
Ultrasound Assisted Particle Size Control by Continuous Seed Generation and Batch Growth


Abstract
:1. Introduction
2. Materials and Methods
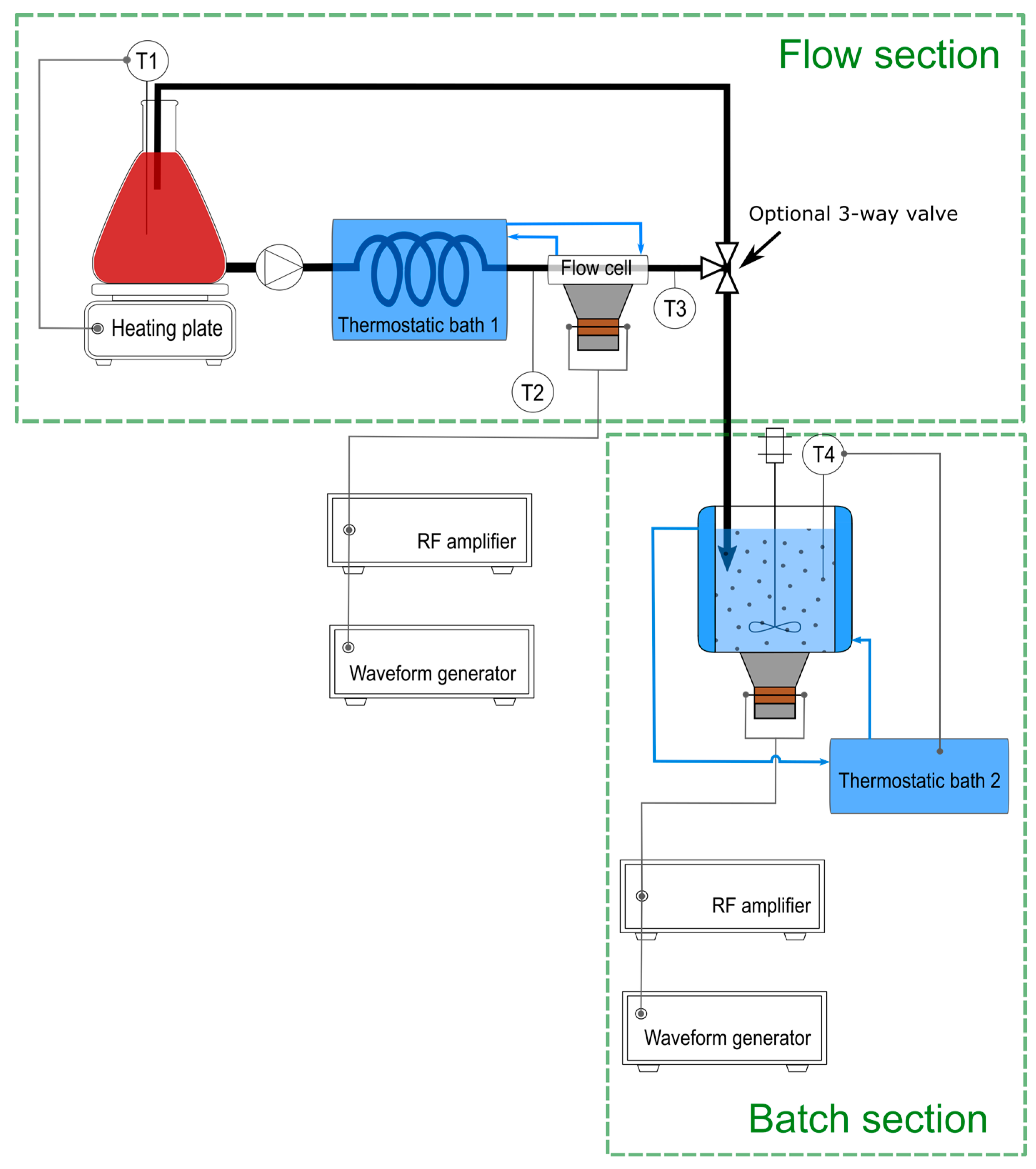
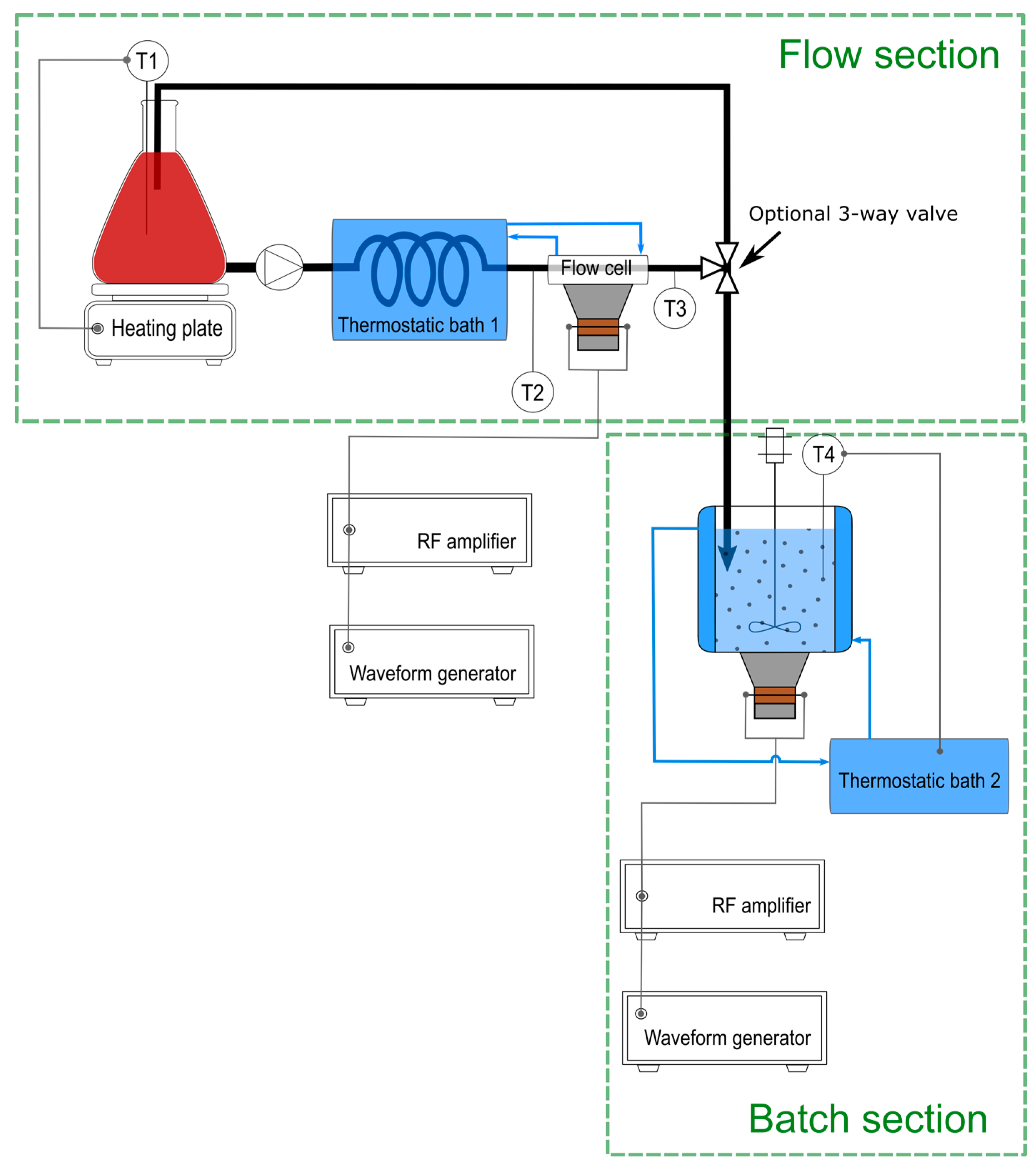
2.1. Experimental Setup
2.2. Calorimetric Power Calibration
2.3. Experimental Procedure Nucleation Temperature
2.4. Experimental Procedure Seed Generation
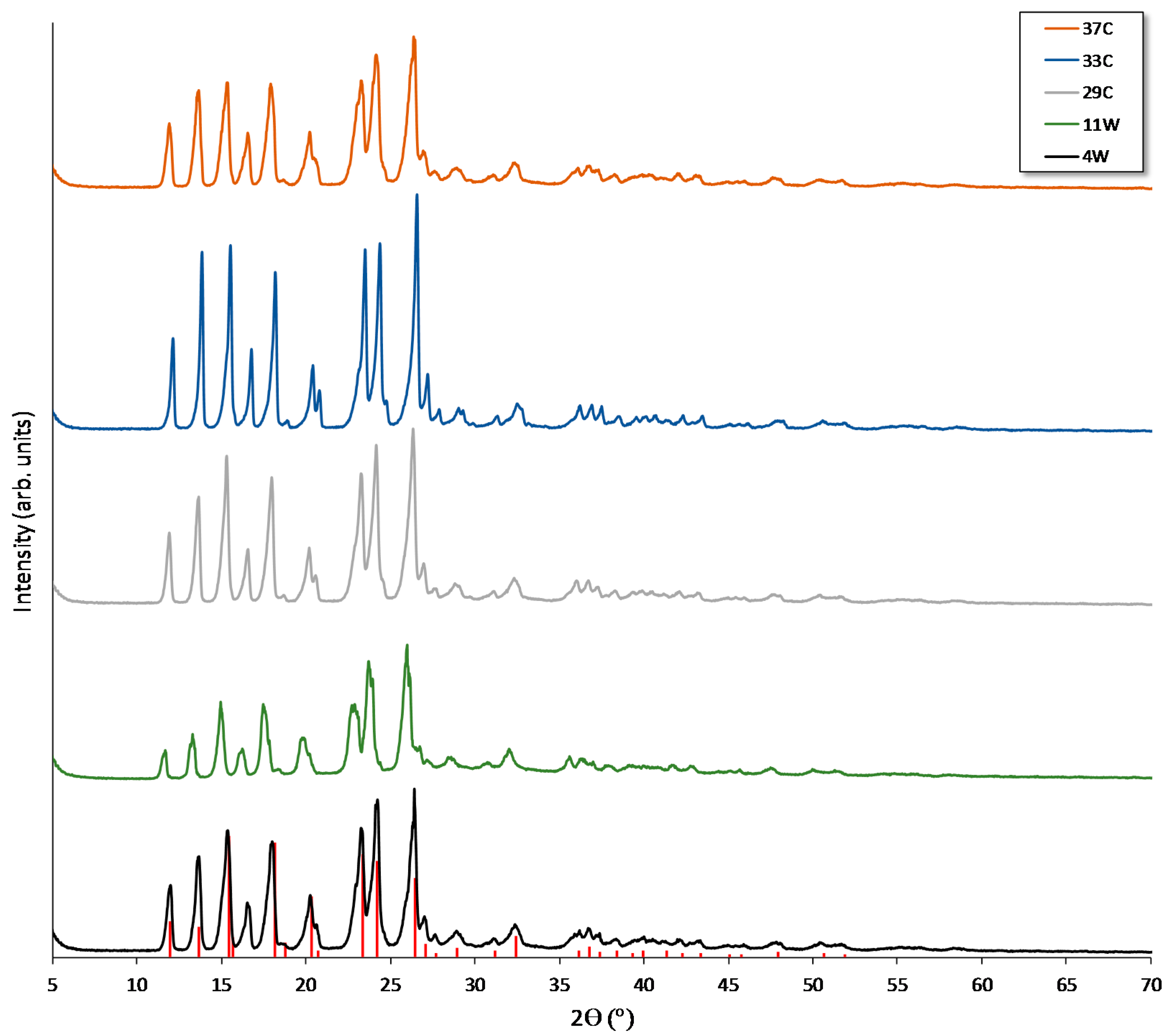
2.5. Analytical Measurements
2.6. Experimental Procedure Batch Growth
2.7. Experimental Procedure Complete Batch Crystallization
3. Results and Discussion
3.1. Particle Size Control During Seed Generation
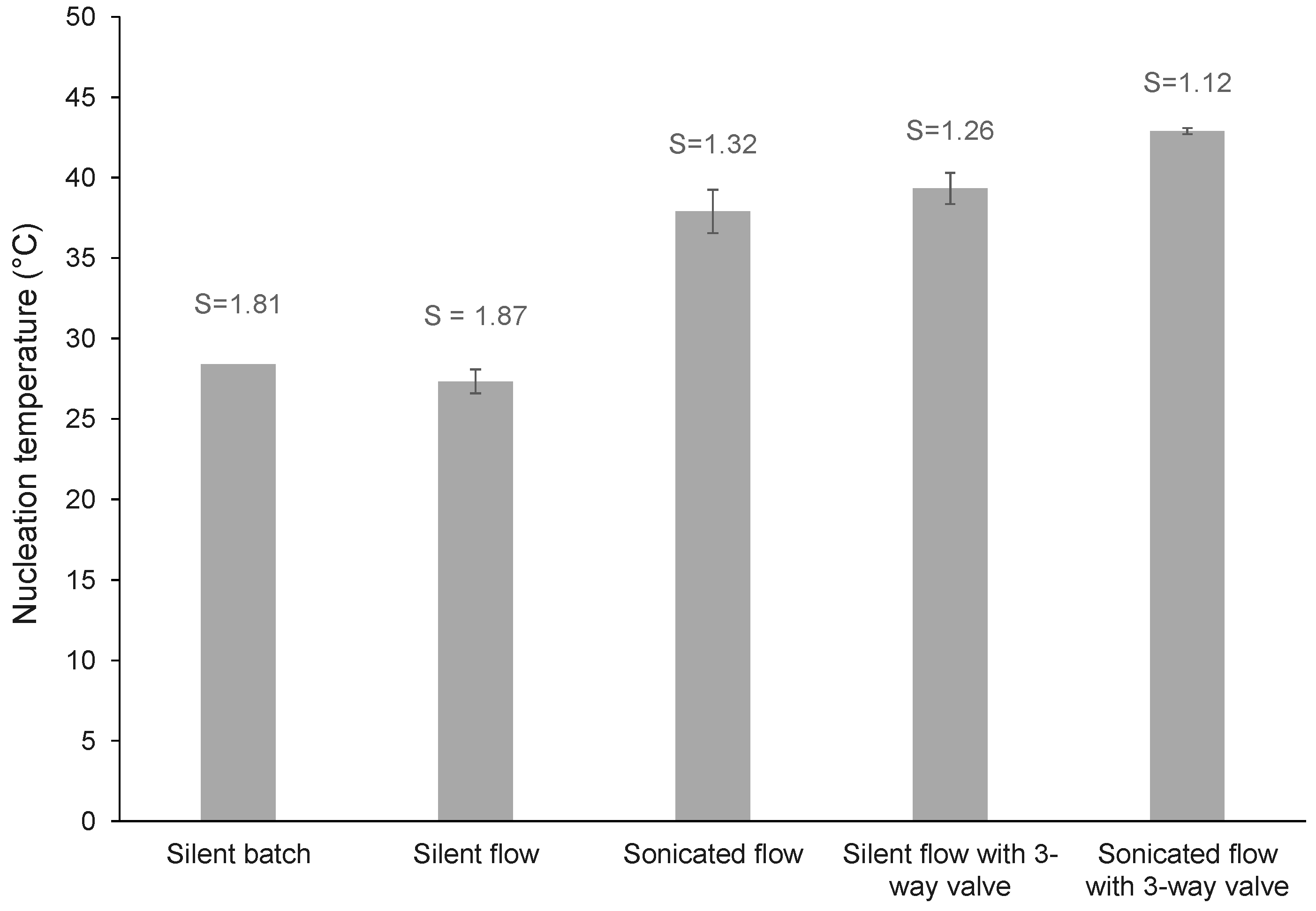
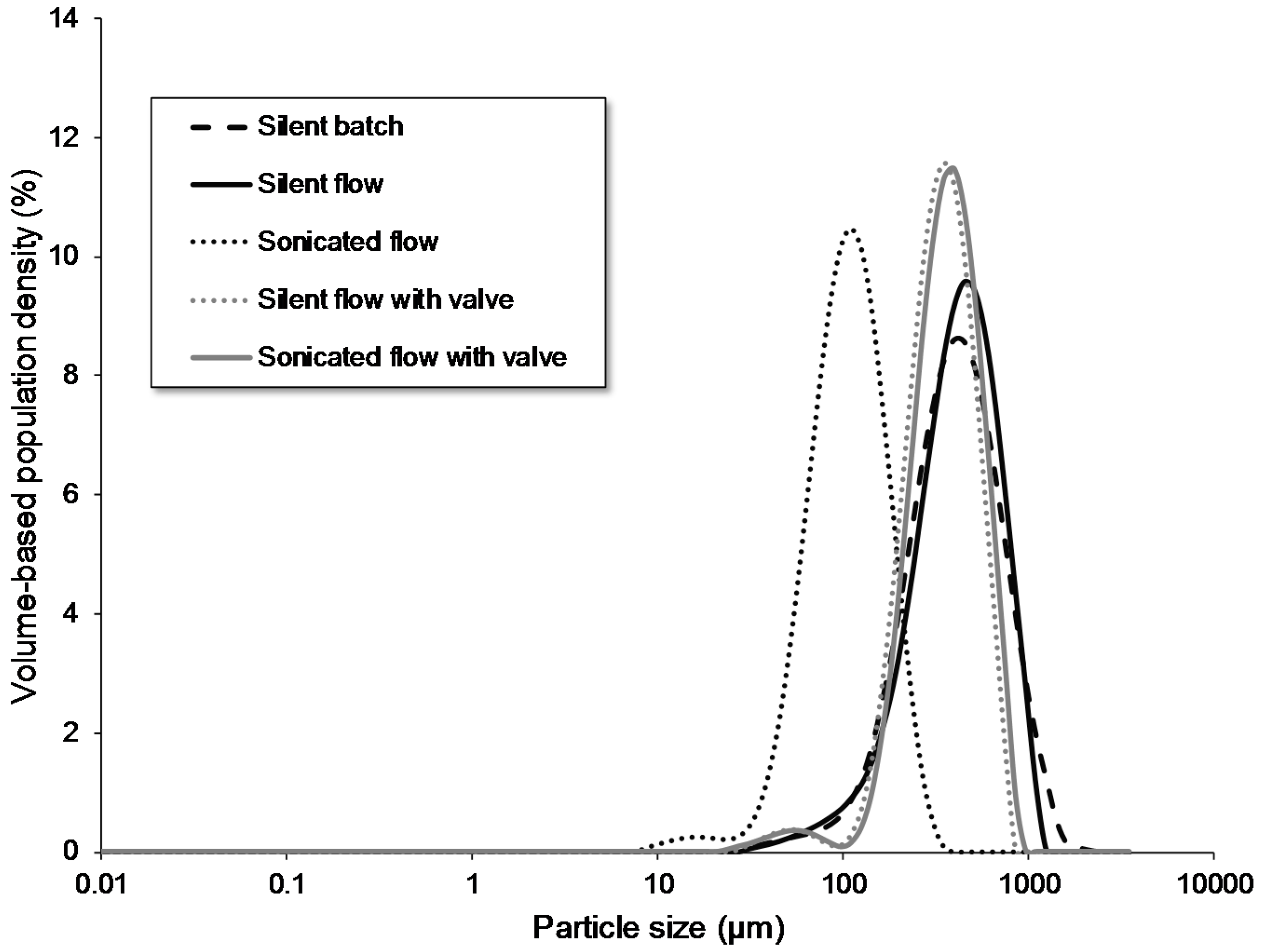
3.1.1. Nucleation in Batch versus Flow
3.1.2. Introduction of a 3-Way Valve
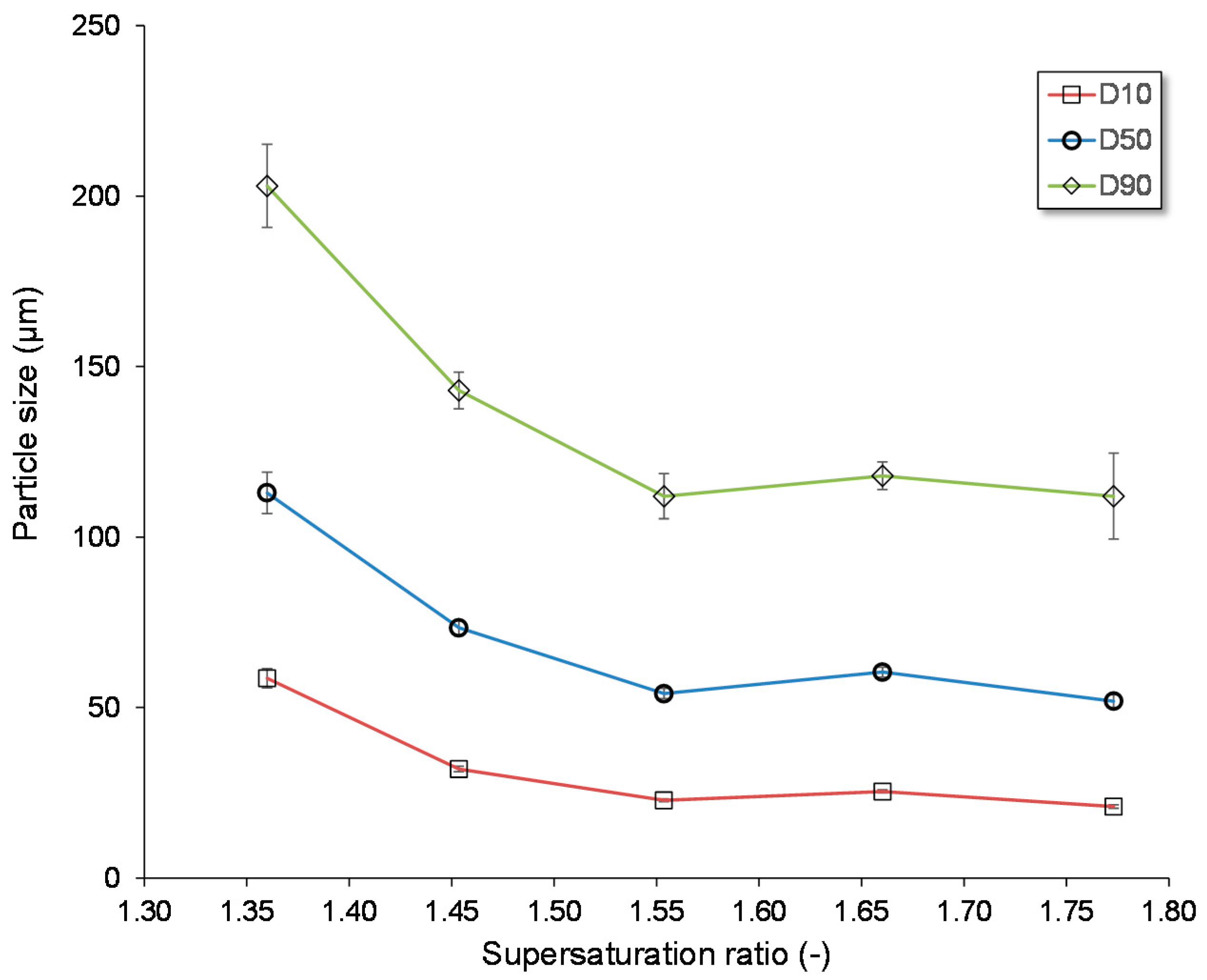
3.1.3. Influence of the Supersaturation Ratio
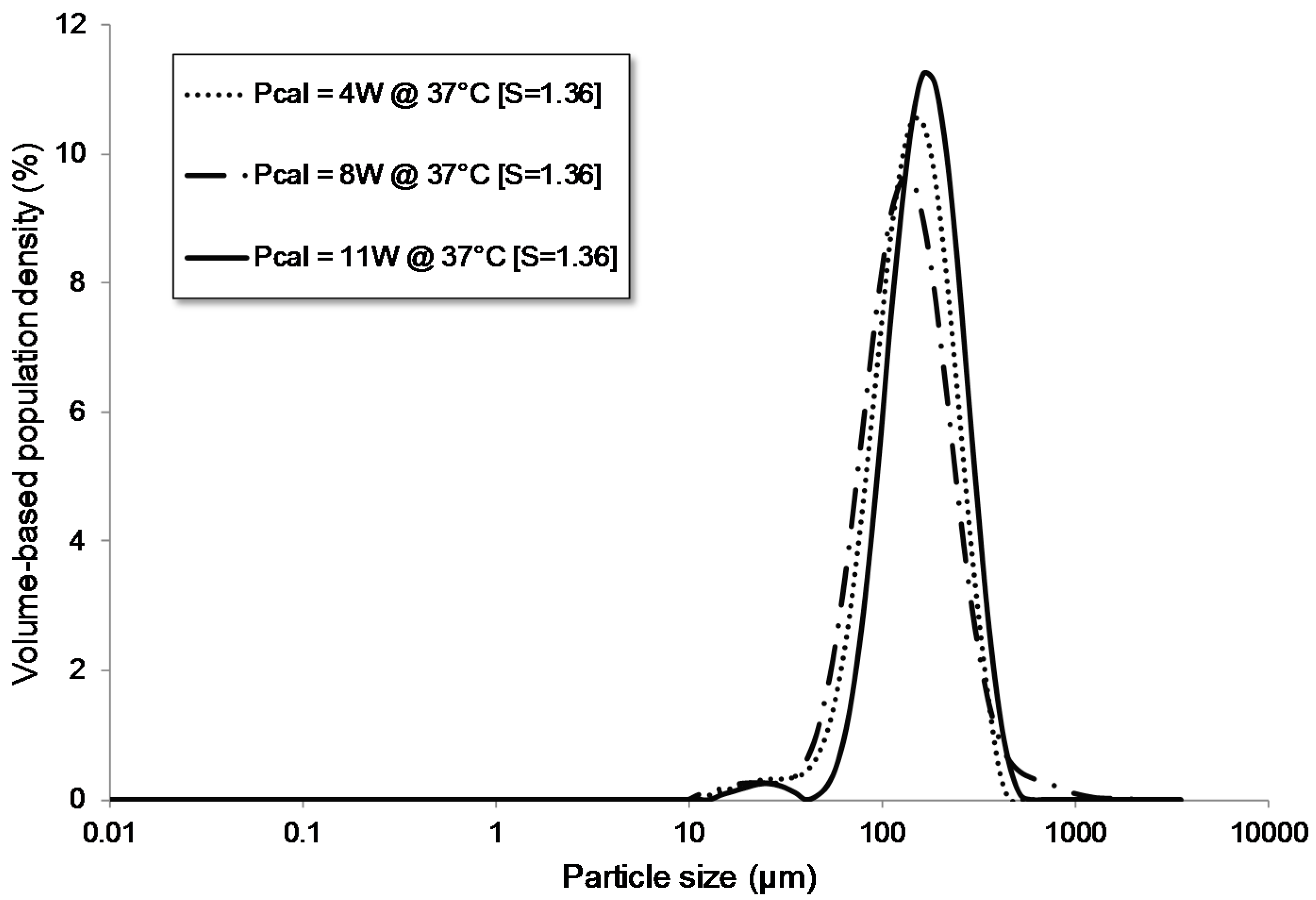
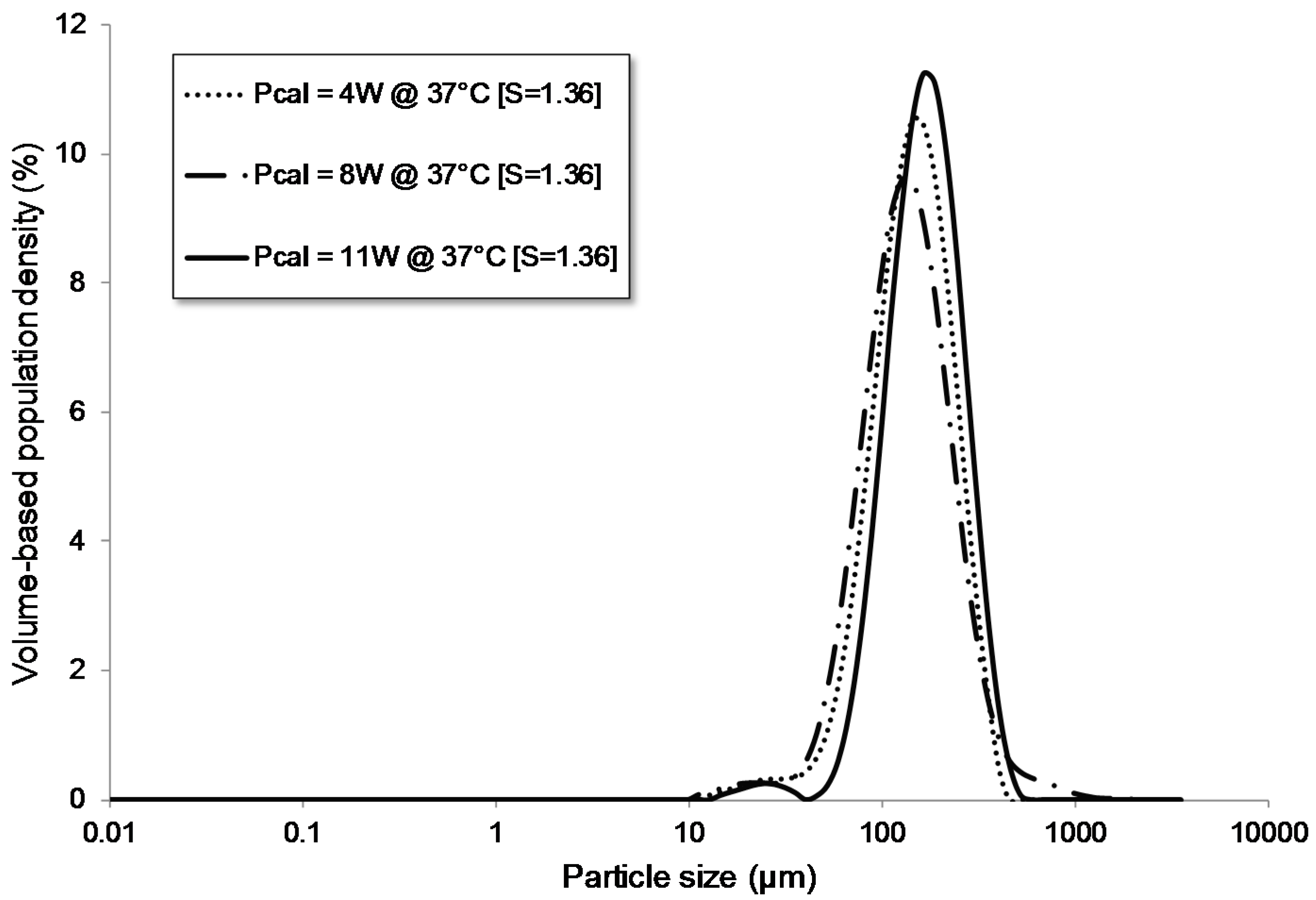
3.1.4. Influence of the Power Level
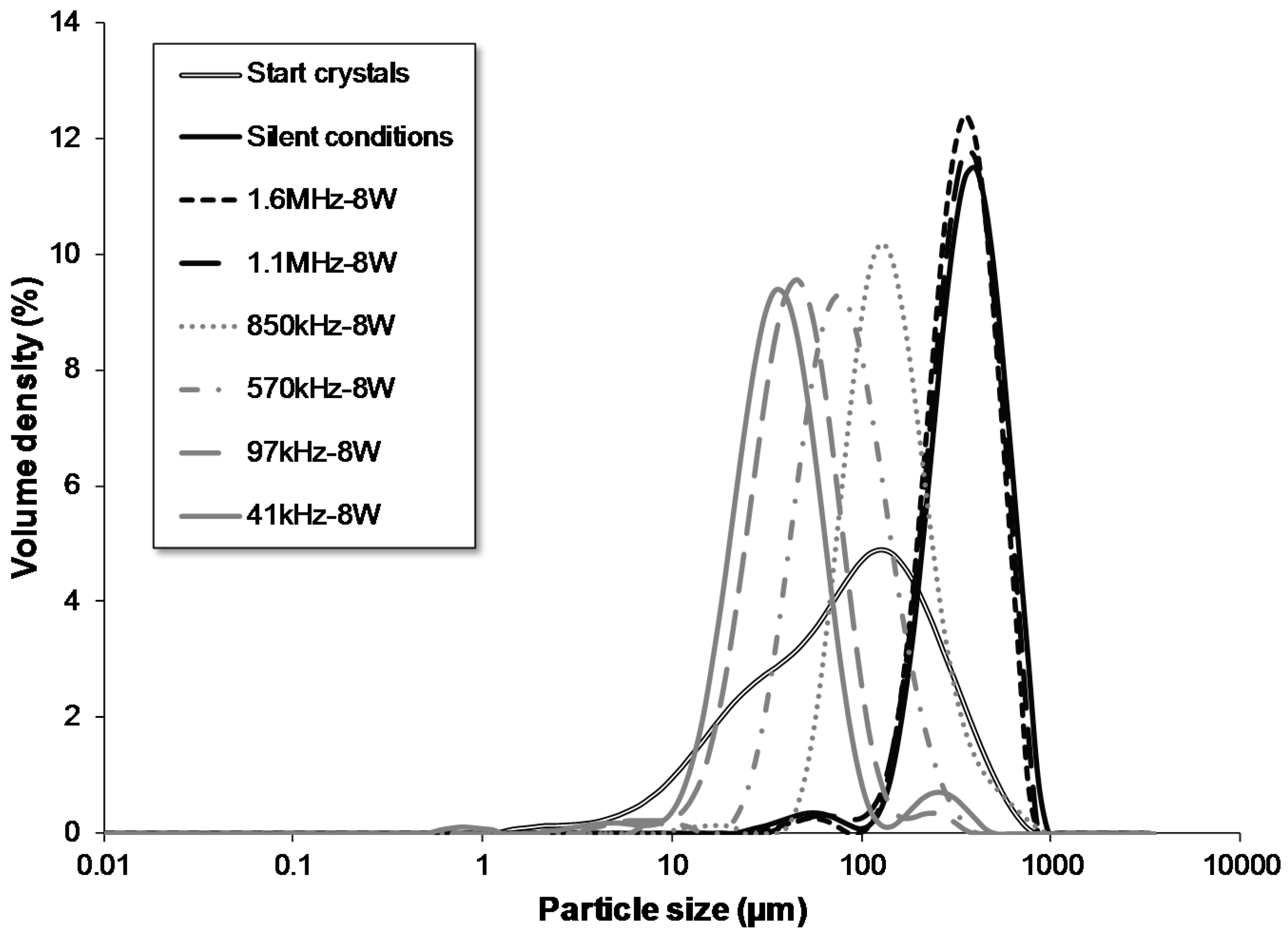
3.2. Particle Size Control in the Growth Section
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eder, R.J.P.; Schmitt, E.K.; Grill, J.; Radl, S.; Gruber-Woelfler, H.; Khinast, J.G. Seed loading effects on the mean crystal size of acetylsalicylic acid in a continuous-flow crystallization device. Cryst. Res. Technol. 2011, 46, 227–237. [Google Scholar] [CrossRef]
- Alvarez, A.J.; Myerson, A.S. Continuous Plug Flow Crystallization of Pharmaceutical Compounds. Cryst. Growth Des. 2010, 10, 2219–2228. [Google Scholar] [CrossRef]
- Castro, F.; Kuhn, S.; Jensen, K.; Ferreira, A.; Rocha, F.; Vicente, A.; Teixeira, J.A. Process intensification and optimization for hydroxyapatite nanoparticles production. Chem. Eng. Sci. 2013, 100, 352–359. [Google Scholar] [CrossRef]
- Eder, R.J.P.; Schrank, S.; Besenhard, M.O.; Roblegg, E.; Gruber-Woelfler, H.; Khinast, J.G. Continuous Sonocrystallization of Acetylsalicylic Acid (ASA): Control of Crystal Size. Cryst. Growth Des. 2012, 12, 4733–4738. [Google Scholar] [CrossRef]
- Besenhard, M.O.; Neugebauer, P.; Ho, C.-D.; Khinast, J.G. Crystal Size Control in a Continuous Tubular Crystallizer. Cryst. Growth Des. 2015, 15, 1683–1691. [Google Scholar] [CrossRef]
- Thorat, A.A.; Dalvi, S.V. Liquid antisolvent precipitation and stabilization of nanoparticles of poorly water soluble drugs in aqueous suspensions: Recent developments and future perspective. Chem. Eng. J. 2012, 181–182, 1–34. [Google Scholar] [CrossRef]
- Lawton, S.; Steele, G.; Shering, P.; Zhao, L.; Laird, I.; Ni, X.-W. Continuous Crystallization of Pharmaceuticals Using a Continuous Oscillatory Baffled Crystallizer. Org. Process. Res. Dev. 2009, 13, 1357–1363. [Google Scholar] [CrossRef]
- Neugebauer, P.; Khinast, J.G. Continuous Crystallization of Proteins in a Tubular Plug-Flow Crystallizer. Cryst. Growth Des. 2015, 15, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Mukai, K.; Cork, D.; Mae, K. Continuous crystallization using a sonicated tubular system for controlling particle size in an API manufacturing process. Chem. Eng. Process. Process. Intensif. 2016, 102, 210–218. [Google Scholar] [CrossRef]
- Siddique, H.; Brown, C.J.; Houson, I.; Florence, A.J. Establishment of a Continuous Sonocrystallization Process for Lactose in an Oscillatory Baffled Crystallizer. Org. Process. Res. Dev. 2015, 19, 1871–1881. [Google Scholar] [CrossRef]
- Jiang, M.; Papageorgiou, C.D.; Waetzig, J.; Hardy, A.; Langston, M.; Braatz, R.D. Indirect Ultrasonication in Continuous Slug-Flow Crystallization. Cryst. Growth Des. 2015, 15, 2486–2492. [Google Scholar] [CrossRef]
- Narducci, O.; Jones, A.G.; Kougoulos, E. Continuous crystallization of adipic acid with ultrasound. Chem. Eng. Sci. 2011, 66, 1069–1076. [Google Scholar] [CrossRef]
- Jordens, J.; Appermont, T.; Gielen, B.; Van Gerven, T.; Braeken, L. Sonofragmentation: Effect of Ultrasound Frequency and Power on Particle Breakage. Cryst. Growth Des. 2016, 16, 6167–6177. [Google Scholar] [CrossRef]
- Sander, J.R.G.; Zeiger, B.W.; Suslick, K.S. Sonocrystallization and sonofragmentation. Ultrason. Sonochem. 2014, 21, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Zeiger, B.W.; Suslick, K.S. Sonofragmentation of molecular crystals. J. Am. Chem. Soc. 2011, 133, 14530–14533. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, K.; Ruether, F.; Schembecker, G. Sonocrystallization and crystallization with gassing of adipic acid. Chem. Eng. Sci. 2010, 65, 1016–1027. [Google Scholar] [CrossRef]
- Harzali, H.; Baillon, F.; Louisnard, O.; Espitalier, F.; Mgaidi, A. Experimental study of sono-crystallisation of ZnSO4·7H2O, and interpretation by the segregation theory. Ultrason. Sonochem. 2011, 18, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Peinado, L.; de los, S.; Castro, M.D.L. The role of ultrasound in pharmaceutical production: Sonocrystallization. J. Pharm. Pharmacol. 2016, 68, 1249–1267. [Google Scholar] [CrossRef] [PubMed]
- Kudo, S.; Takiyama, H. Production of Fine Organic Crystalline Particles by Using Milli Segmented Flow Crystallizer. J. Chem. Eng. Jpn. 2012, 45, 305–309. [Google Scholar] [CrossRef]
- Rossi, D.; Jamshidi, R.; Saffari, N.; Kuhn, S.; Gavriilidis, A.; Mazzei, L. Continuous-Flow Sonocrystallization in Droplet-Based Microfluidics. Cryst. Growth Des. 2015, 15, 5519–5529. [Google Scholar] [CrossRef]
- Suslick, K.S. Sonochemistry. Kirk Othmer Encycl. Chem. Technol. 1998. [Google Scholar] [CrossRef]
- Luque de Castro, M.D.; Priego-Capote, F. Ultrasound-assisted crystallization (sonocrystallization). Ultrason. Sonochem. 2007, 14, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Nii, S.; Takayanagi, S. Growth and size control in anti-solvent crystallization of glycine with high frequency ultrasound. Ultrason. Sonochem. 2014, 21, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.; Cui, Y.; Myerson, A.S. Contact Secondary Nucleation as a Means of Creating Seeds for Continuous Tubular Crystallizers. Cryst. Growth Des. 2013, 13, 2514–2521. [Google Scholar] [CrossRef]
- Jordens, J.; Gielen, B.; Braeken, L.; Van Gerven, T. Determination of the effect of the ultrasonic frequency on the cooling crystallization of paracetamol. Chem. Eng. Process. Process. Intensif. 2014, 84, 38–44. [Google Scholar] [CrossRef]
- Raso, J.; Mañas, P.; Pagán, R.; Sala, F.J. Influence of different factors on the output power transferred into medium by ultrasound. Ultrason. Sonochem. 1999, 5, 157–162. [Google Scholar] [CrossRef]
- Hagenson, L.C.; Doraiswamy, L.K. Comparison of the effects of ultrasound and mechanical agitation on a reacting solid-liquid system. Chem. Eng. Sci. 1998, 53, 131–148. [Google Scholar] [CrossRef]
- Hallez, L.; Touyeras, F.; Hihn, J.Y.; Klima, J. Energetic balance in an ultrasonic reactor using focused or flat high frequency transducers. Ultrason. Sonochem. 2007, 14, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Sutkar, V.S.; Gogate, P.R. Design aspects of sonochemical reactors: Techniques for understanding cavitational activity distribution and effect of operating parameters. Chem. Eng. J. 2009, 155, 26–36. [Google Scholar] [CrossRef]
- Kordylla, A.; Koch, S.; Tumakaka, F.; Schembecker, G. Towards an optimized crystallization with ultrasound: Effect of solvent properties and ultrasonic process parameters. J. Cryst. Growth 2008, 310, 4177–4184. [Google Scholar] [CrossRef]
- Fujiwara, M.; Chow, P.S.; Ma, D.L.; Braatz, R.D. Paracetamol Crystallization Using Laser Backscattering and ATR-FTIR Spectroscopy: Metastability, Agglomeration, and Control. Cryst. Growth Des. 2002, 2, 363–370. [Google Scholar] [CrossRef]
- Kwak, B.-M.; Lee, J.E.; Ahn, J.-H.; Jeon, T.-H. Laser diffraction particle sizing by wet dispersion method for spray-dried infant formula. J. Food Eng. 2009, 92, 324–330. [Google Scholar] [CrossRef]
- Bari, A.H.; Chawla, A.; Pandit, A.B. Sono-crystallization kinetics of K2SO4: Estimation of nucleation, growth, breakage and agglomeration kinetics. Ultrason. Sonochem. 2017, 35, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.; Morris, G.; Hao, H.; Barrett, M.; Glennon, B. In-situ monitoring and characterization of plug flow crystallizers. Chem. Eng. Sci. 2012, 77, 105–111. [Google Scholar] [CrossRef]
- Marchisio, D.L.; Rivautella, L.; Barresi, A.A. Design and scale-up of chemical reactors for nanoparticle precipitation. AIChE J. 2006, 52, 1877–1887. [Google Scholar] [CrossRef]
- Myerson, A. Handbook of Industrial Crystallization; Butterworth-Heinemann: Oxford, UK, 2002. [Google Scholar]
- Méndez del Río, J.R.; Rousseau, R.W. Batch and Tubular-Batch Crystallization of Paracetamol: Crystal Size Distribution and Polymorph Formation. Cryst. Growth Des. 2006, 6, 1407–1414. [Google Scholar] [CrossRef]
- Del Rio, M.; Jose, R. Solubility and Phase Transitions in Batch and Laminar-Flow Tubular Crystallizers; Georgia Institute of Technology: Atlanta, GA, USA, 2004. [Google Scholar]
- Kim, K.-J.; Mersmann, A. Estimation of metastable zone width in different nucleation processes. Chem. Eng. Sci. 2001, 56, 2315–2324. [Google Scholar] [CrossRef]
- Nalajala, V.S.; Moholkar, V.S. Investigations in the physical mechanism of sonocrystallization. Ultrason. Sonochem. 2011, 18, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Kiani, H.; Zhang, Z.; Delgado, A.; Sun, D.-W. Ultrasound assisted nucleation of some liquid and solid model foods during freezing. Food Res. Int. 2011, 44, 2915–2921. [Google Scholar] [CrossRef]
- Wohlgemuth, K.; Kordylla, A.; Ruether, F.; Schembecker, G. Experimental study of the effect of bubbles on nucleation during batch cooling crystallization. Chem. Eng. Sci. 2009, 64, 4155–4163. [Google Scholar] [CrossRef]
- Louisnard, O.; Gomez, F.J.; Grossier, R. Segregation of a liquid mixture by a radially oscillating bubble. J. Fluid Mech. 2007, 577, 385. [Google Scholar] [CrossRef]
- Chow, R.; Blindt, R.; Chivers, R.; Povey, M. The sonocrystallisation of ice in sucrose solutions: Primary and secondary nucleation. Ultrasonics 2003, 41, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Kordylla, A.; Krawczyk, T.; Tumakaka, F.; Schembecker, G. Modeling ultrasound-induced nucleation during cooling crystallization. Chem. Eng. Sci. 2009, 64, 1635–1642. [Google Scholar] [CrossRef]
- Dennehy, R.D. Particle Engineering Using Power Ultrasound1. Org. Process. Res. Dev. 2003, 7, 1002–1006. [Google Scholar] [CrossRef]
- Abbas, A.; Srour, M.; Tang, P.; Chiou, H.; Chan, H.-K.; Romagnoli, J.A. Sonocrystallisation of sodium chloride particles for inhalation. Chem. Eng. Sci. 2007, 62, 2445–2453. [Google Scholar] [CrossRef]
- Kougoulos, E.; Marziano, I.; Miller, P.R. Lactose particle engineering: Influence of ultrasound and anti-solvent on crystal habit and particle size. J. Cryst. Growth 2010, 312, 3509–3520. [Google Scholar] [CrossRef]
- Louhi-Kultanen, M.; Karjalainen, M.; Rantanen, J.; Huhtanen, M.; Kallas, J. Crystallization of glycine with ultrasound. Int. J. Pharm. 2006, 320, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ruecroft, G.; Hipkiss, D.; Ly, T.; Maxted, N.; Cains, P.W. Sonocrystallization: The Use of Ultrasound for Improved Industrial Crystallization. Org. Process. Res. Dev. 2005, 9, 923–932. [Google Scholar] [CrossRef]
- Bučar, D.-K.; Elliott, J.A.; Eddleston, M.D.; Cockcroft, J.K.; Jones, W. Sonocrystallization Yields Monoclinic Paracetamol with Significantly Improved Compaction Behavior. Angew. Chem. 2015, 127, 251–255. [Google Scholar] [CrossRef]
- Kaur Bhangu, S.; Ashokkumar, M.; Lee, J. Ultrasound Assisted Crystallization of Paracetamol: Crystal Size Distribution and Polymorph Control. Cryst. Growth Des. 2016, 16, 1934–1941. [Google Scholar] [CrossRef]
- Wagterveld, R.M.; Miedema, H.; Witkamp, G.-J. Effect of Ultrasonic Treatment on Early Growth during CaCO3 Precipitation. Cryst. Growth Des. 2012, 12, 4403–4410. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section | Transducer Type | f (kHz) | A (mVpp) | ΔT (°C) | Pcal (W) |
|---|---|---|---|---|---|
| Flow | Ultrasonics World MPI-7850D-20_40_60H | 68 | 250 | 0.56 | 4 |
| Ultrasonics World MPI-7850D-20_40_60H | 68 | 570 | 1.21 | 8 | |
| Ultrasonics World MPI-7850D-20_40_60H | 68 | 1200 | 1.63 | 11 | |
| Batch | Ultrasonics World MPI-7850D-20_40_60H | 41 | 318 | - | 8 |
| Ultrasonics World MPI-7850D-20_40_60H | 97 | 484 | - | 8 | |
| Meinhardt E/805/T/M | 570 | 175 | - | 8 | |
| Meinhardt E/805/T/M | 850 | 207 | - | 8 | |
| Meinhardt E/805/T/M | 1130 | 172 | - | 8 | |
| Honda Electronics Co., Ltd.-1.6 MHz | 1600 | 183 | - | 8 |
| Experiment Name | Operation Mode Nucleation | Operation Mode Growth | fnucl | Pcal nucl | fgrowth | Pcal growth | 3-Way Valve | Tus in flow | S | Tnucl | D10 | D50 | D90 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| kHz | W | kHz | W | - | °C | - | °C | µm | µm | µm | |||
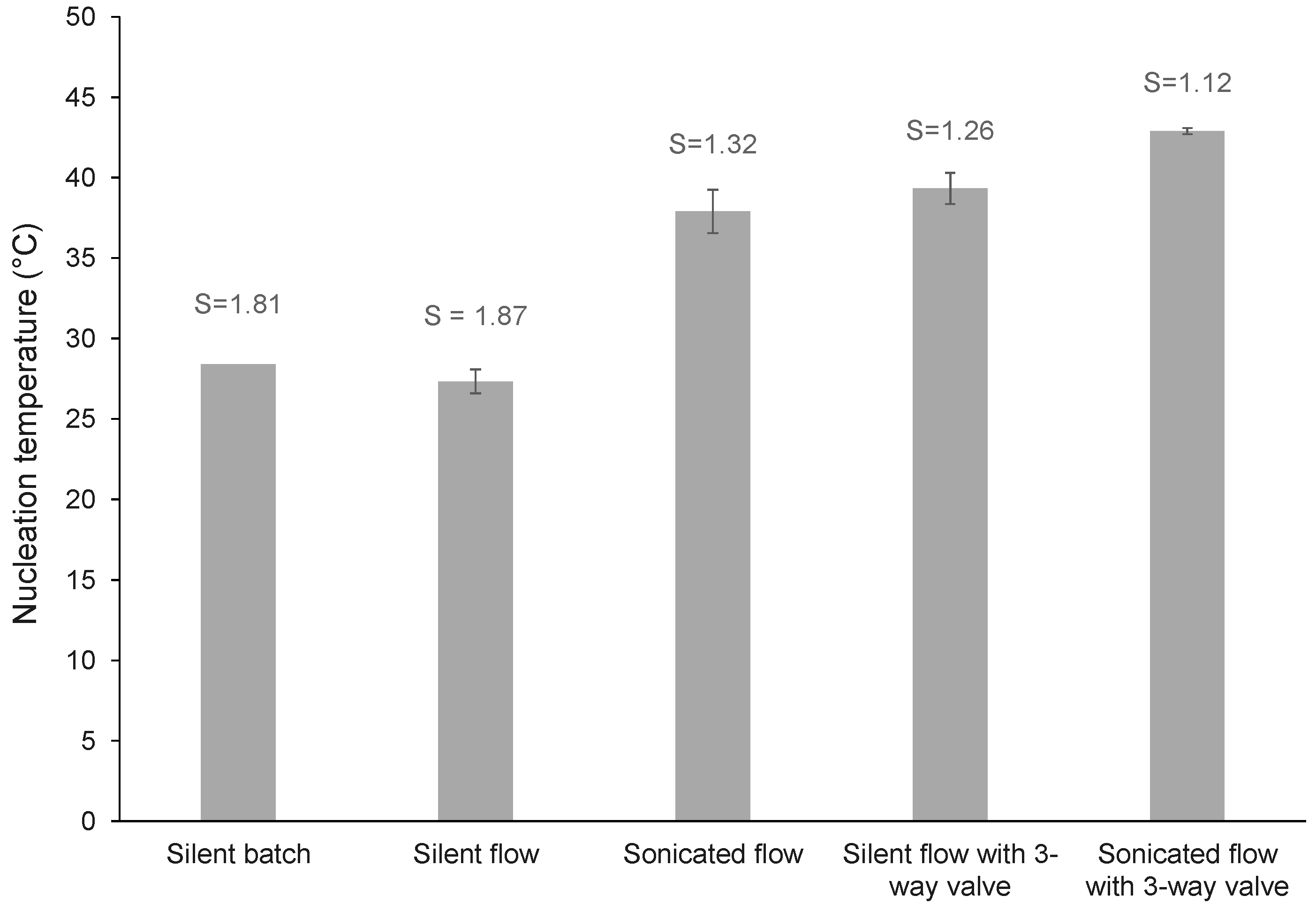
| silent batch | batch | batch | - | - | - | - | - | - | 1.81 | 28.40 | 190 | 429 | 878 |
| silent flow | flow | batch | - | - | - | - | no | - | 1.87 | 27.33 | 197 | 450 | 825 |
| sonicated flow | flow | batch | 68 | 8 | - | - | no | 60.00 | 1.32 | 37.90 | 59 | 113 | 203 |
| silent flow with 3-way valve | flow | batch | - | - | - | - | yes | - | 1.26 | 39.33 | 225 | 410 | 687 |
| Sonicated flow with 3-way valve | flow | batch | 68 | 8 | - | - | yes | 60.00 | 1.12 | 42.89 | 244 | 444 | 746 |
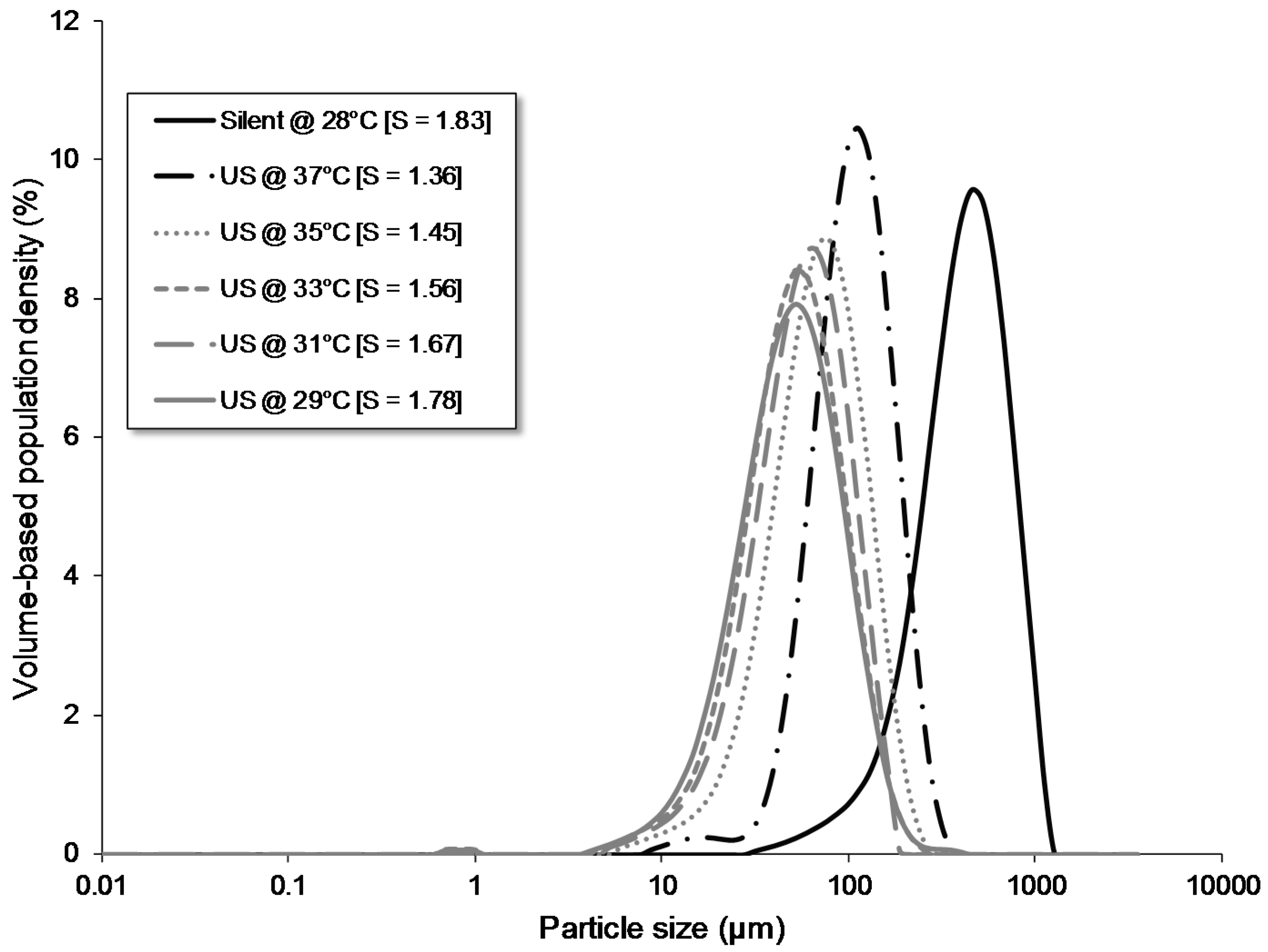
| silent @28 °C | flow | batch | - | - | - | - | no | - | 1.83 | 28.00 | 197 | 450 | 825 |
| US @37 °C | flow | batch | 68 | 8 | - | - | no | 37.00 | 1.36 | 37.00 | 59 | 113 | 203 |
| US @35 °C | flow | batch | 68 | 8 | - | - | no | 35.00 | 1.45 | 35.00 | 32 | 73 | 143 |
| US @33 °C | flow | batch | 68 | 8 | - | - | no | 33.00 | 1.55 | 33.00 | 23 | 54 | 112 |
| US @31 °C | flow | batch | 68 | 8 | - | - | no | 31.00 | 1.66 | 31.00 | 25 | 60 | 118 |
| US @29 °C | flow | batch | 68 | 8 | - | - | no | 29.00 | 1.77 | 29.00 | 21 | 52 | 112 |
| Pcal = 4 W | flow | batch | 68 | 4 | - | - | no | 60.00 | 1.36 | 37.00 | 79 | 154 | 271 |
| Pcal = 8 W | flow | batch | 68 | 8 | - | - | no | 60.00 | 1.36 | 37.00 | 71 | 142 | 282 |
| Pcal = 11 W | flow | batch | 68 | 11 | - | - | no | 60.00 | 1.36 | 37.00 | 99 | 177 | 306 |
| start crystals | flow | batch | 68 | 8 | - | - | yes | 60.00 | 1.12 | 42.5–43.1 | 17 | 94 | 305 |
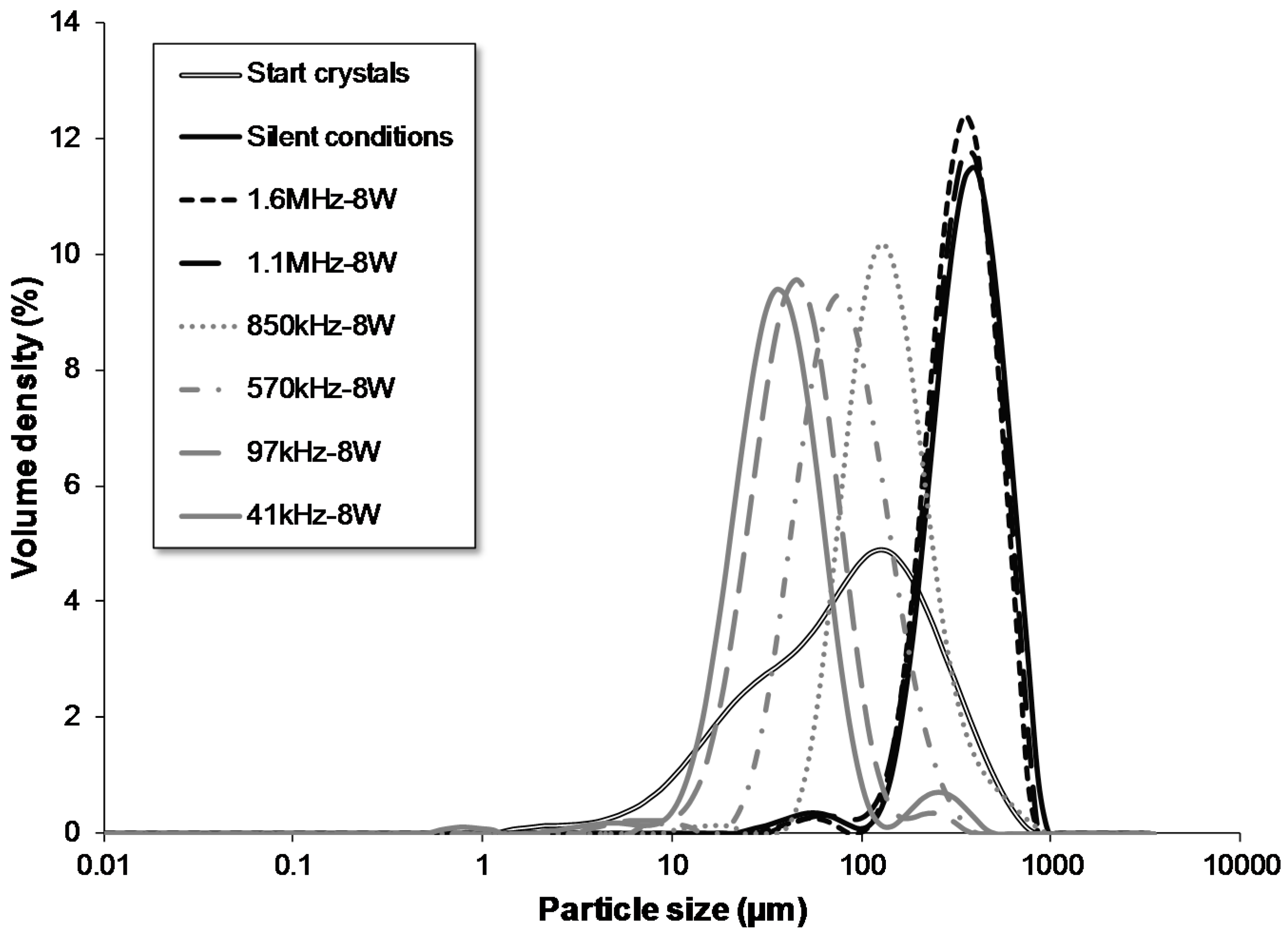
| 1.6 MHz | flow | batch | 68 | 8 | 1600 | 8 | yes | 60.00 | 1.12 | 42.5–43.1 | 211 | 364 | 588 |
| 1.1 MHz | flow | batch | 68 | 8 | 1130 | 8 | yes | 60.00 | 1.12 | 42.5–43.1 | 205 | 372 | 614 |
| 850 kHz | flow | batch | 68 | 8 | 850 | 8 | yes | 60.00 | 1.12 | 42.5–43.1 | 79 | 144 | 295 |
| 570 kHz | flow | batch | 68 | 8 | 570 | 8 | yes | 60.00 | 1.12 | 42.5–43.1 | 42 | 83 | 170 |
| 97 kHz | flow | batch | 68 | 8 | 97 | 8 | yes | 60.00 | 1.12 | 42.5–43.1 | 23 | 47 | 91 |
| 41 kHz | flow | batch | 68 | 8 | 41 | 8 | yes | 60.00 | 1.12 | 42.5–43.1 | 19 | 39 | 79 |
| Setting | Value | Unit |
|---|---|---|
| Refractive index particles | 1.619 | - |
| Absorption index particles | 0.01 | - |
| Density particles | 1.34 | g/mL |
| Refractive index dispersant | 1.375 | - |
| Lower obscuration limit | 10 | % |
| Upper obscuration limit | 30 | % |
| Stirring speed | 2400 | rpm |
| Duration measurement | 10 | s |
| Number of measurements | 5 | - |
| Max. allowed RSD D50 | 3 | % |
| Max. allowed RSD D10 & D90 | 3 | % |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordens, J.; Canini, E.; Gielen, B.; Van Gerven, T.; Braeken, L. Ultrasound Assisted Particle Size Control by Continuous Seed Generation and Batch Growth. Crystals 2017, 7, 195. https://doi.org/10.3390/cryst7070195
Jordens J, Canini E, Gielen B, Van Gerven T, Braeken L. Ultrasound Assisted Particle Size Control by Continuous Seed Generation and Batch Growth. Crystals. 2017; 7(7):195. https://doi.org/10.3390/cryst7070195
Chicago/Turabian StyleJordens, Jeroen, Enio Canini, Bjorn Gielen, Tom Van Gerven, and Leen Braeken. 2017. "Ultrasound Assisted Particle Size Control by Continuous Seed Generation and Batch Growth" Crystals 7, no. 7: 195. https://doi.org/10.3390/cryst7070195
APA StyleJordens, J., Canini, E., Gielen, B., Van Gerven, T., & Braeken, L. (2017). Ultrasound Assisted Particle Size Control by Continuous Seed Generation and Batch Growth. Crystals, 7(7), 195. https://doi.org/10.3390/cryst7070195