Chirality Control in Crystalline Ni(II) Complexes of Thiophosphorylated Thioureas


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General
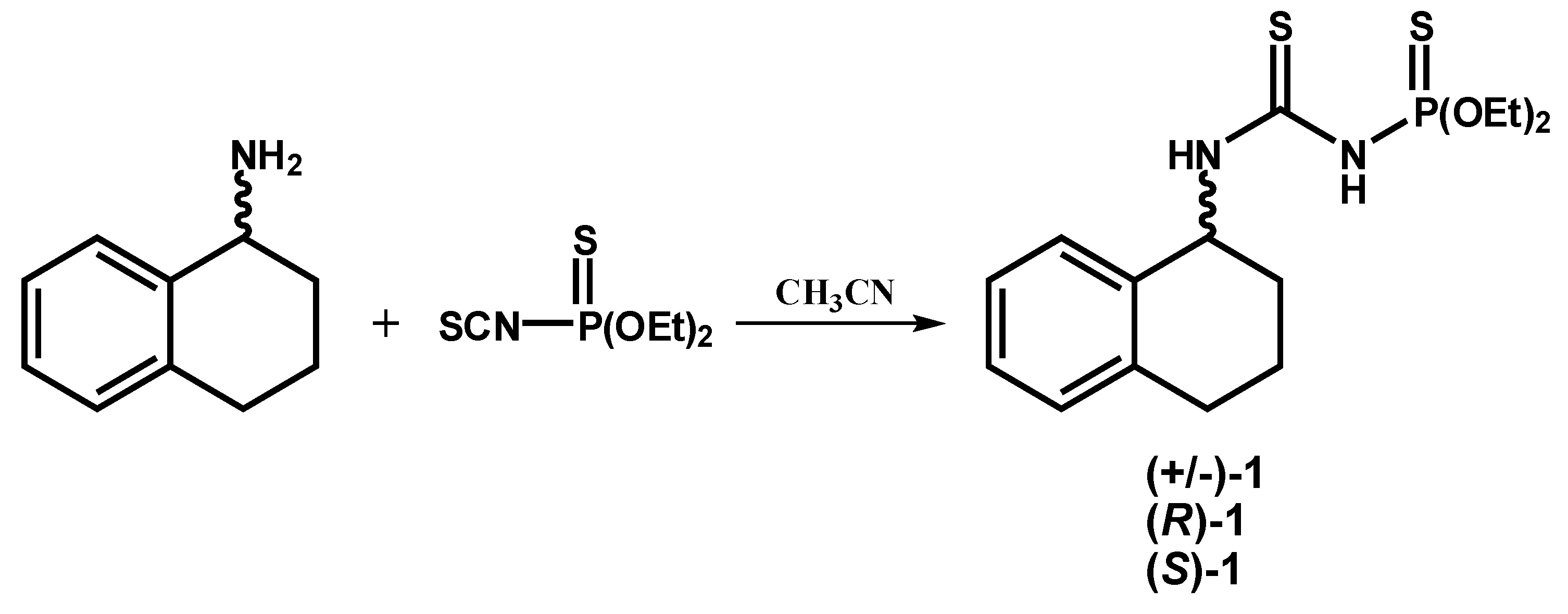
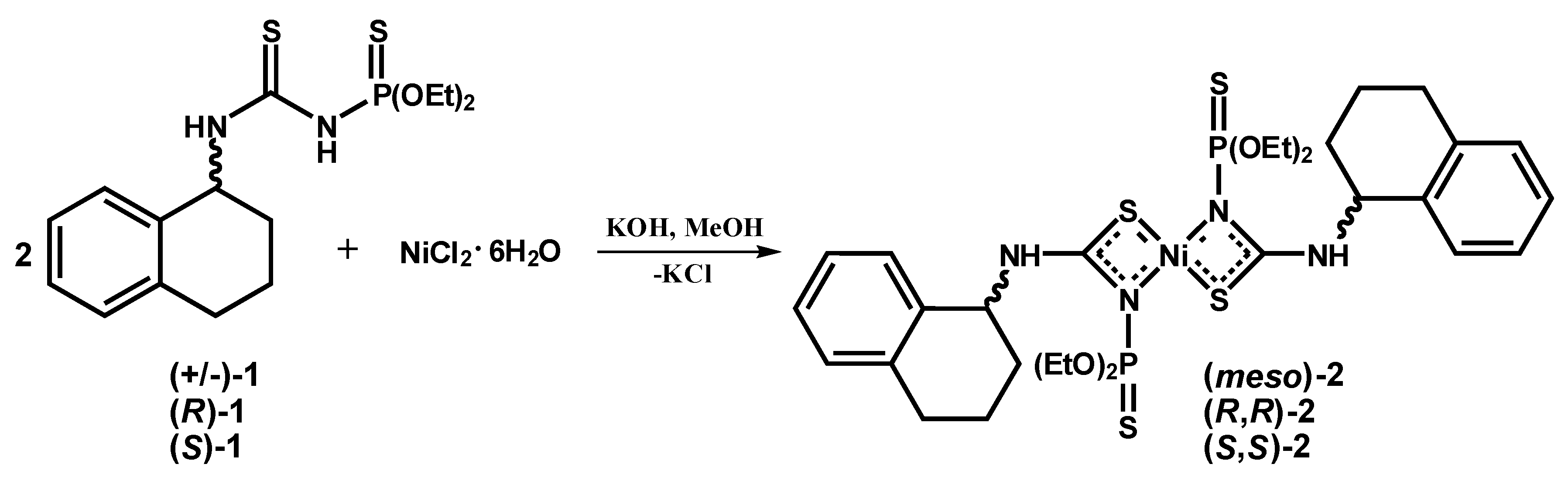
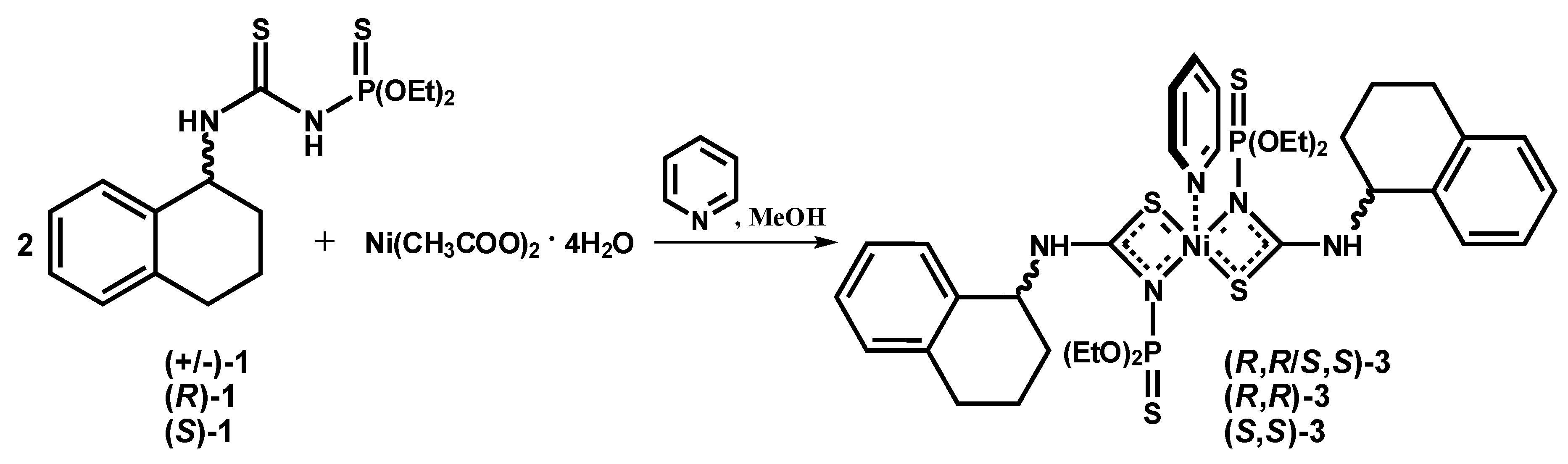
2.1.2. Syntheses
2.2. X-ray Diffraction Study
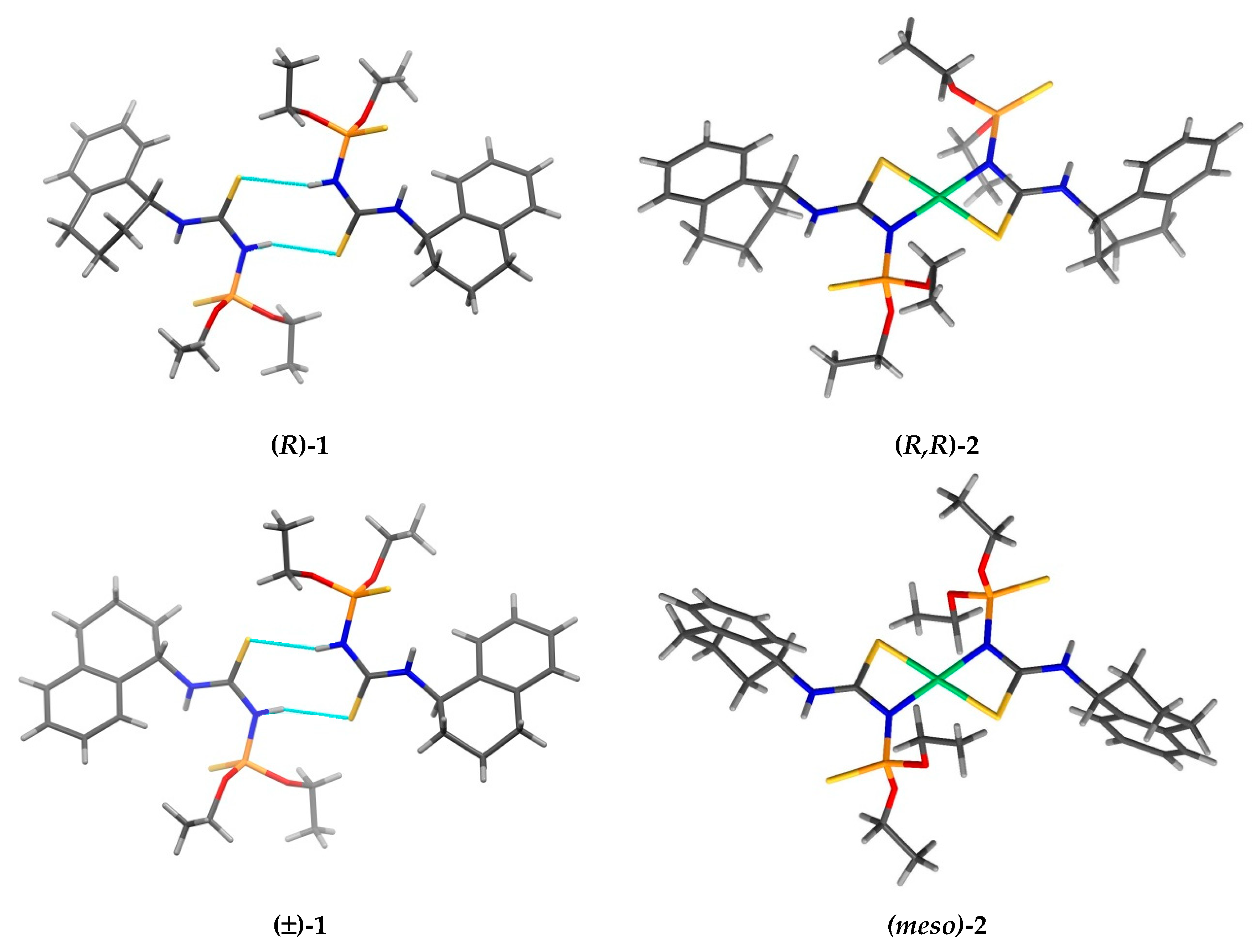
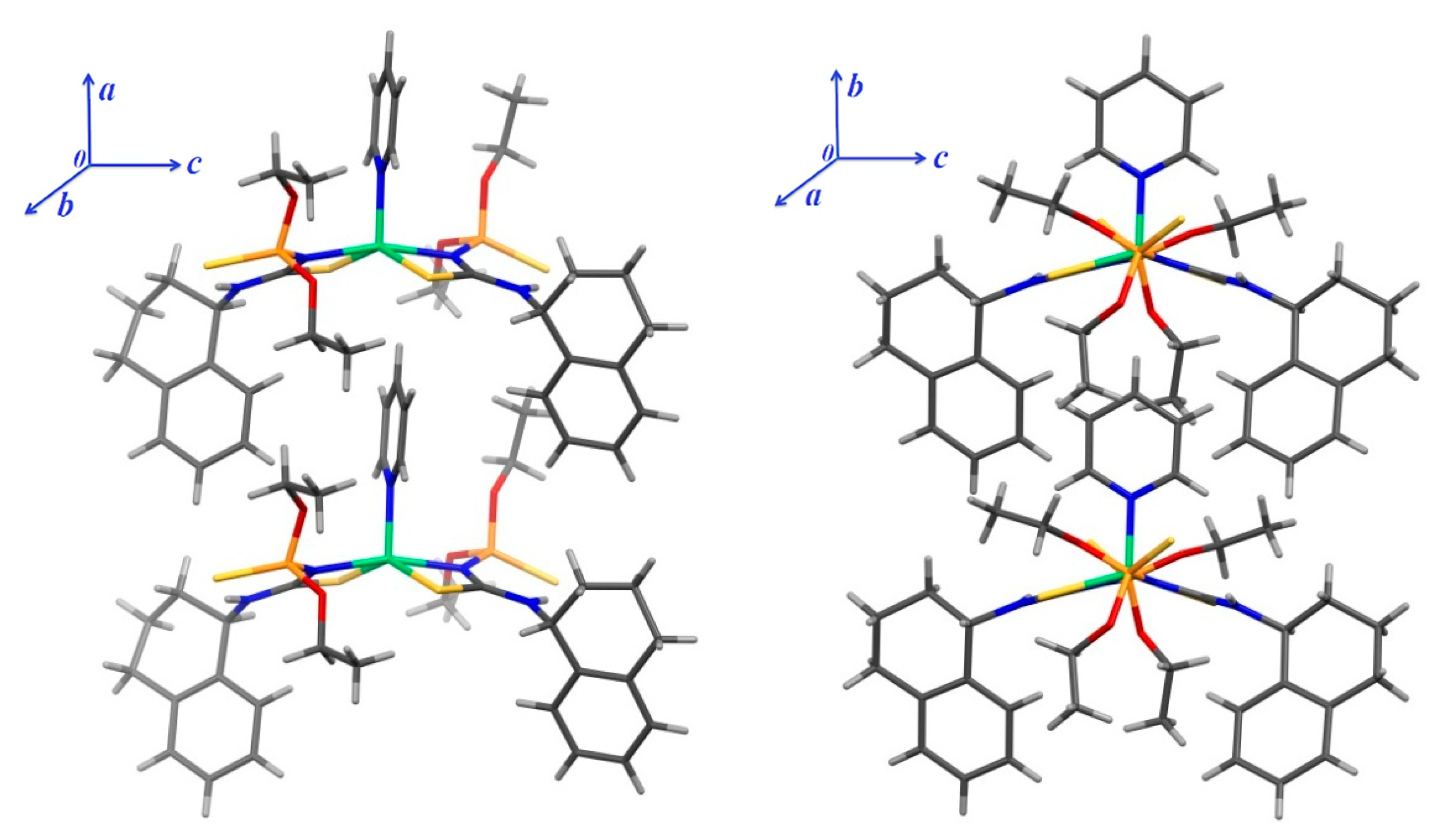
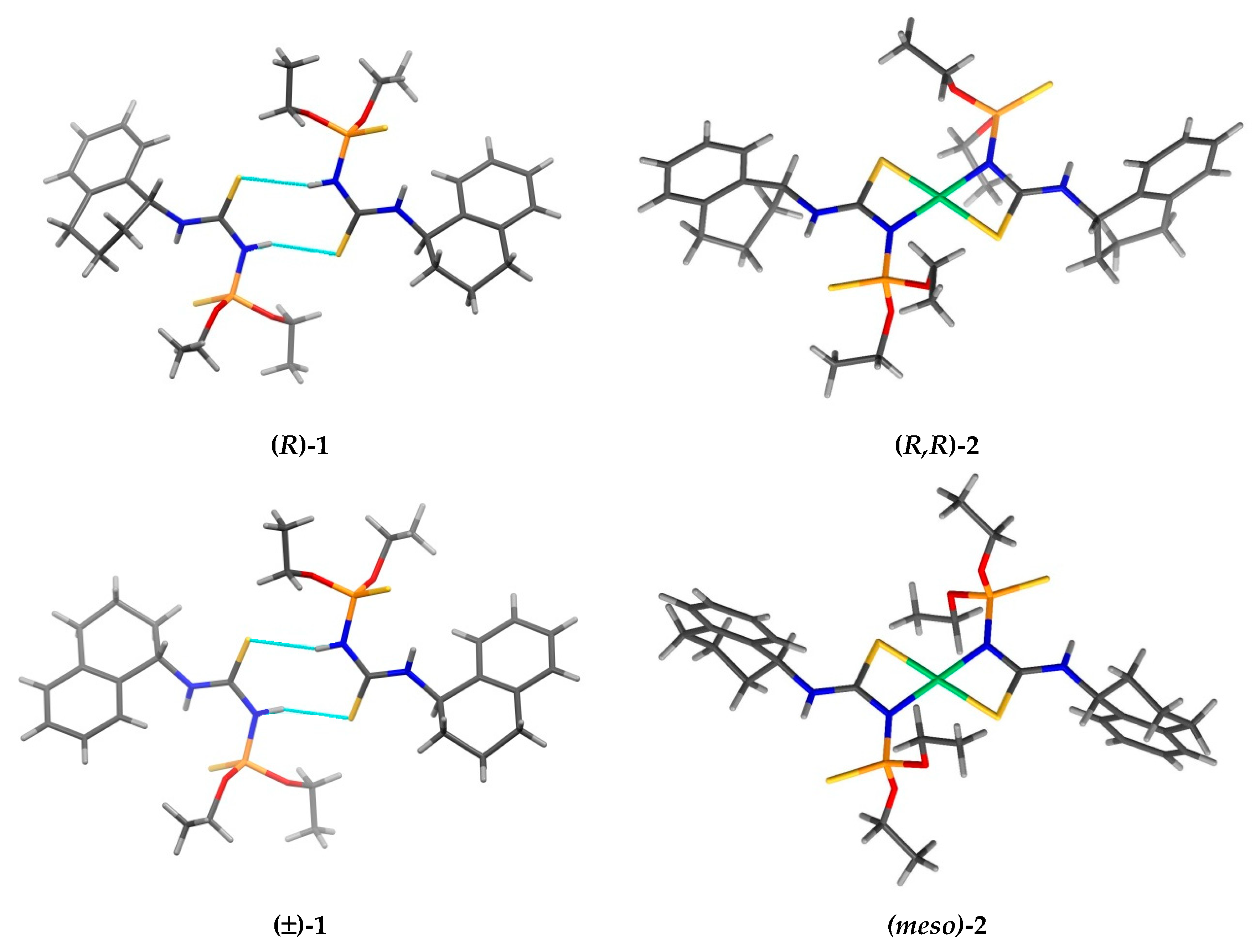
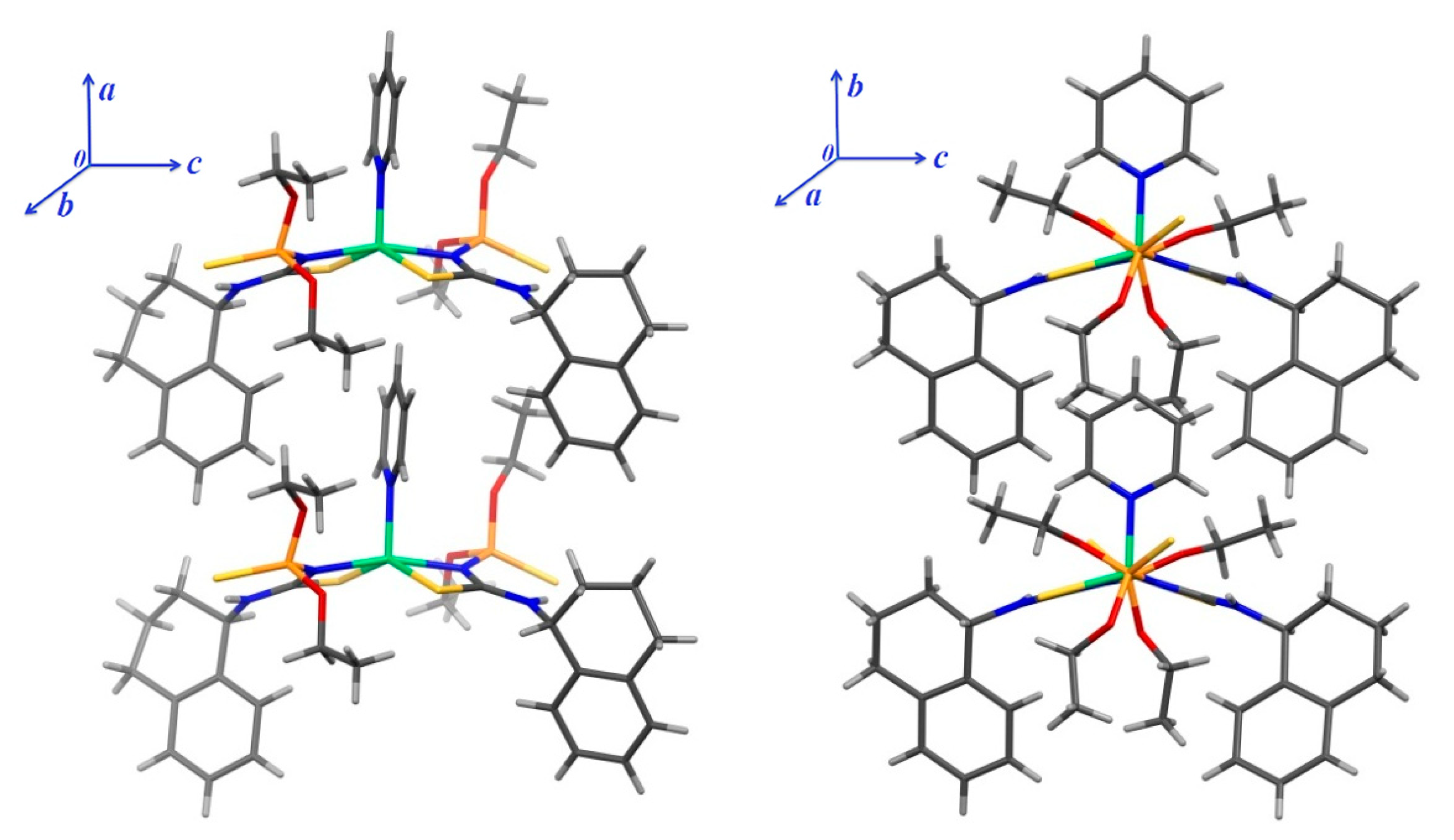
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berthod, A. Chiral recognition mechanism. Anal. Chem. 2006, 78, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Jędrzejewska, H.; Szumna, A. Making a right or left choice: Chiral self-sorting as a tool for the formation of discrete complex structures. Chem. Rev. 2017, 117, 4863–4899. [Google Scholar] [CrossRef] [PubMed]
- Xing, P.; Zhao, Y. Controlling supramolecular chirality in multicomponent self-assembled systems. Acc. Chem. Res. 2018, 51, 2324–2334. [Google Scholar] [CrossRef]
- Song, L.; Shemchuk, O.; Robeyns, K.; Braga, D.; Grepioni, F.; Leyssens, T. Ionic cocrystals of etiracetam and levetiracetam: The importance of chirality for ionic cocrystals. Cryst. Growth Des. 2019, 19, 2446–2454. [Google Scholar] [CrossRef]
- Guijarro, A.; Yus, M. The Origin of Chirality in the Molecules of Life: A Revision from Awareness to the Current Theories and Perspectives of this Unsolved Problem, 1st ed.; Royal Society of Chemistry: Cambridge, UK, 2009; p. 160. [Google Scholar]
- Tsang, M.Y.; Di Salvo, F.; Teixidor, F.; Viñas, C.; Planas, J.G.; Choquesillo-Lazarte, D.; Vanthuyne, N. Is molecular chirality connected to supramolecular chirality? The particular case of chiral 2-pyridyl alcohols. Cryst. Growth Des. 2015, 15, 935–945. [Google Scholar] [CrossRef]
- Scriba, G.K.E. Recognition mechanisms of chiral selectors: An overview. In Chiral Separations. Methods and Protocols; Scriba, G.K.E., Ed.; Humana Press: Totowa, NY, USA, 2019; Volume 1985, pp. 1–33. [Google Scholar]
- Sørensen, H.O.; Larsen, S. Hydrogen bonding in enantiomeric versus racemic mono-carboxylic acids; a case study of 2-phenoxypropionic acid. Acta Crystallogr. B 2003, 59, 132–140. [Google Scholar] [CrossRef]
- Gavezzotti, A.; Lo Presti, L. Theoretical study of chiral carboxylic acids. Structural and energetic aspects of crystalline and liquid states. Cryst. Growth Des. 2015, 15, 3792–3803. [Google Scholar] [CrossRef]
- Metlushka, K.E.; Sadkova, D.N.; Shaimardanova, L.N.; Nikitina, K.A.; Ivshin, K.A.; Islamov, D.R.; Kataeva, O.N.; Alfonsov, A.V.; Kataev, V.E.; Voloshina, A.D.; et al. First coordination polymers on the bases of chiral thiophosphorylated thioureas. Inorg. Chem. Commun. 2016, 66, 11–14. [Google Scholar] [CrossRef]
- Kataeva, O.N.; Metlushka, K.E.; Yamaleeva, Z.R.; Ivshin, K.A.; Kiiamov, A.G.; Lodochnikova, O.A.; Nikitina, K.A.; Sadkova, D.N.; Punegova, L.N.; Voloshina, A.D.; et al. Co-ligand induced chiral recognition of N-thiophosphorylated thioureas in crystalline Ni(II) complexes. Cryst. Growth Des. 2019, 19, 4044–4056. [Google Scholar] [CrossRef]
- Bruker. APEX3 Crystallography Software Suite; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Bruker. SAINT. Crystallography Software Suite; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. A Short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Babashkina, M.G.; Robeyns, K.; Sagan, F.; Szczepanik, D.W.; Seredina, Y.V.; Garcia, Y.; Safin, D.A. Chameleon-like nature of anagostic interactions and its impact on metalloaromaticity in square-planar nickel complexes. Organometallics 2019, 38, 1973–1981. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Robeyns, K.; Mitoraj, M.P.; Kubisiak, P.; Garcia, Y. Influence of the homopolar dihydrogen bonding C-H···H-C on coordination geometry: Experimental and theoretical studies. Chem. Eur. J. 2015, 21, 16679–16687. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Bolte, M.; Mitoraj, M.P.; Klein, A. Complexing cation influences distortion of the ligand in the structure of [M{2-MeO(O)CC6H4NHC(S)NP(S)(OiPr)2}2] (M = ZnII, CdII) complexes: A driving force for the intermolecular aggregation. Dalton Trans. 2015, 44, 14101–14109. [Google Scholar] [CrossRef]
- Babashkina, M.G.; Safin, D.A.; Srebro, M.; Kubisiak, P.; Mitoraj, M.P.; Bolte, M.; Garcia, Y. Crucial influence of the intramolecular hydrogen bond on the coordination mode of RC(S)NHP(S)(OiPr)2 in homoleptic complexes with NiII. Eur. J. Inorg. Chem. 2013, 2013, 545–555. [Google Scholar] [CrossRef]
- Babashkina, M.G.; Safin, D.A.; Srebro, M.; Kubisiak, P.; Mitoraj, M.P.; Bolte, M.; Garcia, Y. Influence of CH2Cl2 for the structure stabilization of the NiII complex [Ni{6-MeO(O)CC6H4NHC(S)NP(S)(OiPr)2-1,5-S,S′}2]·CH2Cl2. CrystEngComm 2012, 14, 370–373. [Google Scholar] [CrossRef]
- Babashkina, M.G.; Safin, D.A.; Robeyns, K.; Garcia, Y. Complexation properties of the crown ether-containing N-thiophosphorylated thiourea towards NiII. Dalton Trans. 2012, 41, 1451–1453. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Bolte, M.; Klein, A. Versatile structures and photophysical properties of poly- and mononuclear CuI complexes with N-thiophosphorylated thioureas RNHC(S)NHP(S)(OiPr)2 and phosphanes. CrystEngComm 2011, 13, 568–576. [Google Scholar] [CrossRef]
- Babashkina, M.G.; Safin, D.A.; Bolte, M.; Srebro, M.; Mitoraj, M.; Uthe, A.; Klein, A.; Köckerling, M. Intramolecular hydrogen bonding controls 1,3-N,S vs. 1,5- S,S′-coordination in NiII complexes of N-thiophosphorylated thioureas RNHC(S)NHP(S)(OiPr)2. Dalton Trans. 2011, 40, 3142–3153. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Bolte, M.; Klein, A. Synthesis and characterization of [H2NC(S)NP(S)(OiPr)2]− complexes of Co(II), Ni(II), Zn(II) and Cd(II). Inorg. Chim. Acta 2011, 365, 32–37. [Google Scholar] [CrossRef]
- Metlushka, K.E.; Sadkova, D.N.; Nikitina, K.A.; Lodochnikova, O.A.; Kataeva, O.N.; Alfonsov, V.A. Ni(II) complex of bisthiophosphorylated thiourea prepared from the Betti base. Russ. J. Gen. Chem. 2017, 87, 2130–2132. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Mitoraj, M.P.; Kubisiak, P.; Robeyns, K.; Bolte, M.; Garcia, Y. An intermolecular pyrene excimer in the pyrene-labeled N-thiophosphorylated thiourea and its nickel(II) complex. Inorg. Chem. Front. 2016, 3, 1419–1431. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | (R)-1 | (±)-1 | (R,R)-2 | (meso)-2 | (R,R)-3 | (R,R/S,S)-3 |
|---|---|---|---|---|---|---|
| Formula | C15H23N2O2PS2 | C15H23N2O2PS2 | C30H44N4NiO4P2S4 | C26H40N4NiO4P2S4 | C35H49N5NiO4P2S4 | C35H49N5NiO4P2S4 |
| CCDC number | 1961490 | 1961489 | 1961491 | 1961492 | 1961494 | 1961493 |
| Color | colorless | colorless | violet | violet | Green | green |
| Habitus | prizm | prizm | prizm | prizm | Prizm | prizm |
| Size (mm) | 0.69 × 0.68 × 0.39 | 0.58 × 0.38 × 0.31 | 0.56 × 0.30 × 0.19 | 0.44 × 0.31 × 0.21 | 0.52 × 0.49 × 0.26 | 0.98 × 0.39 × 0.31 |
| Formula weight | 358.44 | 358.44 | 773.58 | 773.58 | 852.68 | 852.68 |
| T (K) | 150(2) | 150(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| Crystal system | monoclinic | triclinic | monoclinic | monoclinic | monoclinic | orthorhombic |
| Space group | P21 | P-1 | P21 | P21/c | P21 | Pna21 |
| a (Å) | 7.6020(10) | 7.5459(9) | 14.9354(6) | 19.4428(14) | 8.4467(11) | 23.6547(15) |
| b (Å) | 30.256(4) | 8.1735(9) | 7.5352(3) | 12.3486(9) | 20.960(3) | 8.5751(5) |
| c (Å) | 8.2300(11) | 15.2017(16) | 16.8715(7) | 15.5240(11) | 12.2036(17) | 20.0009(13) |
| α (°) | 90 | 92.278(6) | 90 | 90 | 90 | 90 |
| β (°) | 108.423(5) | 90.123(6) | 112.192(2) | 104.448(4) | 107.971(6) | 90 |
| γ (°) | 90 | 107.777(6) | 90 | 90 | 90 | 90 |
| V (Å3) | 1795.9(4) | 892.01(17) | 1758.09(13) | 3609.3(5) | 2055.2(5) | 4057.0(4) |
| Z | 4 | 2 | 2 | 4 | 2 | 4 |
| D Calcd (g m−3) | 1.326 | 1.335 | 1.461 | 1.424 | 1.378 | 1.401 |
| µ (mm−1) | 0.393 | 0.396 | 0.921 | 0.898 | 0.796 | 0.852 |
| Reflection collected | 55072 | 5016 | 30788 | 89973 | 35088 | 65953 |
| Unique reflections | 8927 | 5016 | 7769 | 8932 | 9905 | 9280 |
| Reflections observed | 8899 | 4517 | 6896 | 4555 | 8455 | 8111 |
| Ɵ min, Ɵ max (°) | 1.347, 28.346 | 1.341, 28.390 | 1.303, 27.236 | 1.081, 28.378 | 1.754, 28.375 | 1.722, 27.541 |
| Goodness-of-fit (GOF) on F2 | 1.121 | 1.125 | 1.121 | 1.160 | 0.845 | 1.181 |
| R1, wR2 (I ≥ 2σ(I)) | 0.0228, 0.0606 | 0.0392, 0.0952 | 0.0446, 0.1129 | 0.1345, 0.3012 | 0.0423, 0.1092 | 0.0547, 0.1540 |
| R1, wR2 (all data) | 0.0228, 0.0607 | 0.0454, 0.0970 | 0.0550, 0.1325 | 0.2452, 0.3519 | 0.0552, 0.1297 | 0.0643, 0.1620 |
| Largest peak/hole (e Å−3) | 0.296 | 0.389 | 1.321 | 0.749 | 0.539 | 1.553 |
| −0.248 | −0.266 | −0.943 | −1.132 | −0.859 | −0.575 | |
| Flack | 0.007(6) | 0.007(10) | 0.013(17) | 0.397(8) |
| Crystal | S=P–N–C | S=P–O–C | S=P–O–C | S=P–O–C | S=P–O–C | Pyridine Orientation |
|---|---|---|---|---|---|---|
| P–O–C–C | P–O–C–C | P–O–C–C | P–O–C–C | |||
| (R)-1 | −56.0(2) | −33.2 (2) | −48.1(2) | |||
| (mol A) | 109.2(2) | 172.3(2) | ||||
| (R)-1 | 60.2(2) | 47.0(2) | 33.8(2) | |||
| (mol B) | −172.0(2) | −108.3(2) | ||||
| (±)-1 | −55.2(5) | −49.8(5) | −34.5(4) | |||
| −175.2(5) | 111.3(5) | |||||
| (R,R)-2 | 36.6(5) | 38.6(4) | 49.0(4) | −167.2(4) | 52.0(4) | |
| 36.7(5) | −98.9(5) | −109.2(4) | 106.2(6) | −170.4(4) | ||
| (meso)-2 | 16.4(1) | 57.4(1) | 58.7(8) | −57.4(1) | −58.7(8) | |
| (mol A) | −16.4(1) | −163.1(8) | 165.4(2) | 163.1(8) | −165.4(2) | |
| (meso)-2 | −19.9(1) | −45.7(8) | −56.1(1) | 56.1(1) | 45.7(8) | |
| (mol B) | 19.9(1) | 163.3(2) | −178.1(1) | 178.1(1) | −163.3(2) | |
| (R,R)-3 | 27.1(5) | 39.4(5) | 58.4(5) | 45.5(4) | 58.2(5) | −24.3(4) |
| 1.1(4) | −116.5(5) | −164.2(5) | 87.6(6) | −178.5(4) | −24.8(5) | |
| (R,R/S,S)-3 | −39.1(6) | −52.9(6) | −170.6(5) | 54.2(6) | −178.6(5) | −3.0(5) |
| −44.5(5) | −158.9(5) | 165.2(5) | 172.6(5) | −162.5(7) | −11.7(6) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kataeva, O.; Metlushka, K.; Yamaleeva, Z.; Ivshin, K.; Zinnatullin, R.; Nikitina, K.; Sadkova, D.; Badeeva, E.; Sinyashin, O.; Alfonsov, V. Chirality Control in Crystalline Ni(II) Complexes of Thiophosphorylated Thioureas. Crystals 2019, 9, 606. https://doi.org/10.3390/cryst9120606
Kataeva O, Metlushka K, Yamaleeva Z, Ivshin K, Zinnatullin R, Nikitina K, Sadkova D, Badeeva E, Sinyashin O, Alfonsov V. Chirality Control in Crystalline Ni(II) Complexes of Thiophosphorylated Thioureas. Crystals. 2019; 9(12):606. https://doi.org/10.3390/cryst9120606
Chicago/Turabian StyleKataeva, Olga, Kirill Metlushka, Zilya Yamaleeva, Kamil Ivshin, Ruzal Zinnatullin, Kristina Nikitina, Dilyara Sadkova, Elena Badeeva, Oleg Sinyashin, and Vladimir Alfonsov. 2019. "Chirality Control in Crystalline Ni(II) Complexes of Thiophosphorylated Thioureas" Crystals 9, no. 12: 606. https://doi.org/10.3390/cryst9120606