decompose above 180 GPa.
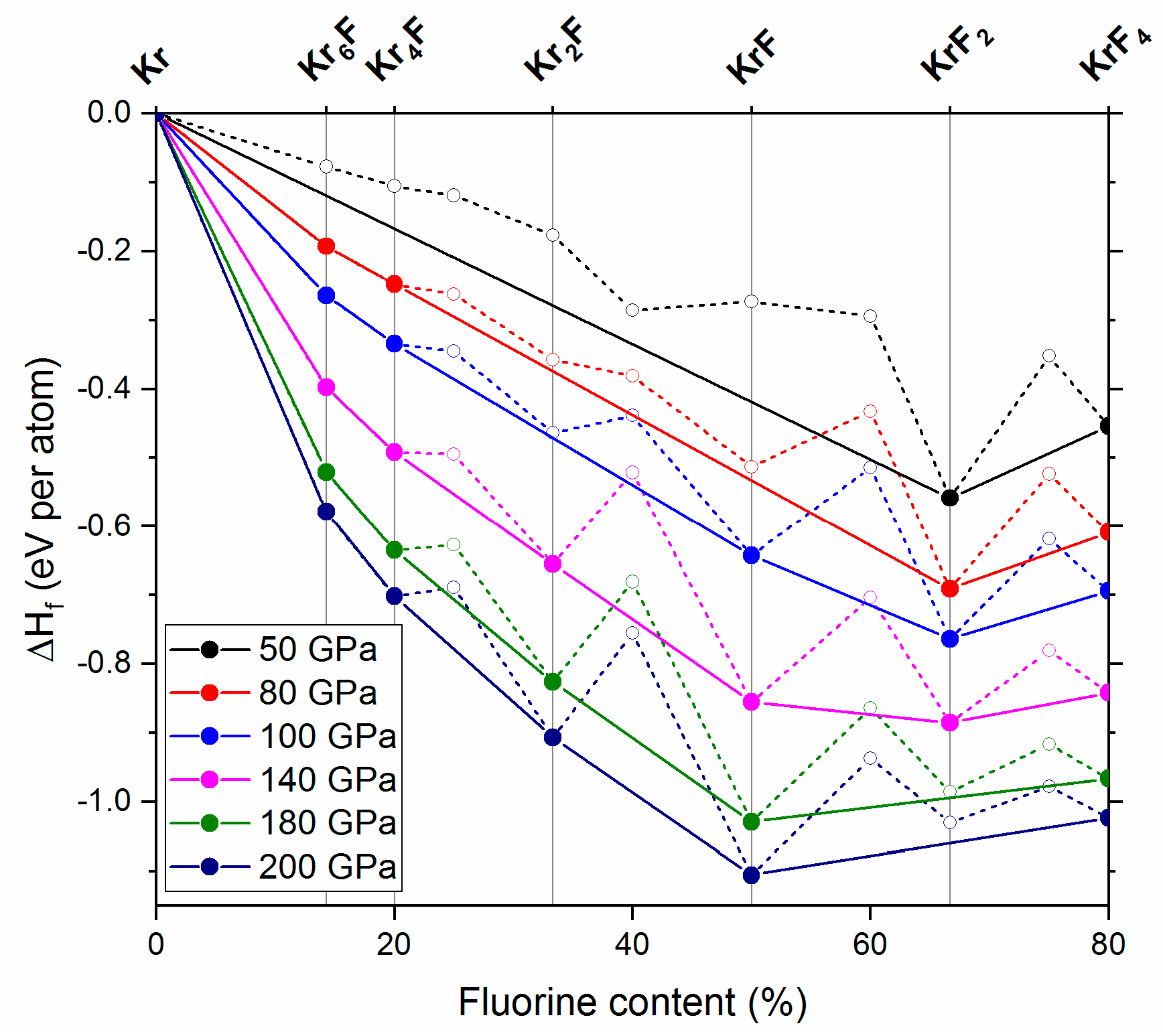
Figure 4.
(a) the I4/mcm structure of Kr2F; (b) the coordination environment of Kr with secondary Kr–Kr contacts marked with dashed lines; (c) ELF function for Kr2F at 150 GPa. Yellow color depicts ELF values above 0.9, cyan—below 0.1, while magenta corresponds to values of 0.4. Shortest Kr–Kr contacts are marked with dashed lines.
Figure 4.
(a) the I4/mcm structure of Kr2F; (b) the coordination environment of Kr with secondary Kr–Kr contacts marked with dashed lines; (c) ELF function for Kr2F at 150 GPa. Yellow color depicts ELF values above 0.9, cyan—below 0.1, while magenta corresponds to values of 0.4. Shortest Kr–Kr contacts are marked with dashed lines.
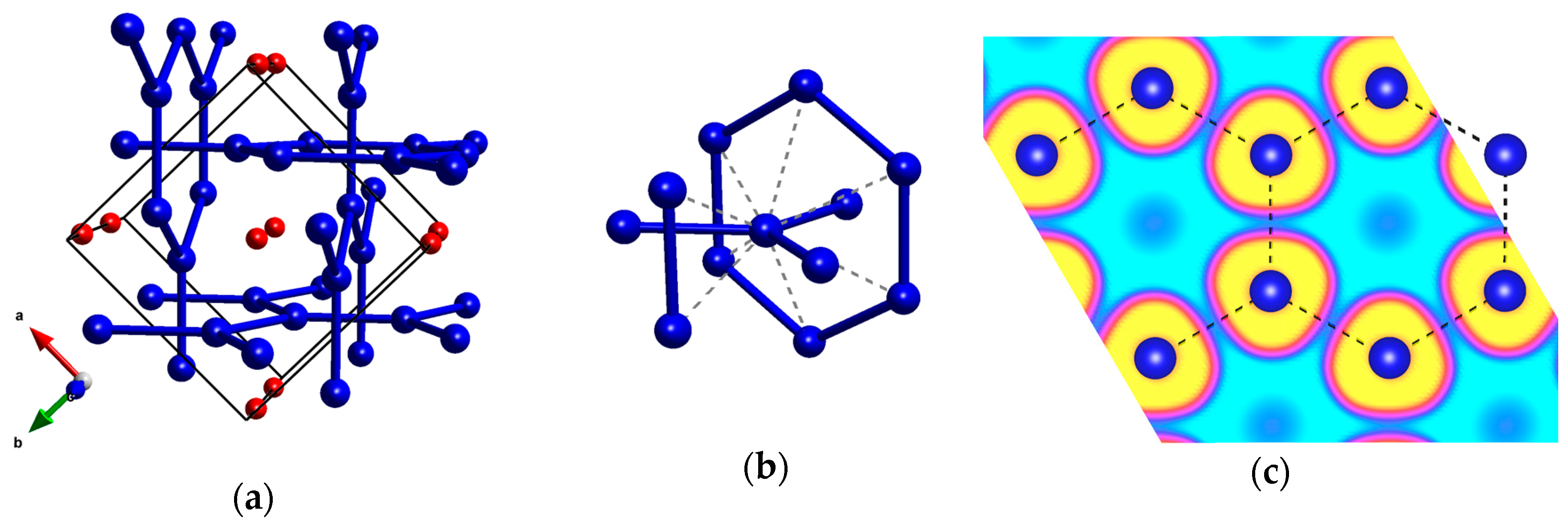
Figure 5.
(a) the P2/m structure of KrF; (b) the coordination environment of Kr with secondary Kr–Kr contacts marked with dashed lines; (c) ELF function for KrF at 150 GPa. Yellow color depicts ELF values above 0.9, cyan—below 0.1, while magenta corresponds to values of 0.4. Shortest Kr–Kr contacts are marked with dashed lines.
Figure 5.
(a) the P2/m structure of KrF; (b) the coordination environment of Kr with secondary Kr–Kr contacts marked with dashed lines; (c) ELF function for KrF at 150 GPa. Yellow color depicts ELF values above 0.9, cyan—below 0.1, while magenta corresponds to values of 0.4. Shortest Kr–Kr contacts are marked with dashed lines.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of the closest Kr–F, Kr–Kr and F–F distances (in Å) for the lowest enthalpy structures of KrmFn compounds at 150 GPa.
Table 1.
Comparison of the closest Kr–F, Kr–Kr and F–F distances (in Å) for the lowest enthalpy structures of KrmFn compounds at 150 GPa.
| Phase | Type | Kr–F | Kr–Kr | F–F |
|---|---|---|---|---|
| Kr | Atomic (3D) | - | 2.68–2.75 | - |
| Kr6F | 2.29–2.39 | 2.58–2.77 | 2.78 | |
| Kr4F | 2.31–2.34 | 2.62–2.70 | 2.68 | |
| Kr2F | 2D | 2.29 | 2.51–2.56; 2.83–2.84 | 2.21 |
| KrF | 1D | 2.27–2.33 | 2.29; 2.78–2.90 | 2.29 |
| KrF2 | Molecular (0D) | 1.80 | 2.66 | 2.19 |
| KrF4 | 1.80 | 3.57 | 2.00 | |
| F2 | - | - | 1.42 |
Table 2.
Comparison of the theoretical thermodynamic stability NgF2, NgF4, and NgF6 (Ng = Ar, Kr, Xe) compounds up to pressures of 200 GPa. Pressures at which a given stoichiometry is stable are given in GPa (amb. —stable at ambient pressure; n.d.—not determined). For NgF2 the numbers in parenthesis correspond to the decomposition pressure given in GPa.
Table 2.
Comparison of the theoretical thermodynamic stability NgF2, NgF4, and NgF6 (Ng = Ar, Kr, Xe) compounds up to pressures of 200 GPa. Pressures at which a given stoichiometry is stable are given in GPa (amb. —stable at ambient pressure; n.d.—not determined). For NgF2 the numbers in parenthesis correspond to the decomposition pressure given in GPa.
| Ng | NgF2 | NgF4 | NgF6 | Ref. |
|---|---|---|---|---|
| Ar | 58 (> 200) | > 200 | n.d. | [30] |
| Kr | amb. (180) | 15 | > 200 | this work |
| Xe | amb. (81) | amb. | amb. | [44] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kurzydłowski, D.; Sołtysiak, M.; Dżoleva, A.; Zaleski-Ejgierd, P. High-Pressure Reactivity of Kr and F2—Stabilization of Krypton in the +4 Oxidation State. Crystals 2017, 7, 329. https://doi.org/10.3390/cryst7110329
AMA Style
Kurzydłowski D, Sołtysiak M, Dżoleva A, Zaleski-Ejgierd P. High-Pressure Reactivity of Kr and F2—Stabilization of Krypton in the +4 Oxidation State. Crystals. 2017; 7(11):329. https://doi.org/10.3390/cryst7110329
Chicago/Turabian StyleKurzydłowski, Dominik, Magdalena Sołtysiak, Aleksandra Dżoleva, and Patryk Zaleski-Ejgierd. 2017. "High-Pressure Reactivity of Kr and F2—Stabilization of Krypton in the +4 Oxidation State" Crystals 7, no. 11: 329. https://doi.org/10.3390/cryst7110329
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.