First-Principles Study on Hydrogen Storage Performance of Transition Metal-Doped Zeolite Template Carbon


Key Laboratory of Engineering Dielectrics and Its Application, Ministry of Education, Heilongjiang Provincial Key Laboratory of Dielectric Engineering, School of Electrical and Electronic Engineering, Harbin University of Science and Technology, Harbin 150080, China
*
Authors to whom correspondence should be addressed.
Crystals 2019, 9(8), 397; https://doi.org/10.3390/cryst9080397
Submission received: 30 June 2019
/
Revised: 27 July 2019
/
Accepted: 29 July 2019
/
Published: 31 July 2019
(This article belongs to the Special Issue New Horizons in Zeolites and Zeolite-Like Materials)
Abstract
:The hydrogen adsorption characteristics and mechanism of transition metal-doped zeolite template carbon (ZTC) as a novel porous material are studied by theoretical calculations employing first-principle all-electron atomic orbital method based on density functional theory. The stability of transition metal atoms (Sc, Ti, and V) decorated on zeolite template carbon is investigated by calculating the absorption binding energy. The adsorption configurations of the doped metal atom and adsorbed hydrogen are obtained from the energy functional minimization of first-principles calculations. The underlying mechanism for improving hydrogen storage performance of ZTC by doping transition metal atoms are explored through analyzing charge/spin populations of metal atoms in combination with the calculated results of hydrogen adsorption quantity and binding energy. To improve the hydrogen storage capability, the Sc, Ti, and V are individually introduced into the ZTC model according to the triplex axisymmetry. The hydrogen storage properties of ZTC decorated with different metal atoms are characterized by the adsorption energy and structure of several hydrogen atoms. The more energetically stable complex system with higher binding energy and adsorbing distance of hydrogen than lithium-doped ZTC can be achieved by doping Sc, Ti, V atoms in ZTC, which is expected to fulfill the substantial safe hydrogen storage by increasing hydrogen capacity with multi-sites doping of transition metal atoms. The present investigation provides a theoretical basis and predictions for the following experimental research and design of porous materials for hydrogen storage.
1. Introduction
At present, hydrogen storage techniques are primarily classified into physical and chemical schemes. For physical hydrogen storage, the hydrogen gas is compressed into a closed container under high pressure at low temperature which accommodates hydrogen in molecular form. There are several basic ways of hydrogen storage by physical interactions, such as liquid hydrogen storage, high-pressure hydrogen storage, activated carbon adsorption storage, etc. [1,2]. Chemical hydrogen storage is fulfilled by chemical bonds formed between hydrogen molecules or between hydrogen atoms and atoms of absorbent material, to uptake hydrogen through several general ways such as metal hydride hydrogen storage, organic liquid hydrogen storage, inorganic hydrogen storage, etc. [3]. The main parameters to measure the performance of hydrogen storage technology are hydrogen volume density, mass fraction, reversibility of hydrogen absorption and release, hydrogen loading and desorption rate, recyclable service life, and safety. Up to now, some research institutions and companies such as International Energy Agency and World Energy Network have proposed hydrogen storage standards. Moreover, an authoritative reference standard of hydrogen storage has been issued by the United States Department of Energy according to the percentage of hydrogen content, the volume density of hydrogen storage, and the kinetics of absorbing/releasing hydrogen. In the advantage of rapid loading, long cycle life, high safety performance, the high-pressure hydrogen storage, liquefaction storage and transportation, and metal hydride hydrogen storage are more suitable for commercial requirements at the present development status. [3,4,5].
Zeolites are hydrated crystalline aluminosilicates, which are constructed by the fundamental framework of oxygen-shared silicon and aluminum tetrahedral combined in three-dimensional space, representing various special performances due to its regular spacial-path structure with molecular size pore and the relatively large internal surface area and micropore volume [6]. Meanwhile, zeolites have some considerable advantages such as simple synthesis, good stability, and low price, and thus can be promised to be applied in gas storage, separation, catalysis and other fields [7]. Although the zeolites for commercial use at present do not meet the standard of hydrogen storage materials, its rich and diverse structural characteristics will provide much broad development in the future energy storage. Therefore, it is of great significance to study the underlying adsorption and diffusion mechanism of hydrogen molecules in zeolites and predict the physical and chemical properties of synthetic materials for the molecular design of new materials in the future [8,9,10].
Due to the great specific surface area and exceedingly uniform microporous structure, the zeolites template carbon (ZTC) nanostructures could render high capacity for absorbing hydrogen [11,12]. Furthermore, the intrinsic cation distribution in zeolites makes it feasible to improve hydrogen storage capability by decorating metal atoms in ZTC, to meet the technical requirements of safe and reliable hydrogen storage with small volume, lightweight, low cost and low density [13,14]. Recently, the density functional (DFT) study on hydrogen storage of lithium modified ZTC has been reported in high interest, in which the lithium atoms are utilized to modify the convex surface of ZTC carbon nanostructures so that this doping structure can be efficiently used as a hydrogen storage medium [15]. Because of the evident difference in electronic structure between transition metal and lithium atoms, especially for the unsaturated d-orbital electrons of transition metal atom which can produce a stronger force similar to coordinating bond than main group metal, it is reasonably suggested that the hydrogen storage performance of ZTC being doped with transition metal atoms will be improved more obviously compared with main-group metal. Triggered by this inspiration, the present paper proposes to modify ZTC by individually doping three representative transition metals, Sc, Ti, and V, to verify that the number and binding energy of adsorbed hydrogen molecules will be ameliorated. The adsorption configuration, energy, and electronic structure are investigated by first-principles calculations for four intrinsic sites of ZTC. The adsorption properties and mechanism of each transition metal element at different sites on ZTC are analyzed by comparing the energy, charge and spin population obtained by first-principles calculations. The optimum adsorption position and quantity of hydrogen molecules around the doped metal atoms are studied to found a theoretical basis for experiments and explore the transition metal-decorated ZTC porous materials applied in hydrogen storage.
2. Modeling and Calculation Methodology
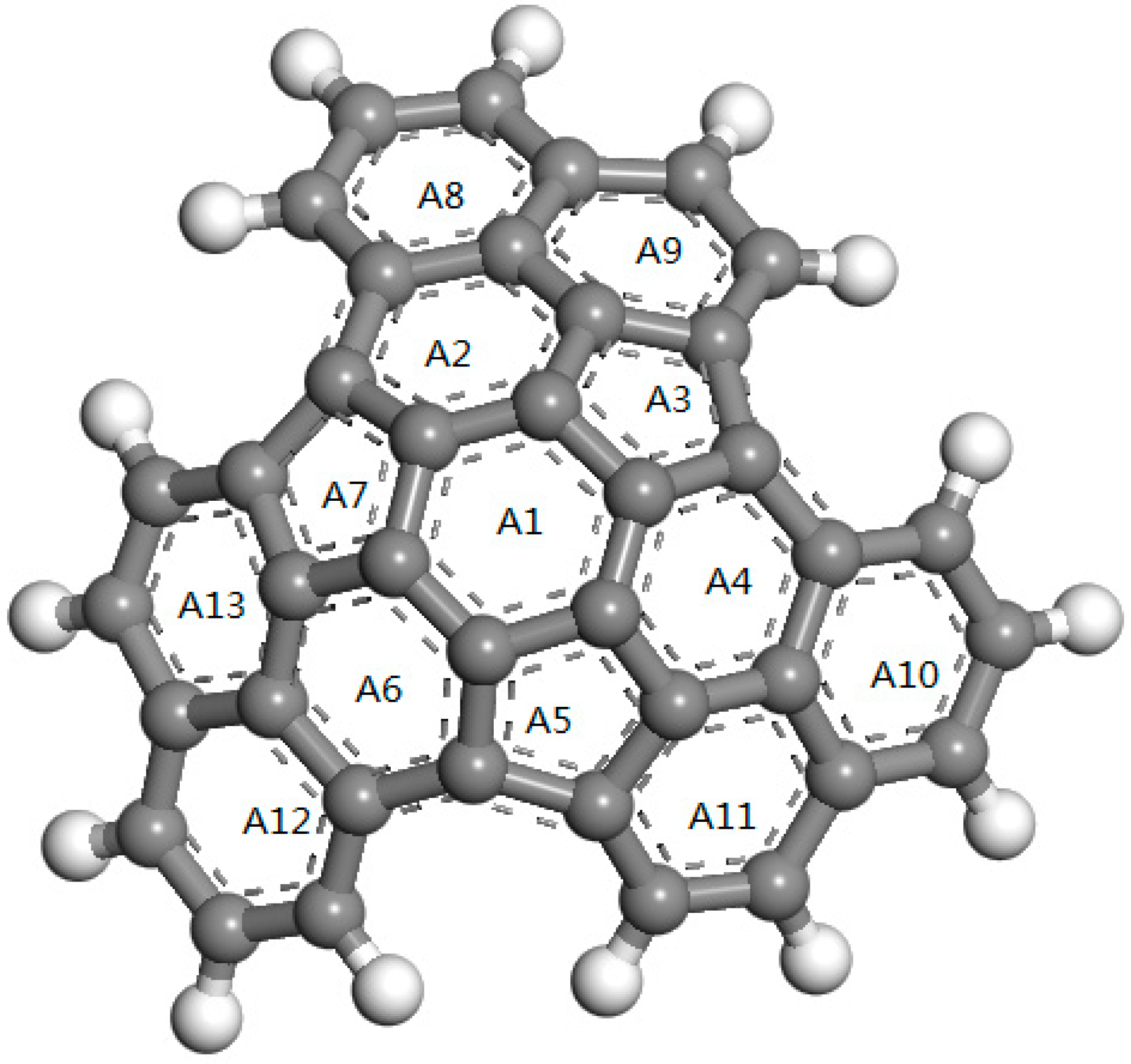
According to the molecular structure of ZTC reported by Nishihara, the C39H15 as a molecular structural unit of ZTC porous material is composed of carbocyclic rings connecting in a three-dimensional arrangement, which exhibits as a cambered surface curving to one side due to containing the five-carbocyclic rings [16]. The molecular structure of C39H15 consists of nine six carbocyclic rings, three five-carbocyclic rings and 15 hydrogen atoms at the edge, formalized by a total of 39 carbon atoms and 15 hydrogen atoms as shown in Figure 1. This pristine ZTC model is initially constructed by subsequently connecting conjugated carbocyclic rings from the central six carbocyclic ring labeled with A1, in which the six and five carbocyclic rings from ZTC center to the edge passivated with hydrogen are symbolized as A3, A5, A7 and A2, A4, A6 respectively as shown in Figure 1. The geometrical optimization by minimizing total energy in first-principles calculations has been performed on the initial model to realize atomic structure relaxation in C39H15. It is illustrated from Figure 1 that the six and five carbocyclic rings are alternatively adjacent to each other and encompass the benzene center to a curved atomic surface in three-fold rotation symmetry.
According to the all-electron numerical orbital first-principles formalism based on spin density functional theory, the atomic structure, energy and electronic structure of transition metal-doped ZTC and their hydrogen adsorption configuration are calculated as implemented by DMol3 code of Materials Studio 8.0 software package (Accelrys Inc., Materials Stutio v8.0.0.843, San Diego, CA, USA) [17,18]. The M11-L form of exchange-correlation-functional in meta-generalized-gradient-approximation is adopted [19], and the electronic eigenstate wave functions are expanded by the double numerical polarized (DNP) basis set with the total orbital cutoff (global orbital cutoff) being set as 5.0 Å so that the error introduced from the finite basis set is minimal enough. The interactions between electron and atomic kernel are described by the all-electron relative (all-electron relativistic) core treatment. The convergence tolerance of the self-consistent field (SCF) iteration is set to 1.0 × 10−6 Ha/atom (1 Ha = 27.2 eV). Based on the spin density functional theory deriving from Dirac’s relativistic quantum mechanics equations, the spin-orbit interaction and spin polarization are calculated by employing individual eigenstate wave functions for the two fundamental states of electronic spin [20]. The geometry optimization by minimizing the total energy is implemented with the conjugated gradient algorithm so that the total energy convergence of less than 1.0 × 10−5 Ha/atom, and the force and atomic displacement acting on atoms respectively less than 0.002 Ha/Å and 0.005 Å can be obtained for the relaxed atomic structure [21].
3. Results and Discussion
After geometric optimization of C39H15, three transition metal atoms are individually decorated on it as modeled by doping single scandium (Sc), titanium (Ti) or alum (V) respectively at various sites to improve the hydrogen storage performance of ZTC. The total energy calculations show that the binding forces from different carbocyclic parts of ZTC partially offset each other, and thus lead to the relative lowest adsorption binding energy of the transition metal atoms on A1 site in the symmetrical center of C39H15, compared with the other sites. Nevertheless, the adsorption binding energy of the transition metals investigated here still meets the requirement of stabilized doping structure by forming coordinating bonds with the interaction similar to chemical adsorption. The calculation results of binding energy and coordination bond length (measured by the averaged distance away from nearing-carbon atoms) between the doped metal atom and ZTC substrate for all kinds of decoration sites (A1, A3, A4, A10) are listed in Table 1. It is reasonable and feasible to fulfill improvement of hydrogen adsorption performance by introducing tightly bridging metal atoms which provide higher binding force to hydrogen molecules than ZTC framework. In comparison, the forces from different carbon rings for the metal atoms decorated on A3, A4 and A10 sites can not completely offset, leading to definitely higher binding energy with the total energy of the whole system being slightly reduced, which makes the coordination of metal atoms to ZTC more firmly and benefits the adsorption and desorption of hydrogen molecules, and thus will increase the recycle times and service life in hydrogen storage application.
The transition metal elements such as Sc, Ti, and V in the same periodic row, the atomic radius of which decreases with the increase of atomic number, present partially unoccupied d-orbitals. Hence, transition metal atoms will interact with the carbon ring of ZTC by forming a stronger force as the coordinate bonding than that for alkali metals such as lithium. According to the SCF electron density distribution, the atomic charge and spin analyzed from the Mulliken population are calculated to investigate the electron transfer and spin distribution of doping atoms in the metal-decorated ZTC, as shown in Figure 2. Taking ZTC doped with Sc (Sc-C39H15) as an example, it is indicated that the atomic charges of Sc atom on A1, A3, A4, and A10 sites are 0.822, 0.592, 0.808 and 0.752e, respectively (e denotes elementary charge), which implies the substantial electron transfer occurs from the Sc atom to the nearest carbon atomic ring for each site. The positive population charges of Sc, Ti and V atoms and the negative charges on the neighboring carbocyclic rings demonstrate the partial Coulomb attribute of these transition metal atoms as coordinating center being attached to ZTC ligands by a relatively strong force as chemical absorption. Besides, partial positive charges populated on Sc, Ti and V dopants on ZTC imply that multiple H2 molecules will bind to the doped metal atoms through electrostatic interactions with remarkably higher adsorption energies than directly absorbed on ZTC, as verified in the following text and being consistent with recently reported results for carbon nitride (C3N) [22].
The electron transfer has been found from Sc, Ti, V atoms to the most of the carbon atoms adjacent to them. The charge population of Ti-C39H15 and V-C39H15 system is similar to that of Sc-C39H15. It is noted that there is no significant difference in electron transfer for the same metal atom at different sites, while the three metal atoms of Sc, Ti, V show discrepancy in charge and spin population more obviously. The electron transfer quantity of the doped metal atom declines with the increase of its atomic number, resulting in the reduced Coulomb force and total energy of the modified ZTC system. Moreover, the atomic spin distribution will affect the magnetic energy, in which the smaller absolute spin value populated on doped metal atoms means the lower energy and higher structural stability of the doping system.
A cluster of metal atoms is individually added at every adsorption site on ZTC convex surface with the binding distance similar to the single atom coordinating on ZTC, which is modeled as the initial molecular structure and relaxed with geometry optimization to investigate the possibility of atomic accumulations for transition metal dopants. The geometrically optimized structures as shown in Figure 3 for a representative cluster of 5 Sc atoms doped on ZTC surface center illustrate that the multiple metal atoms decorated on ZTC will be dispersed on adjacent adsorption sites, verifying that the doped transition metal atoms cannot assemble together, due to the high adsorption forces deriving from the coordinating interactions between dopants and carbon rings of ZTC.
Referencing on the calculation results reported by Frank et al. that six H2 can be effectively adsorbed by doping lithium atoms at the A4 site [19], the hydrogen adsorption model is built by placing 3, 5 and 6 H2 molecules near the doping metal atoms at each site in the geometrically optimized structures of Sc-C39H15,Ti-C39H15 and V-C39H15, and as an initial adsorption configurations to be further implemented by Dmol3 code to carry out geometric optimization and energy calculations for hydrogen adsorption configuration and binding energy. The calculation results will give fundamental evidence that the proposed transition metal decoration is an effective routine to ameliorate hydrogen storage performance of ZTC. The hydrogen adsorption properties for the complex structures of ZTC doped with three kinds of metals are analyzed in comparison, emphasizing hydrogen molecules adsorbed around the metal-doped at A4 site compared with Li doped ZTC.
To investigate adsorption properties of hydrogen molecules on metal-doped ZTC, more than 10 adsorption configurations have been simulated. It is found that H2 molecules tend to symmetrically locate around Sc, Ti or V atoms doped on C39H15 surface. Therefore, when building the initial model of H2 adsorption configuration, the H2 molecules are uniformly loaded around the doped metal atoms in almost symmetrical positions, with a gradually increased loading number of H2 molecules. First-principles geometric optimization has been individually performed for each additional H2 molecule to obtain all the relaxed and stable adsorption structures. Representative adsorption configurations after geometry optimization of three and six H2 molecules at A1-A10 sites of Sc-C39H15 are shown with the top (left) and side (right) views in Figure 4.
Based on the first-principles total energy, the average adsorption binding energy of multiple H2 molecules and the adsorption binding energy of the nth H2 molecule are calculated to evaluate adsorption capacity of ZTC decorated with the transition metal atoms. The average adsorption energy of n H2 molecules is computed by the following formula:
and the adsorption energy of the nth H2 molecule is formalized as:
where Td denotes the doped transition metal atom, E(H2) symbolizes the total energy of an isolated H2 molecule, E(Td-C39H15) signifies the total energy of Td doped ZTC, and E(Td-C39H15+nH2) represents the total energy of Td-doped-ZTC adsorbed with n H2 molecules. The calculated results of the average adsorption energy of 3–6 H2 molecules and adsorption energy of the nth H2 molecule for each representative sites are listed in Table 2 and plotted in Figure 5 respectively.
For the coordination structures of Sc, Ti or V doped ZTC, the average adsorption energies of 3–6 H2 molecules at the A1 site is considerably higher than that at the A3, A4, and A10 sites, all of which are in the physical adsorption range. The average binding energy of H2 molecules absorbed by these transition metal atoms coordinating to ZTC shows a decreasing trend with the increment of H2 uptake. But for Sc-C39H15 with Sc atom decorated at A10 as an example, the adsorption energy of the sixth H2 molecule around Sc is −0.226 eV, and the farthest and nearest H2 molecule is 5.403 Å and 2.036 Å respectively from Sc atom. The large difference in distance between the nearest and farthest H2 molecule indicates the poor binding force so that adsorption capacity has attained to the limit of 5 H2 molecules on Sc-C39H15. When additional hydrogen molecules are added, a stable adsorption structure cannot be formed on this basis.
The adsorption energies of ZTC doped with three transition metals at A3, A4 and A10 sites decrease or even become negative with the increase of the H2 uptake because the H2 molecules being located away from the symmetric center of ZTC cannot efficiently acquire the binding forces from different parts of ZTC. Meanwhile, the transition metal with unoccupied 3d orbitals will increase the adsorption binding energy and capacity of H2 molecules in comparison with alkali metals. Doping transition metal atoms at the A1 site can remarkably increase the adsorption capacity of hydrogen molecules, and even exceed the typical upper limit of 0.5 eV for physical adsorption (as shown in Table 2 and Figure 5). Although it promotes the capability of adsorbing hydrogen molecules, as concerned for the perfect hydrogen storage materials, the excessively intensified binding force for hydrogen adsorption implies the unduly high temperature and low pressure needed for hydrogen desorption in the extraction process, which will, however, increase cost and impede popularization.
The binding energy of doping metal atoms onto ZTC is identical for A3, A4 and A10 sites. For example, in Sc-C39H15, the binding energies of the Sc atom doping on these three sites are 60.41, 60.41 eV and 60.14 eV respectively, which are higher than the lower limit of chemical adsorption as to form the decorated complex structure. The adsorption binding energies of H2 molecules on A3, A4, A10 sites are almost the same after modification by doping transition metal in ZTC, verifying the suggested mechanism described previously. Also taking the Sc-C39H15 system as an example, the average adsorption energy of three H2 molecules at A3, A4 and A10 sites are 0.463, 0.454 and 0.354 eV respectively, and for six H2 molecules are 0.301, 0.271 and 0.221 eV respectively. The average adsorption energy of H2 molecules at A3, A4 and A10 sites of the transition metal doped ZTC varies in 0.25~0.63 eV, which belong to physical adsorption and are significantly higher than the recently reported hydrogen adsorption energy range of 0.17~0.24 eV for the ZTC doped with lithium at the A4 site [15]. It is reasonably suggested that transition metals such as Sc, Ti and V can be effectively doped in ZTC to improve the hydrogen storage performance. Therefore, the metal-carbon complex structures of Sc-C39H15, Ti-C39H15, and V-C39H15 designed and theoretically demonstrated in this paper are prospective candidates as hydrogen storage materials to acquire safety of the hydrogen storage system and facilitate long-distance mass transportation in the external environment.
4. Conclusions
The hydrogen adsorption properties of ZTC modified by doping transition metal atoms (Sc, Ti, V) at various sites are studied by employing the all-electron numerical orbital first-principles calculations based on density functional theory, being analyzed by calculating the binding energy of complex metal-decorated structure and H2 adsorption for four representative absorbing sites according to the three-fold rotational symmetry of ZTC. The transition metal atoms and H2 molecules are subsequently introduced onto A1, A3, A4 and A10 sites in ZTC respectively, and the geometric optimization has been performed to obtain metal coordination structures and H2 multiple-molecule adsorption configurations. The binding energy and atomic Mulliken population of Sc, Ti and V decorated on various sites of ZTC are individually investigated to reveal the interaction attributes and structural stability of Sc-C39H15, Ti-C39H15 and V-C39H15 doping systems. The structural characteristics and charge/spin population analysis show that ZTC structure has a strong binding effect on Sc, Ti and V atoms by forming coordination bonds through its delocalized π electrons donating to the unoccupied d orbitals of the doped transition metal atoms. The average adsorption energy of multiple H2 molecules and the adsorption energy of the sixth H2 molecule indicate that the three metal-ZTC complexes can uptake six H2 molecules at the A1 site while adsorbing up to only five H2 molecules at the A3, A4 and A10 sites. The average H2 adsorption energy varies in the range of 0.25~0.63 eV, which belongs to physical adsorption and is significantly higher than the recent theoretically reported 0.17~0.24 eV at A4 site in the Li-doped ZTC system. It is proven that decorating transition metal onto ZTC can achieve higher hydrogen storage performance than that of a lithium atom, which provides a theoretical basis for developing high-performance hydrogen storage materials by transition metal modification technology.
Author Contributions
Conceptualization, B.H. and W.-F.S.; Data curation, P.-H.L.; Funding acquisition, B.H. and S.-W.S.; Investigation, P.-H.L.; Methodology, W.-F.S.; Software, P.-H.L.; Writing—original draft, B.H.; Writing—review & editing, P.-H.L., W.-F.S. and S.-W.S.
Funding
This research was funded by the University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province (Grant No. UNPYSCT-2016158 and UNPYSCT-2016161) and National Natural Science Foundation of China (Grant No. 51607048).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Sethia, G.; Sayari, A. Activated carbon with optimum pore size distribution for hydrogen storage. Carbon 2016, 99, 289–294. [Google Scholar] [CrossRef]
- Hwang, H.T.; Varm, A.A. Hydrogen storage for fuel cell vehicles. Curr. Opin. Chem. Eng. 2014, 8, 42–48. [Google Scholar] [CrossRef]
- Makepeace, J.W.; He, T.; Weidenthaler, C.; Jensen, T.R.; Chang, F.; Vegge, T.; Ngene, P.; Kojima, Y.; de Jongh, P.E.; Chen, P. Reversible ammonia-based and liquid organic hydrogen carriers for high-density hydrogen storage: Recent progress. Int. J. Hydrog. Energy 2019, 44, 7746–7767. [Google Scholar] [CrossRef]
- Wang, J.G.; Ma, L. Theoretical Study of Hydrogen Storage on Ti-Decorated BC3 Nanostructures. J. Nanosci. Nanotechnol. 2019, 19, 5650–5655. [Google Scholar] [CrossRef]
- Al, S. Theoretical investigations of elastic and thermodynamic properties of LiXH4 Compounds for Hydrogen Storage. Int. J. Hydrog. Energy 2019, 4, 1727–1730. [Google Scholar] [CrossRef]
- Cheng, X.; Li, Z.; He, Y.-L. Effects of temperature and pore structure on the release of methane in zeolite nanochannels. RSC Adv. 2019, 9, 9546–9554. [Google Scholar] [CrossRef] [Green Version]
- Gigli, L.; Arletti, R.; Fois, E.; Tabacchi, G.; Quartieri, S.; Dmitriev, V.; Vezzalini, G. Unravelling the High-Pressure Behaviour of Dye-Zeolite L Hybrid Materials. Crystals 2018, 8, 79. [Google Scholar] [CrossRef]
- Wang, Y.; Ren, F.; Pan, D.; Ma, J. A Hierarchically Micro-Meso-Macroporous Zeolite CaA for Methanol Conversion to Dimethyl Ether. Crystals 2016, 6, 155. [Google Scholar] [CrossRef]
- De Oliveira, L.H.; Meneguin, J.G.; Pereira, M.V.; da Silva, E.A.; Grava, W.M.; do Nascimento, J.F.; Arroyo, P.A. H2S adsorption on NaY zeolite. Microporous Mesoporous Mater. 2019, 284, 247–257. [Google Scholar] [CrossRef]
- Dai, W.; Luo, J.S.; Tang, Y.J.; Wang, C.Y.; Chen, S.J.; Sun, W.G. Simulation of hydrogen adsorption in molecular sieves. Acta Phys. Sin. 2009, 58, 1890. [Google Scholar] [CrossRef]
- Han, S.W.; Bang, J.; Ko, S.H.; Ryoo, R. Variation of nitrogen species in zeolite-templated carbon by low-temperature carbonization of pyrrole and the effect on oxygen reduction activity. J. Mater. Chem. A 2019, 7, 8353–8360. [Google Scholar] [CrossRef]
- Musyoka, N.M.; Rambau, K.M.; Manyala, N.; Ren, J.W.; Langmi, H.W.; Mathe, M.K. Utilization of waste tyres pyrolysis oil vapour in the synthesis of Zeolite Templated Carbons (ZTCs) for hydrogen storage application. J. Environ. Sci. Health Part A 2018, 53, 1022–1028. [Google Scholar] [CrossRef]
- Yang, B.; Zou, J.; Huang, T.; Mao, J.; Zeng, X.; Ding, W. Enhanced hydrogenation and hydrolysis properties of core-shell structured Mg-MOx (M = Al, Ti, and Fe) nanocomposites prepared by arc plasma method. Chem. Eng. J. 2019, 371, 233–243. [Google Scholar] [CrossRef]
- Wu, D.; Ouyang, L.; Wu, C.; Gu, Q.; Wang, H.; Liu, J.; Zhu, M. Phase transition and hydrogen storage properties of Mg17 Ba2 compound. J. Alloys Compd. 2017, 690, 519–522. [Google Scholar] [CrossRef]
- Isidro-Ortega, F.J.; Pacheco-Sanchez, J.H.; Desales-Guzman, L.A. Hydrogen storage on lithium decorated zeolite templated carbon, DFT study. Int. J. Hydrog. Energy 2017, 42, 30704–30717. [Google Scholar] [CrossRef]
- Nishihara, H.; Yang, Q.; Hou, P.; Unno, M.; Yamauchi, S.; Saito, R. A possible buckybowl-like structure of zeolite templated carbon. Carbon 2009, 47, 1220–1230. [Google Scholar] [CrossRef]
- Köhler, C.; Seifert, G.; Frauenheim, T. Density-Functional based calculations for Fen(n ≤ 32). Chem. Phys. 2005, 309, 23–31. [Google Scholar] [CrossRef]
- Andzelm, J.; King-smith, R.D.; Fitzgerald, G. Geometry optimization of solids using delocalized internal coordinates. Chem. Phys. Lett. 2001, 335, 321–326. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics. J. Phys. Chem. Lett. 2012, 3, 117–124. [Google Scholar] [CrossRef]
- Chantis, A.N.; Christensen, N.E.; Svane, A.; Cardona, M. Full-zone analysis of relativistic spin splitting at band anticrossings: The case of zinc-blende semiconductors. Phys. Rev. B 2000, 81, 205025. [Google Scholar] [CrossRef]
- Baker, A.; Kessi, B.; Delley, A. The generation and use ofdelocalized internal coordinates in geometry optimization. J. Chem. Phys. 1996, 105, 192. [Google Scholar] [CrossRef]
- Faye, O.; Hussain, T.; Karton, A.; Szpunar, J. Tailoring the capability of carbon nitride (C3N) nanosheets toward hydrogen storage upon light transition metal decoration. Nanotechnology 2018, 30, 075404. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Molecular structure model with gray and white symbolizing carbon and hydrogen atoms respectively.
Figure 1.
Molecular structure model with gray and white symbolizing carbon and hydrogen atoms respectively.

Figure 2.
Population Analysis of transition metal-doped ZTC (M-C39H15, M=Sc, Ti, V): Mulliken atomic charge (left panel) and spin (right panel).
Figure 2.
Population Analysis of transition metal-doped ZTC (M-C39H15, M=Sc, Ti, V): Mulliken atomic charge (left panel) and spin (right panel).

Figure 3.
The relaxation process from the initial atomic cluster to dispersed adsorption at adjacent sites for multiple Sc atoms being doped around the ZTC surface center, with the gray, white and violet balls representing carbon, hydrogen and scandium atoms respectively.
Figure 3.
The relaxation process from the initial atomic cluster to dispersed adsorption at adjacent sites for multiple Sc atoms being doped around the ZTC surface center, with the gray, white and violet balls representing carbon, hydrogen and scandium atoms respectively.

Figure 4.
Adsorption configurations of three (above panels) and six (below panels) H2 molecules on A1, A3, A4, A10 sites respectively in Sc-doped-ZTC (Sc-C39H15). The top and side views are respectively shown by the left and right parts in each schematic unit, and the gray, white and violet balls identify carbon, hydrogen and scandium atoms respectively.
Figure 4.
Adsorption configurations of three (above panels) and six (below panels) H2 molecules on A1, A3, A4, A10 sites respectively in Sc-doped-ZTC (Sc-C39H15). The top and side views are respectively shown by the left and right parts in each schematic unit, and the gray, white and violet balls identify carbon, hydrogen and scandium atoms respectively.

Figure 5.
The adsorption energy of the 6th H2 molecule for Sc, Ti and V doped ZTC.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The binding energies and coordination bond lengths of Sc, Ti and V atoms being adsorbed on ZTC at various sites.
Table 1.
The binding energies and coordination bond lengths of Sc, Ti and V atoms being adsorbed on ZTC at various sites.
| Sites | Metal | Binding Energy/eV | Coordination Bond Length/Å |
|---|---|---|---|
| A1 | Sc | 4.08 | 2.284 |
| Ti | 4.35 | 2.250 | |
| V | 2.18 | 2.211 | |
| A3 | Sc | 6.41 | 2.368 |
| Ti | 6.24 | 2.305 | |
| V | 5.95 | 2.244 | |
| A4 | Sc | 7.22 | 2.317 |
| Ti | 6.83 | 2.294 | |
| V | 6.59 | 2.281 | |
| A10 | Sc | 7.04 | 2.297 |
| Ti | 6.45 | 2.275 | |
| V | 6.26 | 2.263 |
Table 2.
Average adsorption energy (eV) of 3–6 H2 molecules adsorbed around transition metal at each site.
Table 2.
Average adsorption energy (eV) of 3–6 H2 molecules adsorbed around transition metal at each site.
| Sites | ZTC/Td | 3 | 5 | 6 |
|---|---|---|---|---|
| A1 | ZTC/Sc | 0.708 | 0.424 | 0.364 |
| ZTC/Ti | 0.718 | 0.430 | 0.376 | |
| ZTC/V | 0.720 | 0.436 | 0.390 | |
| A3 | ZTC/Sc | 0.463 | 0.412 | 0.321 |
| ZTC/Ti | 0.524 | 0.414 | 0.311 | |
| ZTC/V | 0.609 | 0.410 | 0.357 | |
| A4 | ZTC/Sc | 0.454 | 0.316 | 0.264 |
| ZTC/Ti | 0.463 | 0.312 | 0.251 | |
| ZTC/V | 0.544 | 0.414 | 0.327 | |
| A10 | ZTC/Sc | 0.354 | 0.314 | 0.224 |
| ZTC/Ti | 0.544 | 0.362 | 0.283 | |
| ZTC/V | 0.626 | 0.418 | 0.338 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Han, B.; Lv, P.-H.; Sun, W.-F.; Song, S.-W. First-Principles Study on Hydrogen Storage Performance of Transition Metal-Doped Zeolite Template Carbon. Crystals 2019, 9, 397. https://doi.org/10.3390/cryst9080397
AMA Style
Han B, Lv P-H, Sun W-F, Song S-W. First-Principles Study on Hydrogen Storage Performance of Transition Metal-Doped Zeolite Template Carbon. Crystals. 2019; 9(8):397. https://doi.org/10.3390/cryst9080397
Chicago/Turabian StyleHan, Bai, Peng-Hao Lv, Wei-Feng Sun, and Shu-Wei Song. 2019. "First-Principles Study on Hydrogen Storage Performance of Transition Metal-Doped Zeolite Template Carbon" Crystals 9, no. 8: 397. https://doi.org/10.3390/cryst9080397
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.