A Robust Framework Based on Polymeric Octamolybdate Anions and Copper(II) Complexes of Tetradentate N-donor Ligands

 ,
, 
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Methods
2.2. Synthesis of [Cu(cyclam)]2[Mo8O26]·1.5H2O (1) and [Cu(cyclam)]2[Mo8O26] (1a)
2.3. X-ray Crystallography
3. Results and Discussion
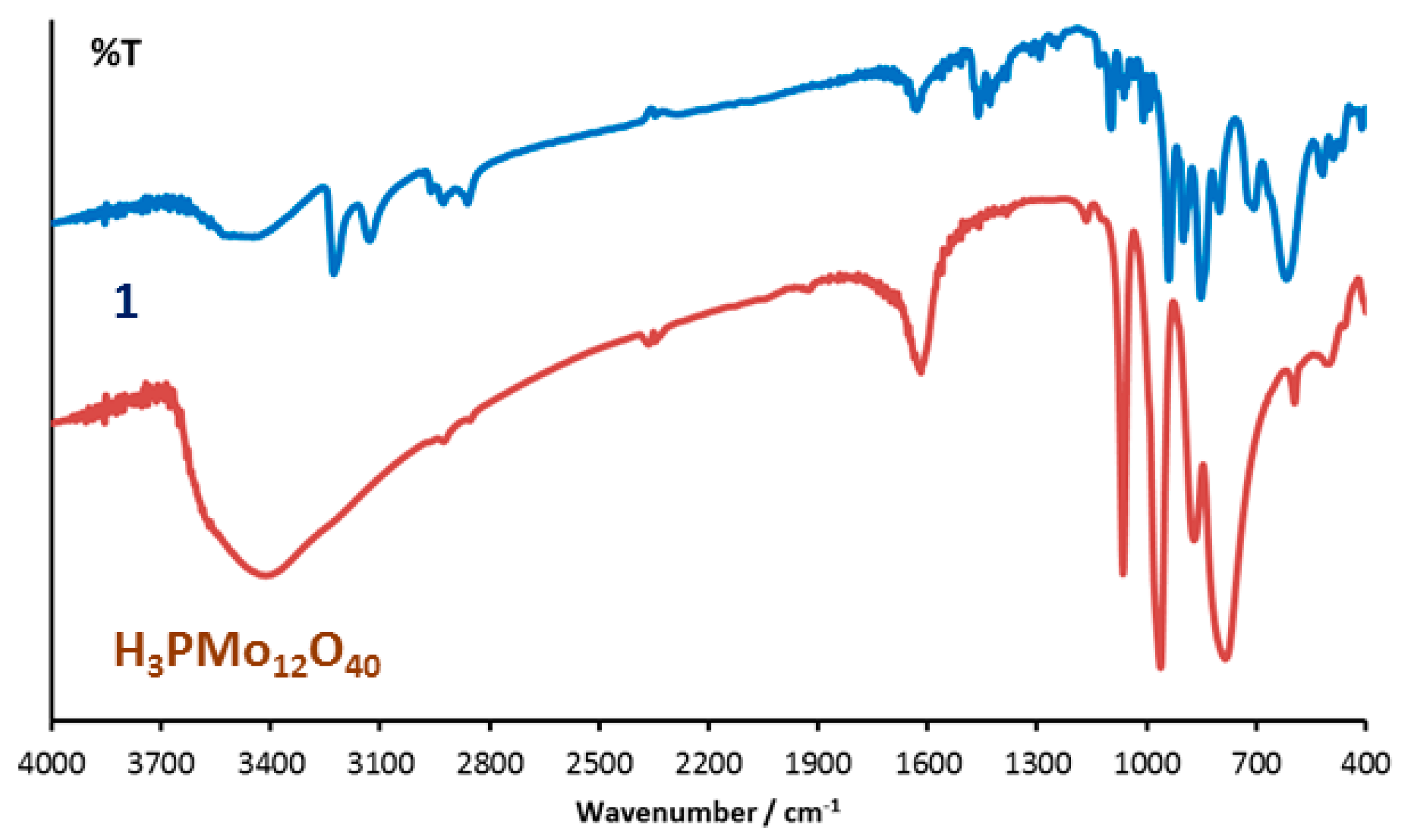
3.1. Synthesis and Infrared Spectroscopy
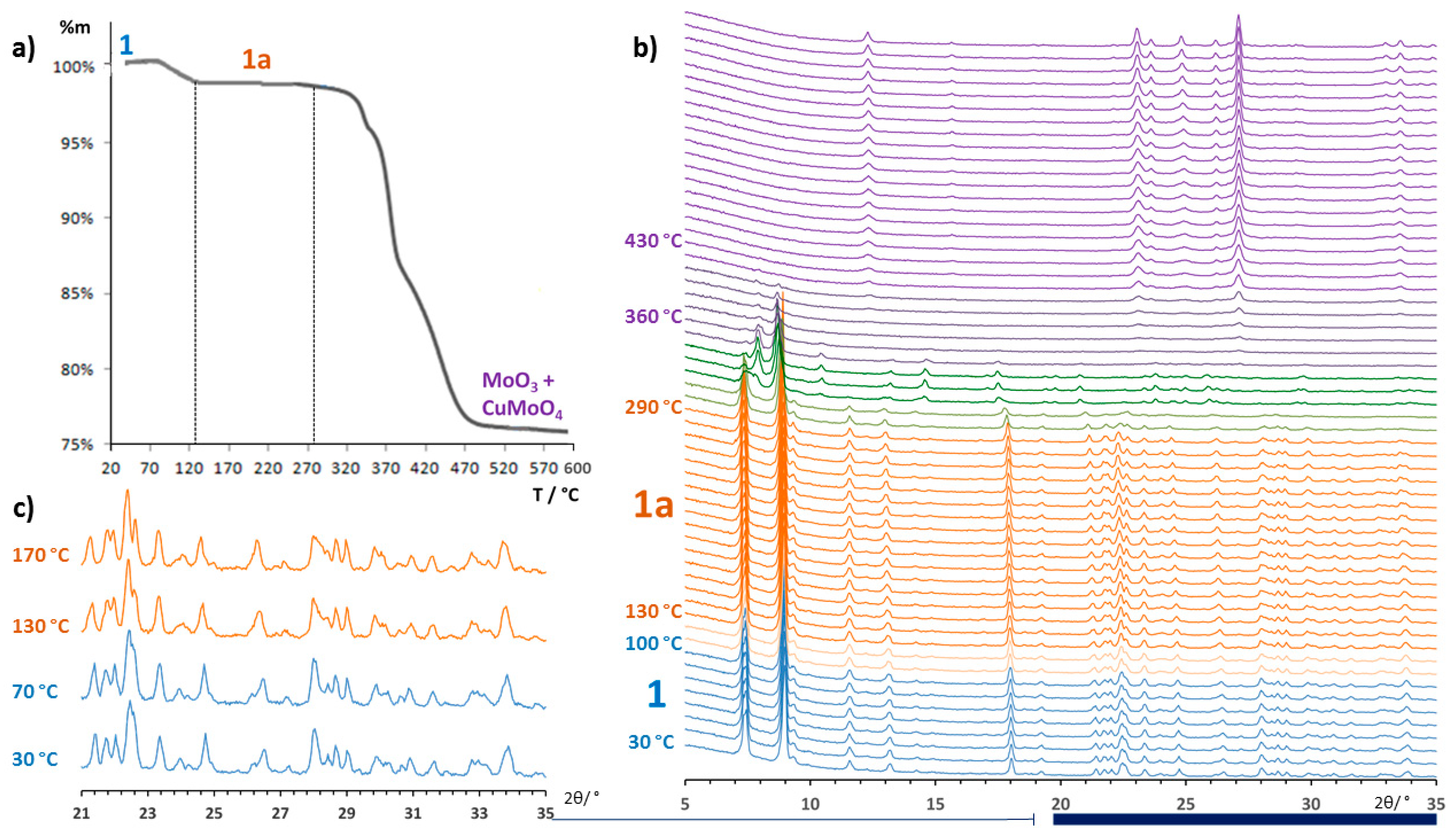
3.2. Thermostructural Analysis
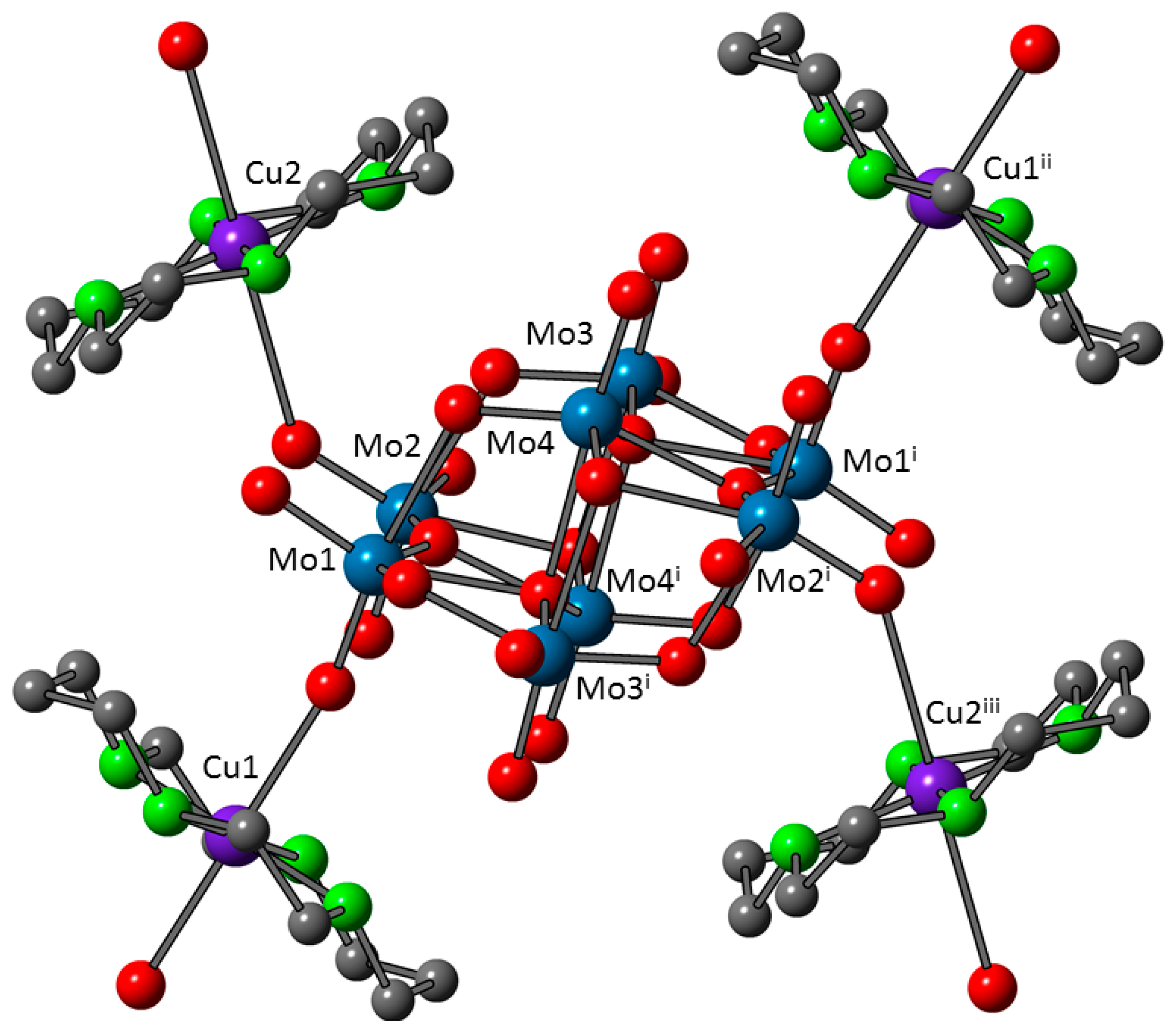
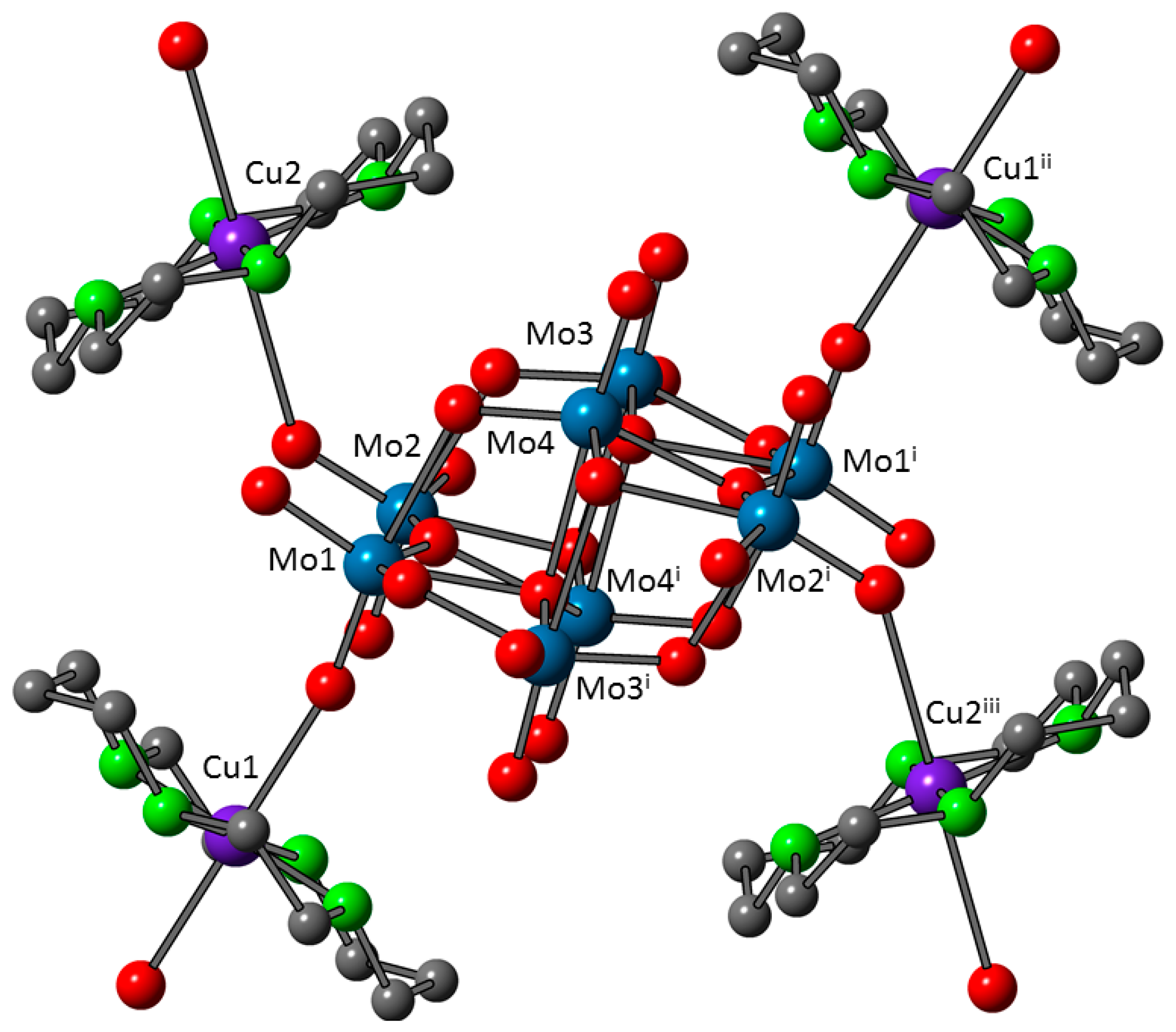
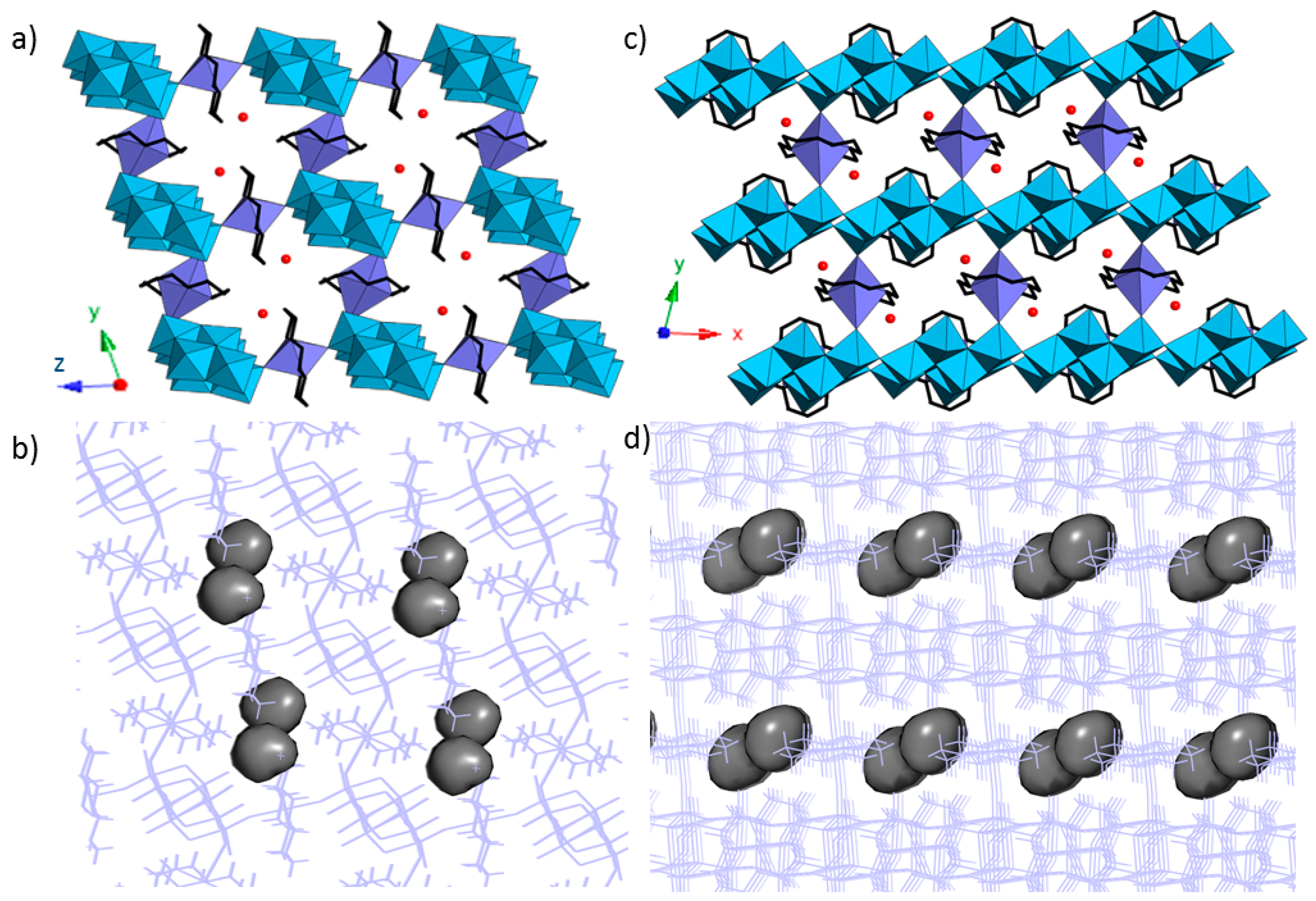
3.3. Crystal Structures of 1 and 1a
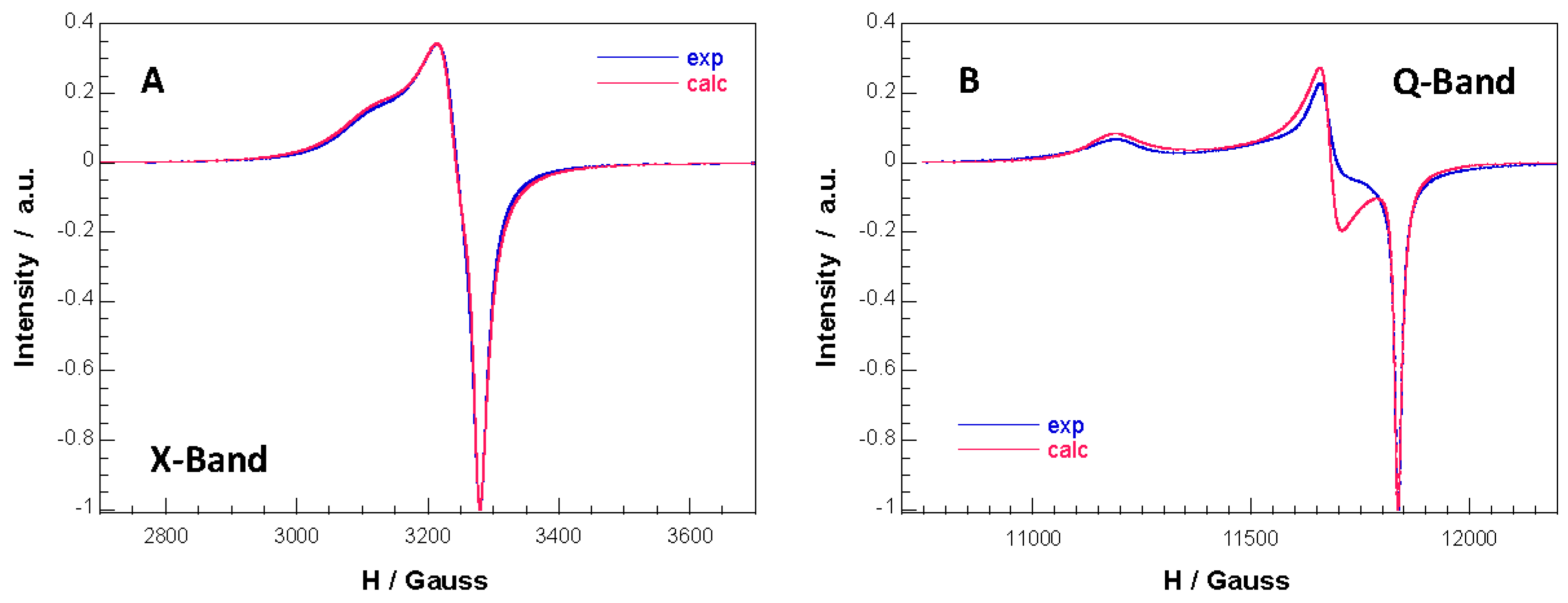
3.4. Electron Paramagnetic Resonance Spectroscopy
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Farrusseng, D. Metal-Organic Frameworks: Applications from Catalysis to Gas. Storage; Wiley-VCH Press: Weinheim, Germany, 2011. [Google Scholar]
- Kitagawa, S.; Kitaura, R.; Noro, S. Functional porous coordination polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef] [PubMed]
- Cote, A.P.; Benin, A.I.; Ockwig, N.W.; O'Keeffe, M.; Matzger, A.J.; Yaghi, O.M. Porous, crystalline, covalent organic frameworks. Science 2005, 310, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-C.; Long, J.R.; Yaghi, O.M. Introduction to metal-organic frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Ding, X.S.; Jiang, D.L. Covalent organic frameworks. Chem. Soc. Rev. 2012, 41, 6010–6022. [Google Scholar] [CrossRef] [PubMed]
- Maurin, G.; Serre, C.; Cooper, A.; Férey, G. The new age of MOFs and of their porous-related solids. Chem. Soc. Rev. 2017, 46, 3104–3107. [Google Scholar] [CrossRef] [PubMed]
- Chughtai, A.H.; Ahmad, N.; Hussein, H.A.; Laypkovc, A.; Verpoort, F. Metal–organic frameworks: Versatile heterogeneous catalysts for efficient catalytic organic transformations. Chem. Soc. Rev. 2015, 44, 6804–6849. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-B.; Liang, J.; Wang, X.-S.; Cao, R. Multifunctional metal-organic framework catalysts: Synergistic catalysis and tandem reactions. Chem. Soc. Rev. 2017, 46, 126–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Yu, T.; Shi, Z.; Wang, Z. The preparation of metal-organic frameworks and their biomedical application. Int. J. Nanomed. 2016, 11, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Mínguez-Espallargas, G.; Coronado, E. Magnetic functionalities in MOFs: From the framework to the pore. Chem. Soc. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.S.; Ogiwara, N.; Chen, K.J.; Madden, D.G.; Pham, T.; Forrest, K.; Space, B.; Horike, S.; Perry, J.J., IV; Kitagawa, S.; et al. Crystal engineering of a family of hybrid ultramicroporous materials based upon interpenetration and dichromate linkers. Chem. Sci. 2016, 7, 5470–5476. [Google Scholar] [CrossRef]
- Scott, H.S.; Bajpai, A.; Chen, K.J.; Pham, T.; Forrest, K.; Space, B.; Perry, J.J., IV; Zaworotko, M.J. Novel mode of 2-fold interpenetration observed in a primitive cubic network of formula [Ni(1,2-bis(4-pyridyl)acetylene)2(Cr2O7)]n. Chem. Commun. 2015, 51, 14832–14835. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.H.; Elsaidi, S.K.; Wojtas, L.; Pham, T.; Forrest, K.A.; Tudor, B.; Space, B.; Zaworotko, M.J. Highly selective CO2 uptake in uninodal 6-connected “mmo” nets based upon MO42– (M = Cr, Mo) pillars. J. Am. Chem. Soc. 2012, 134, 19556–19559. [Google Scholar] [CrossRef] [PubMed]
- Pope, M.T. Heteropoly and Isopoly Oxometalates; Springer Press: Berlin, Germany, 1983. [Google Scholar]
- Pope, M.T.; Müller, A. Polyoxometalates: From Platonic Solids to Anti-Retroviral Activity; Kluwer Press: Dordrecht, The Netherlands, 1994. [Google Scholar]
- Sécheresse, F. Polyoxometalate Chemistry: Some Recent Trends; World Scientific Press: Singapore, 2013. [Google Scholar]
- Miras, H.N.; Vilà-Nadal, L.; Cronin, L. Polyoxometalate based open-frameworks (POM-OFs). Chem. Soc. Rev. 2014, 43, 5679–5699. [Google Scholar] [CrossRef] [PubMed]
- Du, D.-Y.; Qin, J.-S.; Li, S.-L.; Su, Z.-M.; Lan, Y.-Q. Recent advances in porous polyoxometalate-based metal–organic framework materials. Chem. Soc. Rev. 2014, 43, 4615–4632. [Google Scholar] [CrossRef] [PubMed]
- Vilà-Nadal, L.; Cronin, L. Design and synthesis of polyoxometalate-framework materials from cluster precursors. Nat. Rev. Mater. 2017, 2, 17054. [Google Scholar] [CrossRef]
- Uchida, S.; Mizuno, N. Design and syntheses of nano-structured ionic crystals with selective sorption properties. Coord. Chem. Rev. 2007, 251, 2537–2546. [Google Scholar] [CrossRef]
- Uchida, S.; Hosono, R.; Eguchi, R.; Kawahara, R.; Osuga, R.; Kondo, J.N.; Hibino, M.; Mizuno, N. Proton conduction in alkali metal ion-exchanged porous ionic crystals. Phys. Chem. Chem. Phys. 2017, 19, 29077–29083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sadakane, M.; Noro, S.-I.; Murayama, T.; Kamachi, T.; Yoshisawa, K.; Ueda, W. Selective carbon dioxide adsorption of ε-Keggin-type zincomolybdate-based purely inorganic 3D frameworks. J. Mater. Chem. A 2015, 3, 746–755. [Google Scholar] [CrossRef]
- Boyd, T.; Mitchell, S.G.; Gabb, D.; Long, D.-L.; Song, Y.-F.; Cronin, L. POMzites: A family of zeolitic polyoxometalate frameworks from a minimal building block library. J. Am. Chem. Soc. 2017, 139, 5930–5938. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Hill, C.L. A coordination network that catalyzes O2-based oxidations. J. Am. Chem. Soc. 2007, 129, 15094–15095. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Wang, Y.-X.; Wang, R.-H.; Cui, C.-Y.; Tian, C.-B.; Yang, G.-Y. Designed assembly of heterometallic cluster organic frameworks based on Anderson-type polyoxometalate clusters. Angew. Chem. Int. Ed. 2016, 55, 6462–6466. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-T.; Zhang, J.; Yang, G.-Y. Designed synthesis of POM–organic frameworks from {Ni6PW9} building blocks under hydrothermal conditions. Angew. Chem. Int. Ed. 2008, 47, 3909–3913. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-S.; Du, D.-Y.; Guan, W.; Bo, X.-J.; Li, Y.-F.; Guo, L.-P.; Su, Z.-M.; Wang, Y.-Y.; Lan, Y.-Q.; Zhou, H.-C. Ultrastable polymolybdate-based metal–organic frameworks as highly active electrocatalysts for hydrogen generation from water. J. Am. Chem. Soc. 2015, 137, 7169–7177. [Google Scholar] [CrossRef] [PubMed]
- Martín-Caballero, J.; Wéry, A.S.J.; Reinoso, S.; Artetxe, B.; San Felices, L.; El Bakkali, B.; Trautwein, G.; Alcañiz-Monge, J.; Vilas, J.L.; Gutiérrez-Zorrilla, J.M. A robust open framework formed by decavanadate clusters and copper(II) complexes of macrocyclic polyamines: Permanent microporosity and catalytic oxidation of cycloalkanes. Inorg. Chem. 2016, 55, 4970–4979. [Google Scholar] [CrossRef] [PubMed]
- Martín-Caballero, J.; Wéry, A.S.J.; Artetxe, B.; Reinoso, S.; San Felices, L.; Vilas, J.L.; Gutiérrez-Zorrilla, J.M. Sequential single-crystal-to-single-crystal transformations promoted by gradual thermal dehydration in a porous metavanadate hybrid. CrystEngComm 2015, 17, 8915–8925. [Google Scholar] [CrossRef]
- Martín-Caballero, J.; Artetxe, B.; Reinoso, S.; San Felices, L.; Castillo, O.; Beobide, G.; Vilas, J.L.; Gutiérrez-Zorrilla, J.M. Thermally-triggered crystal dynamics and permanent porosity in the first heptatungstate-metalorganic three-dimensional hybrid framework. Chem. Eur. J. 2017, 23, 14962–14974. [Google Scholar] [CrossRef] [PubMed]
- Reinoso, S.; Artetxe, B.; San Felices, L.; Gutiérrez-Zorrilla, J.M. Single-crystal-to-single-crystal transformations in stimuli responsive compounds based on polyoxometalate clusters. In Polyoxometalates: Properties, Structure and Synthesis; Roberts, A.P., Ed.; Nova Science Press: Hauppauge, NY, USA, 2016; pp. 143–212. [Google Scholar]
- Müller, A.; Krickemeyer, E.; Meyer, J.; Bögge, H.M.; Peters, F.; Plass, W.; Diemann, E.; Dillinger, S.; Nonnenbruch, F.; Randerath, M.; Menke, C. [Mo154(NO)14O420(OH)28(H2O)70](25 ± 5)−: A water-soluble big wheel with more than 700 atoms and a relative molecular mass of about 24000. Angew. Chem. Int. Ed. Engl. 1995, 34, 2122–2124. [Google Scholar] [CrossRef]
- Müller, A.; Krickemeyer, E.; Bögge, H.; Schmidtmann, M.; Peters, F. Organizational forms of matter: An inorganic super fullerene and keplerate based on molybdenum oxide. Angew. Chem. Int. Ed. 1998, 37, 3359–3363. [Google Scholar] [CrossRef]
- Müller, A.; Beckmann, E.; Bögge, H.; Schmidtmann, M.; Dress, A. Inorganic chemistry goes protein size: A mo368 nano-hedgehog initiating nanochemistry by symmetry breaking. Angew. Chem. Int. Ed. 2002, 41, 1132–1167. [Google Scholar] [CrossRef]
- Blazevic, A.; Rompel, A. The Anderson–Evans polyoxometalate: From inorganic building blocks via hybrid organic–inorganic structures to tomorrows “Bio-POM”. Coord. Chem. Rev. 2016, 307, 42–64. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, S.; Yu, H.; Su, X.; Zhang, M.; Zhang, F.; Han, J. Cs4Mo5P2O22: A first Strandberg-type POM with 1D straight chains of polymerized [Mo5P2O23]6− units and moderate second harmonic generation response. Chem. Commun. 2013, 49, 306–308. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro. Software System, Version 171.37.34; Agilent Technologies UK, Ltd.: Oxford, UK, 2012. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H.J. Olex2—A complete package for Molecular Crystallography. J. Appl. Crystallogr. 2009, 42, 339–342. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX Program Features. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Brown, I.D.; Alternatt, D. Bond-Valence Parameters obtained from a systematic analysis of the inorganic crystal structure database. Acta Crystallogr. 1985, 41, 244–247. [Google Scholar] [CrossRef]
- Llunell, M.; Casanova, D.; Cirera, J.; Bofill, J.M.; Alemany, P.; Alvarez, S.; Pinsky, M.; Avnir, D. SHAPE v1.1b; Universitat de Barcelona: Barcelona, Spain; The Hebrew University of Jerusalem: Jerusalem, Israel, 2005. [Google Scholar]
- Kihlborg, L. Least squares refinement of the crystal structure of molybdenum trioxide. Ark. Kemi 1963, 21, 357–364. [Google Scholar]
- Nassau, K.; Abrahams, S.C. The growth and properties of single crystal cupric molybdate. J. Cryst. Growth 1968, 2, 136–140. [Google Scholar] [CrossRef]
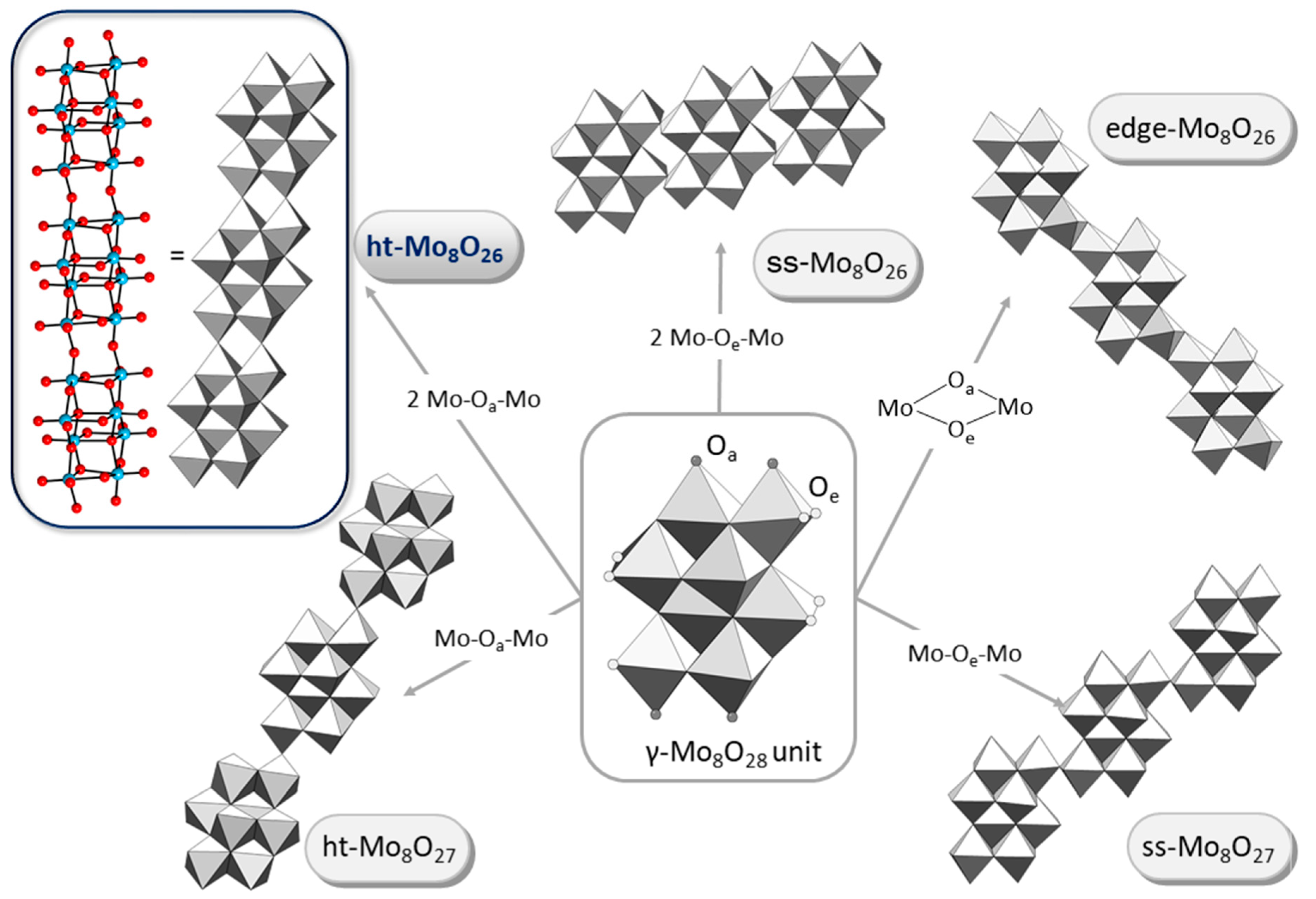
- Bridgeman, A.J. The electronic structure and stability of the isomers of octamolybdate. J. Phys. Chem. A 2002, 106, 12151–12160. [Google Scholar] [CrossRef]
- Xiao, D.; Hou, Y.; Wang, E.; Wang, S.; Li, Y.; Xu, L.; Hu, C. Hydrothermal synthesis and characterization of an unprecedented η-type octamolybdate: [{Ni(phen)2}2(Mo8O26)]. Inorg. Chim. Acta 2004, 357, 2525–2531. [Google Scholar] [CrossRef]
- Allis, D.G.; Burkholder, E.; Zubieta, J. A new octamolybdate: Observation of the θ-isomer in [Fe(tpyprz)2]2[Mo8O26]·3.7H2O (tpyprz¼tetra-2-pyridylpyrazine). Polyhedron 2004, 23, 1145–1152. [Google Scholar] [CrossRef]
- Yue, Z.-C.; Du, H.-J.; Niu, Y.-Y.; Jin, G.-X. An unprecedented ι-type octamolybdate: [TbI1]2[(β-Mo8O26)0.5(ι-Mo8O26)] directed by a new tricationic template. CrystEngComm 2013, 15, 9844–9848. [Google Scholar] [CrossRef]
- Wang, L.; Yin, P.; Zhang, J.; Hao, J.; Lv, C.; Xiao, F.; Wei, Y. χ-Octamolybdate [MoV4MoVI4O24]4−: An unusual small polyoxometalate in partially reduced form from nonaqueous solvent reduction. Chem. Eur. J. 2011, 17, 4796–4801. [Google Scholar] [CrossRef] [PubMed]
- McCarron, E.M., III; Harlow, R.L. The synthesis and structure of tetrasodium tetramethoxyoctamolybdate-methanol complex (Na4[Mo8O24(OCH3)4].8MeOH): A novel isopolymolybdate that decomposes with the loss of formaldehyde. J. Am. Chem. Soc. 1983, 105, 6179–6181. [Google Scholar] [CrossRef]
- Ito, T.; Mikurube, K.; Hasegawa, K.; Matsumoto, T.; Kosaka, K.; Naruke, H.; Koguchi, S. Structural variation in polyoxomolybdate hybrid crystals comprising ionic-liquid surfactants. Crystals 2014, 4, 42–52. [Google Scholar] [CrossRef]
- Coué, V.; Dessapt, R.; Bujoli-Doeuff, M.; Evain, M.; Jobic, S. Synthesis, characterization and photochromic properties of hybrid organic-inorganic materials based on molybdate, DABCO, and pyperazine. Inorg. Chem. 2007, 46, 2824–2835. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.-C.; Shen, L.-X.; Wu, H.-H.; Li, X.-H.; Niu, Y.-Y. pH-dependent assembly of metal–organic hybrid compounds based on octamolybdates and a new flexible multidentate ligand. CrystEngComm 2013, 15, 9938–9948. [Google Scholar] [CrossRef]
- Zhai, Q.-G.; Wu, X.-Y.; Chen, S.-M.; Zhao, Z.-G.; Lu, C.-Z. Construction of Ag/1,2,4-triazole/polyoxometalates hybrid family varying from diverse supramolecular assemblies to 3-D rod-packing framework. Inorg. Chem. 2007, 46, 5046–5058. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Wu, H.; Yang, J.; Liu, Y.-Y.; Liu, B.; Liu, Y.-Y.; Ma, J.F. pH-Dependent assembly of 1D to 3D octamolybdate hybrid materials based on a new flexible bis-[(pyridyl)-benzimidazole] ligand. Cryst. Growth Des. 2011, 11, 2920–2927. [Google Scholar] [CrossRef]
- Wang, X.; Sun, J.; Lin, H.; Chang, Z.; Wang, X.; Liu, G. A series of Anderson-type polyoxometalate-based metal-organic complexes: Their pH-dependent electrochemical behaviour, and as electrocatalysts and photocatalysts. Dalton Trans. 2016, 45, 12465–12478. [Google Scholar] [CrossRef] [PubMed]
- Du, H.-J.; Shu, Z.-Z.; Niu, Y.-Y.; Song, L.-S.; Zhu, Y. Unprecedented polymeric 1/∞ [Mo8O26]4− chains and four novel organic-inorganic hybrids based on Mo-POMs and azaheterocycles templates. J. Solid State Chem. 2012, 190, 296–302. [Google Scholar] [CrossRef]
- Hubbard, D.J.; Johnston, A.R.; Casalongue, H.S.; Sarjeant, A.N.; Norquist, A.J. Synthetic approaches for noncentrosymmetric molybdates. Inorg. Chem. 2008, 47, 8518–8525. [Google Scholar] [CrossRef] [PubMed]
- Thorn, K.J.; Sarjeant, A.N.; Norquist, A.J. (C4H14N2)2[Mo8O26]·2H2O: A new octamolybdate salt. Acta Crystallogr. 2005, 61, m1665–m1667. [Google Scholar]
- Casalongue, H.S.; Choyke, S.J.; Sarjeant, A.N.; Schrier, J.; Norquist, A.J. Charge density matching in templated molybdates. J. Solid State Chem. 2009, 182, 1297–1303. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Tan, K.; Guan, W.; Li, S.-L.; Yang, G.-S.; Shao, K.-Z.; Yan, L.-K.; Su, S.-M. Inorganic-organic hybrid compounds based on the co-existence of different isomers or forms of polymolybdate. CrystEngComm 2010, 12, 3684–3690. [Google Scholar] [CrossRef]
- Evain, M.; Petricek, V.; Coué, V.; Dessapt, R.; Bujoli-Doeuff, M.; Jobic, S. Commensurate (C6H14N2)2[Mo8O26]·4H2O and incommensurate (C6H14N2)2[Mo8O26]·4.66H2O: A structural versatility linked to solvent content. Acta Crystallogr. 2006, 62, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Bosnich, B.; Poon, C.K.; Tobe, M.L. Complexes of cobalt(III) with a cyclic tetradentate secondary amine. Inorg. Chem. 1965, 4, 1102–1108. [Google Scholar] [CrossRef]
- Bakaj, M.; Zimmer, M. Conformational analysis of copper(II) 1,4,8,11-tetraazacyclotetradecane macrocyclic systems. J. Mol. Struct. 1999, 508, 59–72. [Google Scholar] [CrossRef]
- Iturrospe, A.; Artetxe, B.; Reinoso, S.; San Felices, L.; Vitoria, P.; Lezama, L.; Gutiérrez-Zorrilla, J.M. Copper(II) complexes of tetradentate pyridil ligands supported on Keggin polyoxometalates: Single-crystal to single-crystal transformations promoted by reversible dehydration processes. Inorg. Chem. 2013, 52, 3084–3093. [Google Scholar] [CrossRef] [PubMed]
- Hathaway, B.J. A New Look at the stereochemistry and electronic properties of complexes of the copper(II) ion. Struct. Bonding 1984, 57, 55–118. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | 1 | 1a |
|---|---|---|
| Formula | C20H51Cu2Mo8N8O27.5 | C20H48Cu2Mo8N8O26 |
| FW (g mol−1) | 1738.3 | 1711.26 |
| Crystal System | Triclinic | Triclinic |
| Space Group | P-1 | P-1 |
| a (Å) | 9.6366(5) | 9.6460(5) |
| b (Å) | 10.3864(6) | 10.3541(7) |
| c (Å) | 12.3411(6) | 12.2349(8) |
| α (˚) | 73.222(4) | 73.882(6) |
| β (˚) | 83.260(4) | 84.087(4) |
| γ (˚) | 76.865(5) | 76.875(5) |
| V (Å3) | 1149.92(11) | 1142.22(12) |
| Z | 1 | 1 |
| ρcalcd (g cm−3) | 2.510 | 2.488 |
| μ (mm−1) | 3.102 | 3.118 |
| Collected Reflections | 7539 | 8043 |
| Unique Reflections (Rint) | 4062 (0.035) | 4362 (0.031) |
| Observed Reflections [I > 2σ(I)] | 3394 | 3527 |
| Parameters | 304 | 293 |
| R(F)a [I > 2σ(I)] | 0.038 | 0.026 |
| wR(F2)a [all data] | 0.104 | 0.065 |
| GoF | 1.059 | 0.950 |
| Formula | Ref. | Formula | Ref. |
|---|---|---|---|
| ht | ss | ||
| (H3tib)2[Co.(H2O)2(Mo8O26)2]·2H2O | [55] | [Ag2(trz)2]2[Mo8O26] | [56] |
| (H4bbpbm)[Mo8O26]·2H2O | [57] | [Cu(3-dpye)(Mo8O26)(H2O)8]·2H2O | [58] |
| [Cu(die)2(Mo8O26)] | [59] | [Cu(H2bbpbm)(Mo8O26)]·3H2O | [57] |
| (H2dmeen)2[Mo8O26] | [60] | (H2dmen)2[Mo8O26]·2H2O | [61] |
| edge | (H2dmpip)2[Mo8O26] | [62] | |
| {[Ni(H2bpim)3(Mo8O26)2(H2O)2]·2H2O | [63] | (H2dabco)2[Mo8O26]·4H2O | [64] |
| (bmim)2[Mo8O26] | [59] | (H2dabco)2[Mo8O26]·4.66H2O | [64] |
| 1 | 1a | |
|---|---|---|
| Cu1 | ||
| Cu1-N1A | 2.011(8) | 2.008(8) |
| Cu1-N1A i | 2.011(8) | 2.008(8) |
| Cu1-N4A | 2.026(8) | 2.036(8) |
| Cu1-N4A i | 2.026(8) | 2.036(8) |
| Cu1-O1L | 2.299(5) | 2.287(6) |
| Cu1-O1L i | 2.299(5) | 2.287(6) |
| CSM (Oh) | 0.562 | 0.477 |
| Cu2 | ||
| Cu2-N1B | 2.000(5) | 2.007(8) |
| Cu2-N1B ii | 2.000(5) | 2.007(8) |
| Cu2-N4B | 2.013(6) | 2.007(6) |
| Cu2-N4B ii | 2.013(6) | 2.007(6) |
| Cu2-O2L | 2.725(8) | 2.742(12) |
| Cu2-O2L ii | 2.725(8) | 2.742(12) |
| CSM (Oh) | 2.442 | 2.569 |
| 1 | 1a | |
|---|---|---|
| N1A···O3 i | 2.915(8) | 2.901(8) |
| N4A···O4 i | 3.494(7) | 3.401(8) |
| N1B ···O1 | 3.089(7) | 3.068(8) |
| N1B···O14 | 3.416(6) | 3.403(7) |
| N4B···O23 ii | 2.845(7) | 2.827(8) |
| C2A···O2 | 3.324(9) | 3.240(9) |
| C5A···O1w | 3.366(13) | |
| C6A···O4 iii | 3.498(8) | 3.394(9) |
| C7A···O1 | 3.513(9) | 3.486(8) |
| C2B···O13 ii | 3.154(8) | 3.151(8) |
| C3B···O1 | 3.133(9) | 3.139(9) |
| C5B···O2L | 3.299(8) | 3.287(9) |
| C6B···O14 | 3.349(7) | |
| C7B ···O13 iv | 3.273(9) | 3.251(10) |
| O1w···O2 v | 2.919(10) | |
| O1w···O4 | 2.982(10) |
| X-Band | Q-Band | |
|---|---|---|
| g1 | 2.159 | 2.168 |
| g2 | 2.077 | 2.076 |
| g3 | 2.049 | 2.048 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dissem, N.; Artetxe, B.; San Felices, L.; Lezama, L.; Haddad, A.; Gutiérrez-Zorrilla, J.M. A Robust Framework Based on Polymeric Octamolybdate Anions and Copper(II) Complexes of Tetradentate N-donor Ligands. Crystals 2018, 8, 20. https://doi.org/10.3390/cryst8010020
Dissem N, Artetxe B, San Felices L, Lezama L, Haddad A, Gutiérrez-Zorrilla JM. A Robust Framework Based on Polymeric Octamolybdate Anions and Copper(II) Complexes of Tetradentate N-donor Ligands. Crystals. 2018; 8(1):20. https://doi.org/10.3390/cryst8010020
Chicago/Turabian StyleDissem, Nour, Beñat Artetxe, Leire San Felices, Luis Lezama, Amor Haddad, and Juan M. Gutiérrez-Zorrilla. 2018. "A Robust Framework Based on Polymeric Octamolybdate Anions and Copper(II) Complexes of Tetradentate N-donor Ligands" Crystals 8, no. 1: 20. https://doi.org/10.3390/cryst8010020
APA StyleDissem, N., Artetxe, B., San Felices, L., Lezama, L., Haddad, A., & Gutiérrez-Zorrilla, J. M. (2018). A Robust Framework Based on Polymeric Octamolybdate Anions and Copper(II) Complexes of Tetradentate N-donor Ligands. Crystals, 8(1), 20. https://doi.org/10.3390/cryst8010020