Spherical Polybutylene Terephthalate (PBT)—Polycarbonate (PC) Blend Particles by Mechanical Alloying and Thermal Rounding

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental
2.2.1. Co-Grinding in a Planetary Ball Mill
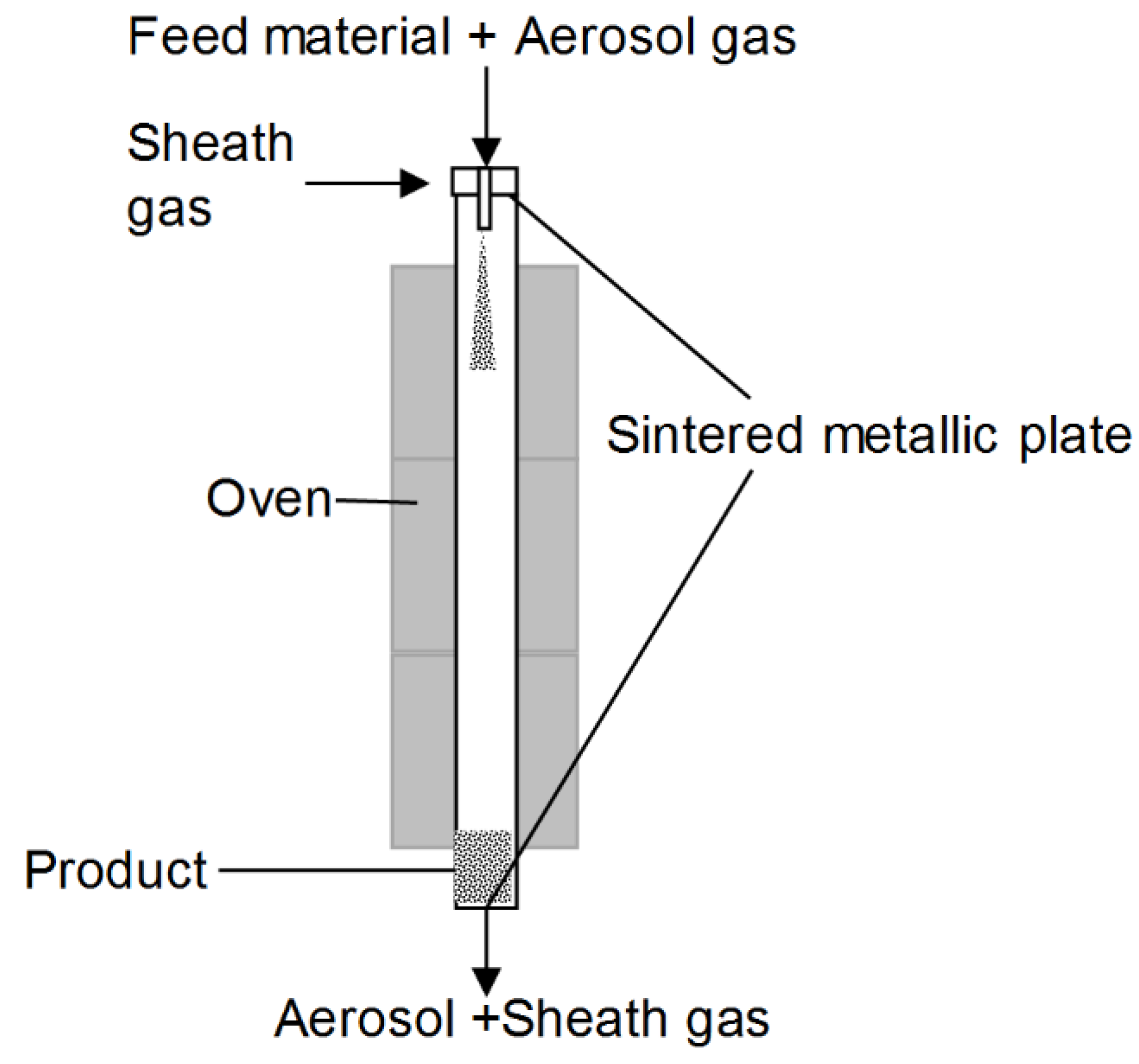
2.2.2. Thermal Rounding
2.3. Characterization
2.3.1. Laser Diffraction Particle Sizing
2.3.2. Scanning Electron Microscopy
2.3.3. Raman Microscopy
2.3.4. Polarized Light Microscopy
2.3.5. Infrared Spectroscopy
2.3.6. Differential Scanning Calorimetry
3. Results and Discussion
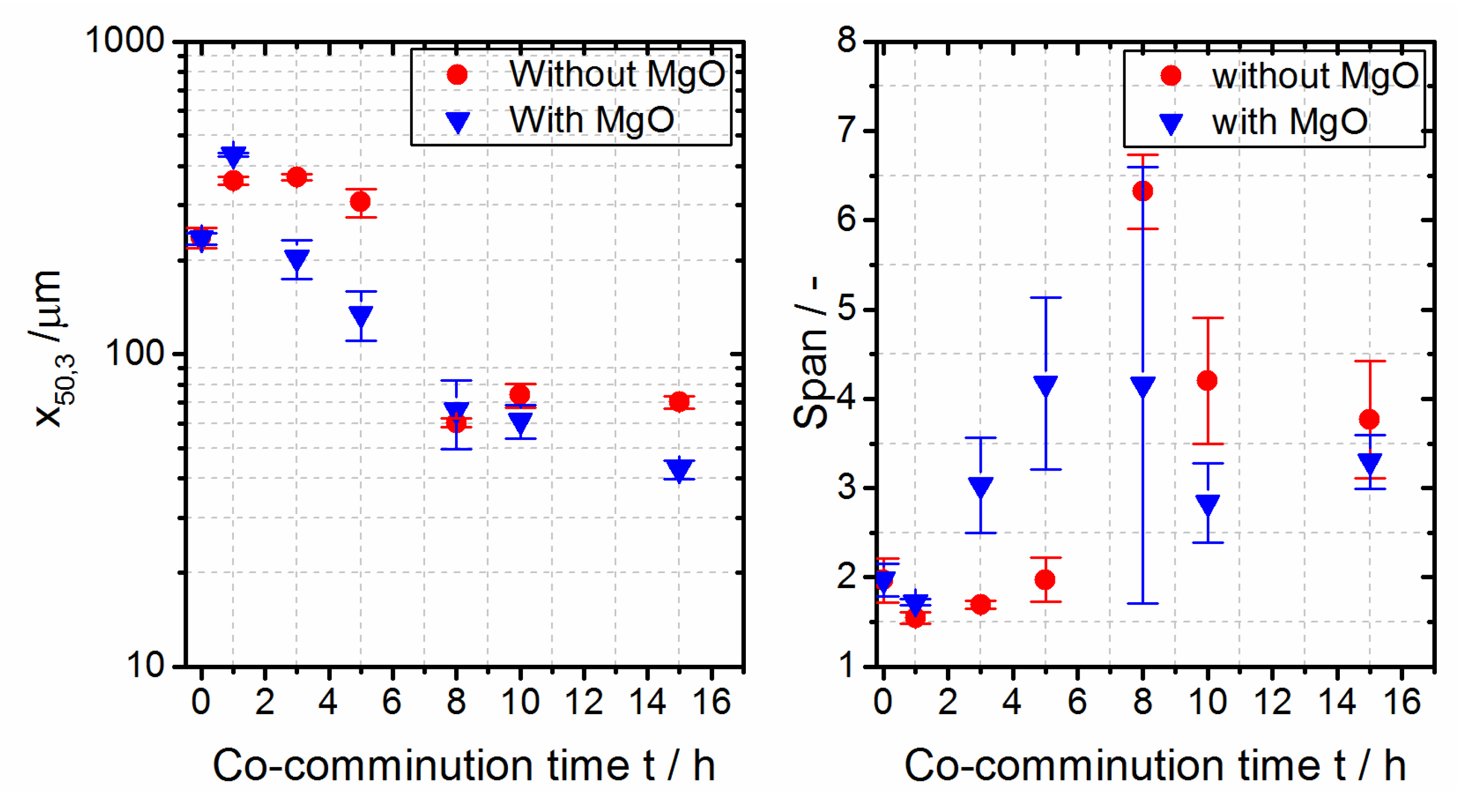
3.1. Co-Grinding and Rounding of PBT and PC
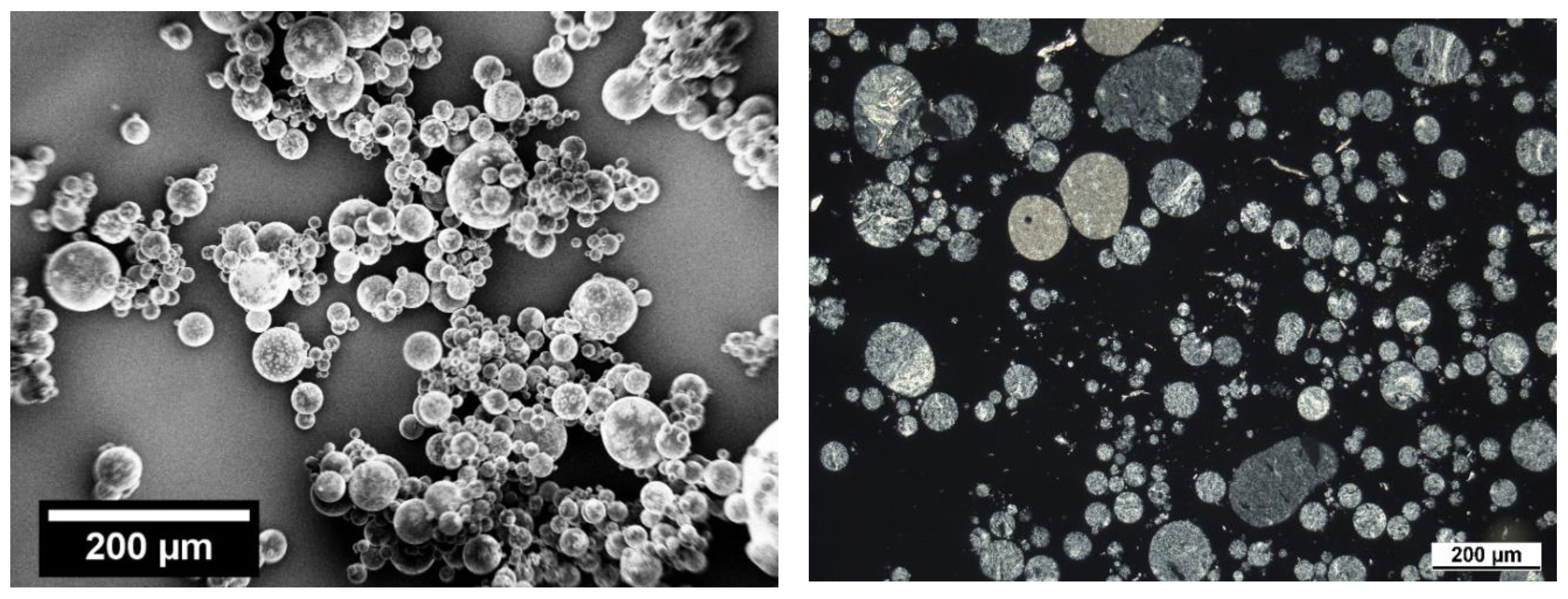
3.2. Rounding
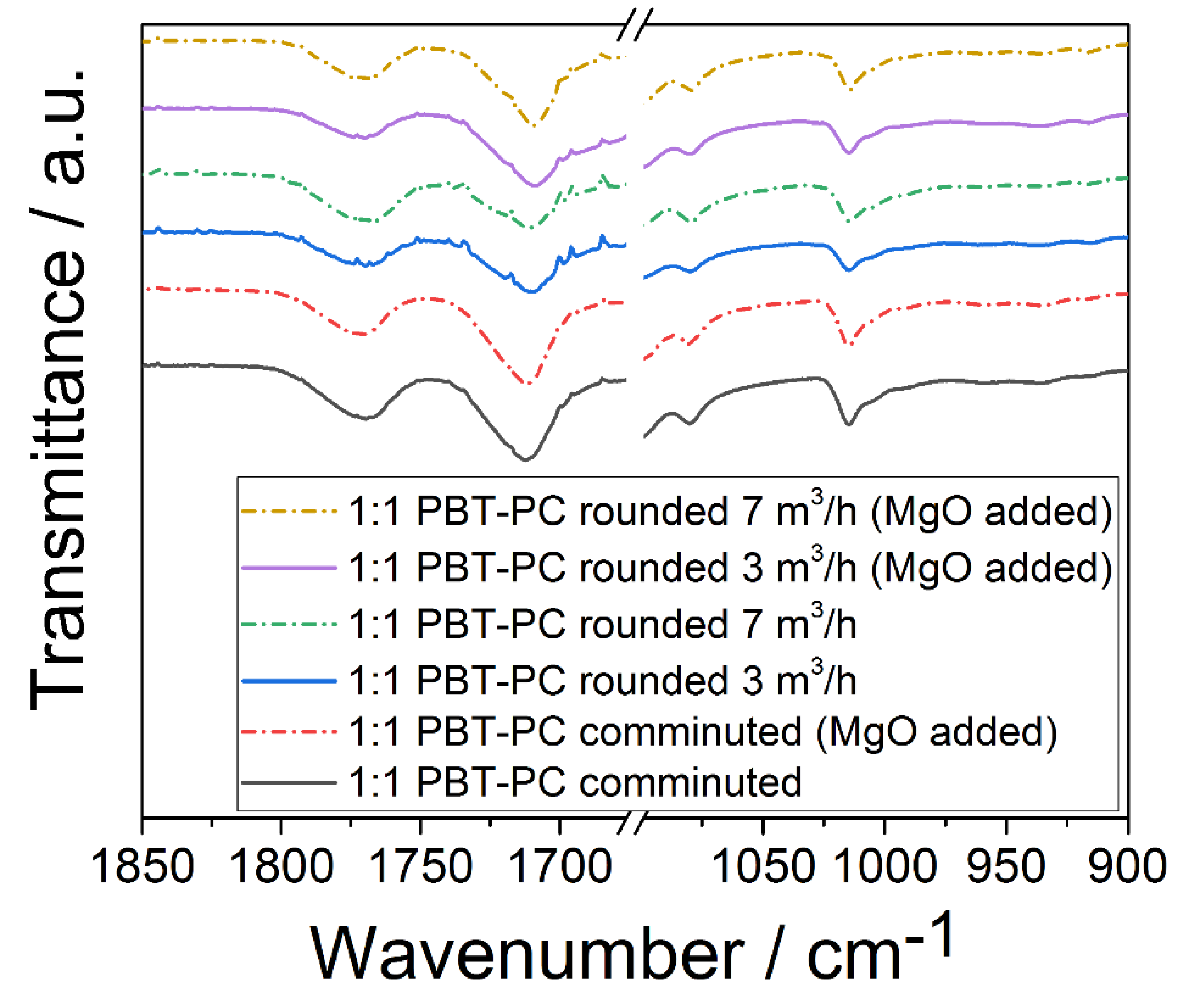
3.3. Vibrational Spectroscopy
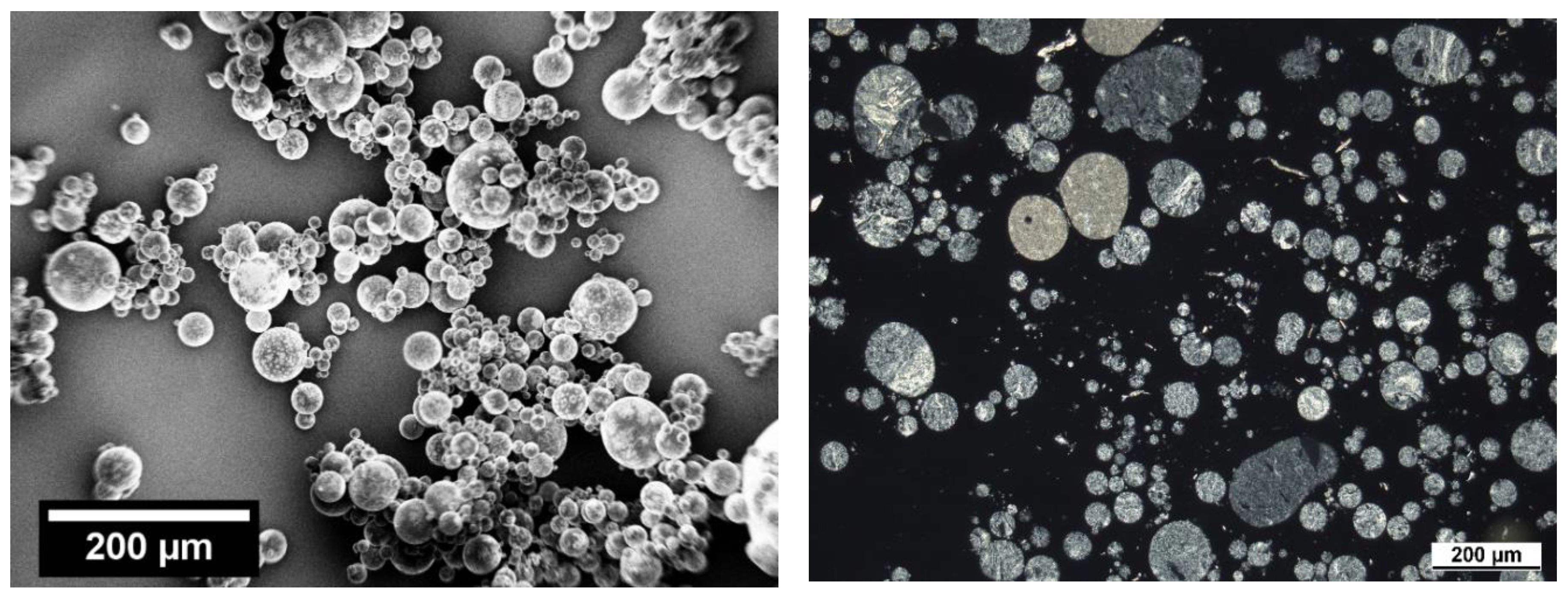
3.4. Staining/SEM Imaging
3.5. Polarized Light Microscopy
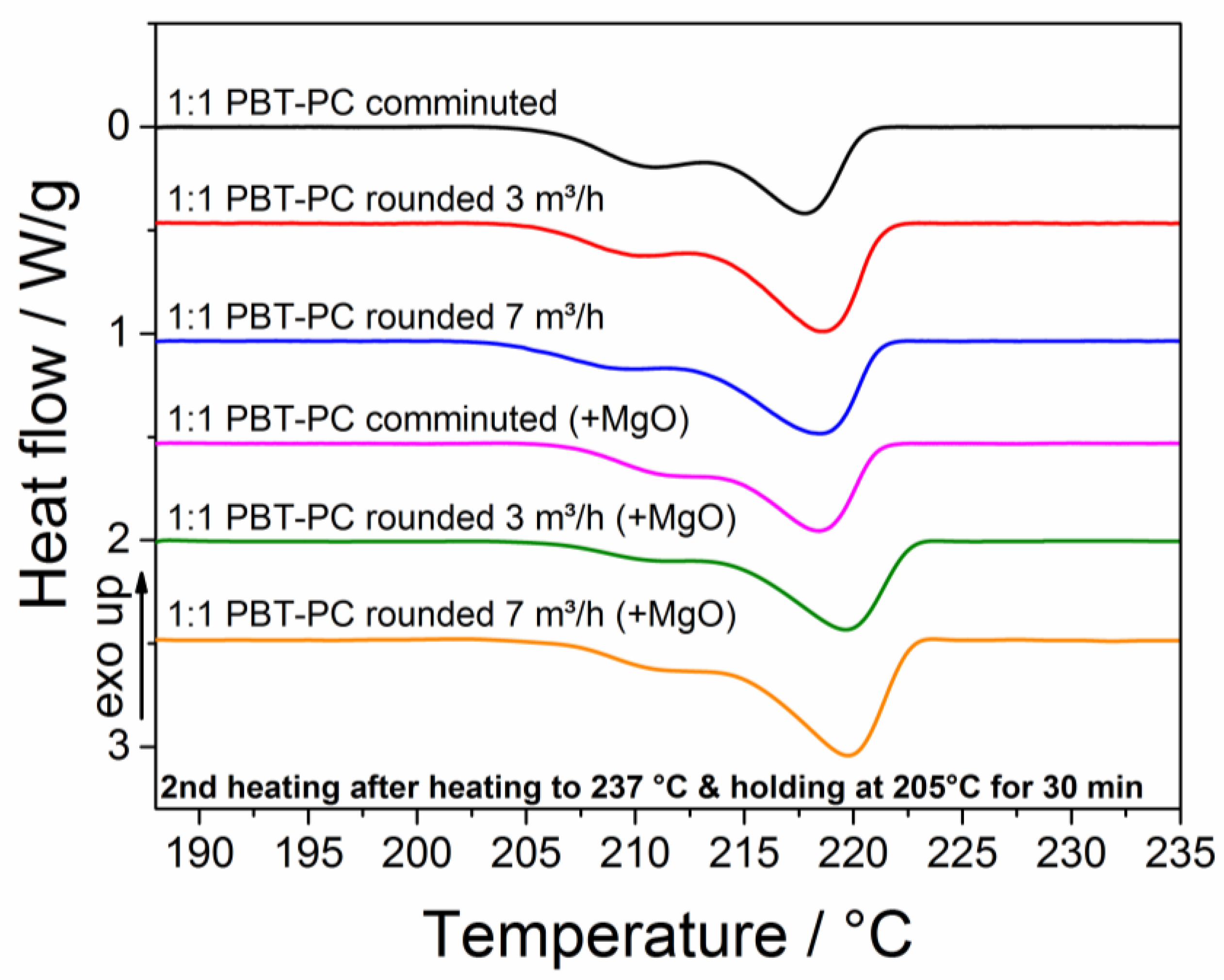
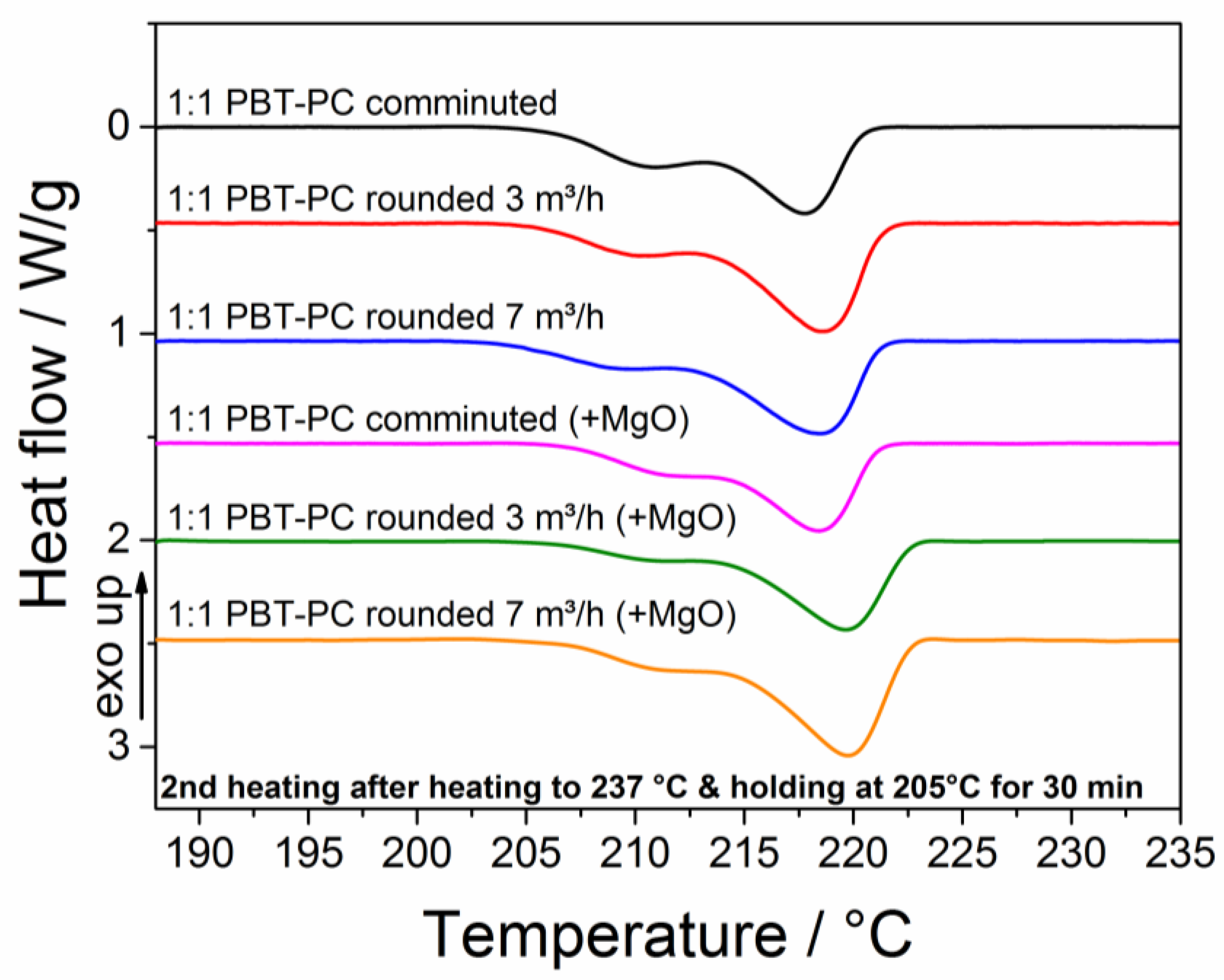
3.6. Thermal Characterization
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gibson, I.; Rosen, D.; Stucker, B. Additive Manufacturing Technologies, 2nd ed.; Springer: New York, NY, USA, 2015; ISBN 978-1-4939-2113-3. [Google Scholar]
- Ligon, S.C.; Liska, R.; Stampfl, J.; Gurr, M.; Mülhaupt, R. Polymers for 3D Printing and Customized Additive Manufacturing. Chem. Rev. 2017, 117, 10212–10290. [Google Scholar] [CrossRef]
- Kruth, J.P.; Mercelis, P.; Van Vaerenbergh, J.; Froyen, L.; Rombouts, M. Binding mechanisms in selective laser sintering and selective laser melting. Rapid Prototyp. J. 2005, 11, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Amado, A.; Wegener, K. Polymer powders for selective laser sintering (SLS). In Proceedings of the 30th International Conference of the Polymer Processing Society, Cleveland, OH, USA, 6–12 June 2014; p. 160009. [Google Scholar]
- Haworth, B.; Hopkinson, N.; Hitt, D.; Zhong, X. Shear viscosity measurements on Polyamide-12 polymers for laser sintering. Rapid Prototyp. J. 2013, 19, 28–36. [Google Scholar] [CrossRef]
- Zhao, M.; Wudy, K.; Drummer, D. Crystallization kinetics of polyamide 12 during Selective laser sintering. Polymers 2018, 10. [Google Scholar] [CrossRef]
- Wohlers, T.T. Wohlers Report 2016; Wohlers Associates: Fort Collins, CO, USA, 2016; ISBN 978-0-9913332-2-6. [Google Scholar]
- Schmidt, J.; Sachs, M.; Fanselow, S.; Zhao, M.; Romeis, S.; Drummer, D.; Wirth, K.E.; Peukert, W. Optimized polybutylene terephthalate powders for selective laser beam melting. Chem. Eng. Sci. 2016, 156, 1–10. [Google Scholar] [CrossRef]
- Schmidt, J.; Plata, M.; Tröger, S.; Peukert, W. Production of polymer particles below 5 μm by wet grinding. Powder Technol. 2012, 228, 84–90. [Google Scholar] [CrossRef]
- Sachs, M.; Friedle, M.; Schmidt, J.; Peukert, W.; Wirth, K.-E. Characterization of a downer reactor for particle rounding. Powder Technol. 2017, 316, 357–366. [Google Scholar] [CrossRef]
- Fanselow, S.; Emamjomeh, S.E.; Wirth, K.E.; Schmidt, J.; Peukert, W. Production of spherical wax and polyolefin microparticles by melt emulsification for additive manufacturing. Chem. Eng. Sci. 2016, 141, 282–292. [Google Scholar] [CrossRef]
- Kloos, S.; Peukert, W.; Schmidt, J. Production of polymer agglomerates by spray drying. In Proceedings of the 6th International Conference on Additive Technologies, Nürnberg, Germany, 29–30 November 2016; pp. 147–151. [Google Scholar]
- Mys, N.; Verberckmoes, A.; Cardon, L. Processing of Syndiotactic Polystyrene to Microspheres for Part Manufacturing through Selective Laser Sintering. Polymers 2016, 8, 383. [Google Scholar] [CrossRef]
- Kloos, S.; Dechet, M.A.; Peukert, W.; Schmidt, J. Production of spherical semi-crystalline polycarbonate microparticles for Additive Manufacturing by liquid-liquid phase separation. Powder Technol. 2018, 335, 275–284. [Google Scholar] [CrossRef]
- Wang, S.-J.; Liu, J.-Y.; Chu, L.-Q.; Zou, H.; Zhang, S.-J.; Wu, C.-J. Preparation of polypropylene microspheres for selective laser sintering via thermal-induced phase separation: Roles of liquid-liquid phase separation and crystallization. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 320–329. [Google Scholar] [CrossRef]
- Drummer, D.; Wudy, K.; Kühnlein, F.; Drexler, M. Polymer Blends for Selective Laser Sintering: Material and Process Requirements. Phys. Procedia 2012, 39, 509–517. [Google Scholar] [CrossRef]
- Salmoria, G.V.; Leite, J.L.; Ahrens, C.H.; Lago, A.; Pires, A.T.N. Rapid manufacturing of PA/HDPE blend specimens by selective laser sintering: Microstructural characterization. Polym. Test. 2007, 26, 361–368. [Google Scholar] [CrossRef]
- Salmoria, G.V.; Leite, J.L.; Paggi, R.A.; Lago, A.; Pires, A.T.N. Selective laser sintering of PA12/HDPE blends: Effect of components on elastic/plastic behavior. Polym. Test. 2008, 27, 654–659. [Google Scholar] [CrossRef]
- Salmoria, G.V.; Leite, J.L.; Vieira, L.F.; Pires, A.T.N.; Roesler, C.R.M. Mechanical properties of PA6/PA12 blend specimens prepared by selective laser sintering. Polym. Test. 2012, 31, 411–416. [Google Scholar] [CrossRef]
- Leite, J.L.; Salmoria, G.V.; Paggi, R.A.; Ahrens, C.H.; Pouzada, A.S. A study on morphological properties of laser sintered functionally graded blends of amorphous thermoplastics. Int. J. Mater. Prod. Technol. 2010, 39, 205. [Google Scholar] [CrossRef]
- Salmoria, G.V.; Lauth, V.R.; Cardenuto, M.R.; Magnago, R.F. Characterization of PA12/PBT specimens prepared by selective laser sintering. Opt. Laser Technol. 2018, 98, 92–96. [Google Scholar] [CrossRef]
- Greiner, S.; Wudy, K.; Lanzl, L.; Drummer, D. Selective laser sintering of polymer blends: Bulk properties and process behavior. Polym. Test. 2017, 64, 136–144. [Google Scholar] [CrossRef]
- Schultz, J.P.; Martin, J.P.; Kander, R.G.; Suchicital, C.T.A. Selective Laser Sintering of Nylon 12-PEEK Blends Formed by Cryogenic Mechanical Alloying. In Proceedings of the SFF Symposium, Austin, TX, USA, 7–9 August 2000; pp. 119–124. [Google Scholar]
- Martin, J.P.; Kander, R.G. Characterization of Mechanically Alloyed Polymer Blends for Selective Laser Sintering. In Proceedings of the SFF Symposium, Austin, TX, USA, 7–9 August 2000; pp. 92–99. [Google Scholar]
- Suryanarayana, C. Mechanical alloying and milling. Prog. Mater. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Smith, A.P.; Spontak, R.J.; Ade, H.; Smith, S.D.; Koch, C.C. High-Energy Cryogenic Blending and Compatibilizing of Immiscible Polymers. Adv. Mater. 1999, 11, 1277–1281. [Google Scholar] [CrossRef]
- Molina-Boisseau, S.; Le Bolay, N. Fine grinding of polymers in a vibrated bead mill. Powder Technol. 1999, 105, 321–327. [Google Scholar] [CrossRef]
- Schmidt, J.; Sachs, M.; Blümel, C.; Winzer, B.; Toni, F.; Wirth, K.-E.; Peukert, W. A novel process route for the production of spherical LBM polymer powders with small size and good flowability. Powder Technol. 2014, 261, 78–86. [Google Scholar] [CrossRef]
- Schmidt, J.; Gómez Bonilla, J.; Sachs, M.; Lanzl, L.; Wudy, K.; Wirth, K.-E.; Drummer, D.; Peukert, W. Production of polybutylene terephthalate glass composite powders and characterization for processing in selective laser sintering. In Proceedings of the 26th Annual International Solid Freeform Fabrication Symposium, Austin, TX, USA, 10–12 August 2015; pp. 989–997. [Google Scholar]
- Santos, J.M.R.C.A.; Guthrie, J.T. Polymer blends: The PC–PBT case. J. Mater. Chem. 2006, 16, 237–245. [Google Scholar] [CrossRef]
- Lin, G.-P.; Lin, L.; Wang, X.-L.; Chen, L.; Wang, Y.-Z. PBT/PC Blends Compatibilized and Toughened via Copolymers in Situ Formed by MgO-Catalyzed Transesterification. Ind. Eng. Chem. Res. 2015, 54, 1282–1291. [Google Scholar] [CrossRef]
- Schmid, M. Laser Sintering with Plastics; Carl Hanser Verlag GmbH & Co. KG: München, Germany, 2018; ISBN 978-1-56990-683-5. [Google Scholar]
- Gath, C.; Drummer, D. Curcuit board application to additive manufactured components by laser-direct-structuring. In Proceedings of the 2016 12th International Congress Molded Interconnect Devices (MID), Würzburg, Germany, 28–29 September 2016; pp. 1–6. [Google Scholar]
- Pompe, G.; Häußler, L. Investigations of transesterification in PC/PBT melt blends and the proof of immiscibility of PC and PBT at completely suppressed transesterification. J. Polym. Sci. Part B Polym. Phys. 1997, 35, 2161–2168. [Google Scholar] [CrossRef]
- Hopfe, I.; Pompe, G.; Eichhorn, K.J.; Häußler, L. FTIR spectroscopy of PC/PBT melt blends: Influence of crystallite morphology and copolyester content. J. Mol. Struct. 1995, 349, 443–446. [Google Scholar] [CrossRef]
- Tattum, S.B.; Cole, D.; Wilkinson, A.N. Controlled Transesterification and Its Effects on Structure Development in Polycarbonate–Poly(Butylene Terephthalate) Melt Blends. J. Macromol. Sci. Part B 2000, 39, 459–479. [Google Scholar] [CrossRef]
- Hopfe, I.; Pompe, G.; Eichhorn, K.-J. Ordered structures and progressive transesterification in PC/PBT melt blends studied by FT i.r. spectroscopy combined with d.s.c. and n.m.r. Polymer 1997, 38, 2321–2327. [Google Scholar] [CrossRef]
- Wilkinson, A.N.; Cole, D.; Tattum, S.B. The effects of transesterification on structure development in PC-PBT blends. Polym. Bull. 1995, 35, 751–757. [Google Scholar] [CrossRef]
- Delimoy, D.; Bailly, C.; Devaux, J.; Legras, R. Morphological studies of polycarbonate-poly(butylene terephthalate) blends by transmission electron microscopy. Polym. Eng. Sci. 1988, 28, 104–112. [Google Scholar] [CrossRef]
- Devaux, J.; Godard, P.; Mercier, J.P.; Touillaux, R.; Dereppe, J.M. Bisphenol-A polycarbonate–poly(butylene terephthalate) transesterification. II. Structure analysis of the reaction products by IR and 1H and 13C NMR. J. Polym. Sci. Polym. Phys. Ed. 1982, 20, 1881–1894. [Google Scholar] [CrossRef]
- Schmidt, J.; Romeis, S.; Peukert, W. Production of PBT/PC particle systems by wet grinding. In Proceedings of the AIP Conference Proceedings, PPS 32, Lyon, France, 25–29 July 2016; Volume 1914, p. 050003. [Google Scholar]
- Abdellaoui, M.; Gaffet, E. The physics of mechanical alloying in a planetary ball mill: Mathematical treatment. Acta Metall. Mater. 1995, 43, 1087–1098. [Google Scholar] [CrossRef]
- Enayati, M.H.; Sadeghian, Z.; Salehi, M.; Saidi, A. The effect of milling parameters on the synthesis of Ni3Al intermetallic compound by mechanical alloying. Mater. Sci. Eng. A 2004, 375–377, 809–811. [Google Scholar] [CrossRef]
- Daiko, Y.; Schmidt, J.; Kawamura, G.; Romeis, S.; Segets, D.; Iwamoto, Y.; Peukert, W. Mechanochemically induced sulfur doping in ZnO via oxygen vacancy formation. Phys. Chem. Chem. Phys. 2017, 19, 13838–13845. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, P.P.; Manna, I.; Talapatra, S.; Pabi, S.K. Mathematical analysis of milling mechanics in a planetary ball mill. Mater. Chem. Phys. 2001, 68, 85–94. [Google Scholar] [CrossRef]
- Mio, H.; Kano, J.; Saito, F. Scale-up method of planetary ball mill. Chem. Eng. Sci. 2004, 59, 5909–5916. [Google Scholar] [CrossRef]
- Burmeister, C.; Titscher, L.; Breitung-Faes, S.; Kwade, A. Dry grinding in planetary ball mills: Evaluation of a stressing model. Adv. Powder Technol. 2018, 29, 191–201. [Google Scholar] [CrossRef]
- Sachs, M.; Schmidt, J.; Peukert, W.; Wirth, K.-E. Design and scale-up of a semi-industrial downer-reactor for the rounding of irregular polymer particles. In Proceedings of the AIP Conference Proceedings, Bandung, Indonesia, 27–28 July 2018; Volume 1713, p. 070003. [Google Scholar]
- Zhu, J.-X.; Yu, Z.-Q.; Jin, Y.; Grace, J.R.; Issangya, A. Cocurrent downflow circulating fluidized bed (downer) reactors—A state of the art review. Can. J. Chem. Eng. 1995, 73, 662–677. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.-X.; Bergougnou, M.A. Flow development in a gas-solids downer fluidized bed. Can. J. Chem. Eng. 1999, 77, 194–198. [Google Scholar] [CrossRef]
- Brown, G.M.; Butler, J.H. New method for the characterization of domain morphology of polymer blends using ruthenium tetroxide staining and low voltage scanning electron microscopy (LVSEM). Polymer 1997, 38, 3937–3945. [Google Scholar] [CrossRef]
- Trent, J.S. Ruthenium tetraoxide staining of polymers: New preparative methods for electron microscopy. Macromolecules 1984, 17, 2930–2931. [Google Scholar] [CrossRef]
- Wudy, K.; Drexler, M.; Lanzl, L.; Drummer, D. Analysis of time dependent thermal properties for high rates in selective laser sintering. Rapid Prototyp. J. 2018. [Google Scholar] [CrossRef]
- Golovoy, A.; Cheung, M.-F.; Carduner, K.R.; Rokosz, M.J. Control of transesterification in polyester blends. Polym. Eng. Sci. 1989, 29, 1226–1231. [Google Scholar] [CrossRef]
- Naito, M.; Hayakawa, O.; Nakahira, K.; Mori, H.; Tsubaki, J.I. Effect of particle shape on the particle size distribution measured with commercial equipment. Powder Technol. 1998, 100, 52–60. [Google Scholar] [CrossRef]
- Han, Q.; Setchi, R.; Evans, S.L. Characterisation and milling time optimisation of nanocrystalline aluminium powder for selective laser melting. Int. J. Adv. Manuf. Technol. 2017, 88, 1429–1438. [Google Scholar] [CrossRef]
- Trass, O.; Ramesh Koka, V.; Papachristodoulou, G. Particle Shapes Produced by Comminution in the Szego Mill. Part. Part. Syst. Charact. 1995, 12, 158–165. [Google Scholar] [CrossRef]
- Romeis, S.; Schmidt, J.; Peukert, W. Mechanochemical aspects in wet stirred media milling. Int. J. Miner. Process. 2016, 156, 24–31. [Google Scholar] [CrossRef]
- Arencón, D.; Velasco, J.I. Fracture toughness of polypropylene-based particulate composites. Materials 2009, 2, 2046–2094. [Google Scholar] [CrossRef]
- Titscher, L.; Breitung-Faes, S.; Kwade, A. Experimentelle Parameterstudie zur Trockenzerkleinerung in Planetenkugelmühlen. Chem. Ing. Tech. 2016, 88, 1524–1529. [Google Scholar] [CrossRef]
- Pabst, W.; Berthold, C.; Gregorová, E. Size and shape characterization of oblate and prolate particles. J. Eur. Ceram. Soc. 2007, 27, 1759–1762. [Google Scholar] [CrossRef]
- Hogg, R. A Spheroid Model for the Role of Shape in Particle Size Analysis. KONA Powder Part. J. 2015, 32, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Dadbakhsh, S.; Verbelen, L.; Vandeputte, T.; Strobbe, D.; Van Puyvelde, P.; Kruth, J.-P. Effect of Powder Size and Shape on the SLS Processability and Mechanical Properties of a TPU Elastomer. Phys. Procedia 2016, 83, 971–980. [Google Scholar] [CrossRef]
- Sachs, M.; Schmidt, J.; Toni, F.; Blümel, C.; Winzer, B.; Peukert, W.; Wirth, K.-E. Rounding of Irregular Polymer Particles in a Downer Reactor. Procedia Eng. 2015, 102, 542–549. [Google Scholar] [CrossRef]
- Meyer, K.; Zimmermann, I. Effect of glidants in binary powder mixtures. Powder Technol. 2004, 139, 40–54. [Google Scholar] [CrossRef]
- Schweiger, A.; Zimmermann, I. A new approach for the measurement of the tensile strength of powders. Powder Technol. 1999, 101, 7–15. [Google Scholar] [CrossRef]
- Califice, A.; Michel, F.; Dislaire, G.; Pirard, E. Influence of particle shape on size distribution measurements by 3D and 2D image analyses and laser diffraction. Powder Technol. 2013, 237, 67–75. [Google Scholar] [CrossRef]
- Mühlenweg, H.; Hirleman, E.D. Laser Diffraction Spectroscopy: Influence of Particle Shape and a Shape Adaptation Technique. Part. Part. Syst. Charact. 1998, 15, 163–169. [Google Scholar] [CrossRef]
- Agimelen, O.S.; Mulholland, A.J.; Sefcik, J. Modelling of artefacts in estimations of particle size of needle-like particles from laser diffraction measurements. Chem. Eng. Sci. 2017, 158, 445–452. [Google Scholar] [CrossRef]
- Pellow-Jarman, M.; Hendra, P.; Hetem, M.J. Poly(butylene terephthalate) polycarbonate transesterification: Monitoring its progress with Fourier transform Raman spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1995, 51, 2107–2116. [Google Scholar] [CrossRef]
- Lee, S.-N.; Stolarski, V.; Letton, A.; Laane, J. Studies of bisphenol-A–polycarbonate aging by Raman difference spectroscopy. J. Mol. Struct. 2000, 521, 19–24. [Google Scholar] [CrossRef]
- Maruyama, Y.; Kanematsu, W. Confocal volume in laser Raman microscopy depth profiling. J. Appl. Phys. 2011, 110, 103107. [Google Scholar] [CrossRef]
- Palmer, A.; Poulin-Dandurand, S.; Brisse, F. On the structure of poly(tetramethylene terephthalate). Structural, infrared, and Raman studies of three tetramethylene glycol dibenzoate derivatives, models for poly(tetramethylene terephthalate). Can. J. Chem. 1985, 63, 3079–3088. [Google Scholar] [CrossRef] [Green Version]
- Apai, G.; McKenna, W.P. Surface analysis of polycarbonate thin films by High-Resolution Electron Energy Loss Spectroscopy. Negative ion resonances and surface vibrations. Langmuir 1991, 7, 2266–2272. [Google Scholar] [CrossRef]
- Aikin, B.J.M.; Courtney, T.H. The kinetics of composite particle formation during mechanical alloying. Metall. Trans. A 1993, 24, 647–657. [Google Scholar] [CrossRef]
- Hsiao, B.S.; Wang, Z.; Yeh, F.; Gao, Y.; Sheth, K. Time-resolved X-ray studies of structure development in poly(butylene terephthalate) during isothermal crystallization. Polymer 1999, 40, 3515–3523. [Google Scholar] [CrossRef]
- Righetti, M.C.; Munari, A. Influence of branching on melting behavior and isothermal crystallization of poly(butylene terephthalate). Macromol. Chem. Phys. 1997, 198, 363–378. [Google Scholar] [CrossRef]
- Pillin, I.; Pimbert, S.; Feller, J.-F.; Levesque, G. Crystallization kinetics of poly(butylene terephthalate) (PBT): Influence of additives and free carboxylic acid chain ends. Polym. Eng. Sci. 2001, 41, 178–191. [Google Scholar] [CrossRef]
- Devaux, J.; Godard, P.; Mercier, J.P. Bisphenol-A polycarbonate–poly(butylene terephthalate) transesterification. IV. Kinetics and mechanism of the exchange reaction. J. Polym. Sci. Polym. Phys. Ed. 1982, 20, 1901–1907. [Google Scholar] [CrossRef]
- Van De Grampel, H.T. Blends and Alloys of Engineering Thermoplastics. Rapra Rev. Rep. 1991, 5, 132. [Google Scholar]
- Wilkinson, A.N.; Tattum, S.B.; Ryan, A.J. Melting, reaction and recrystallization in a reactive PC-PBT blend. Polymer 1997, 38, 1923–1928. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Pan, R.; Wunderlich, B. Thermal analysis of poly(butylene terephthalate) for heat capacity, rigid-amorphous content, and transition behavior. Die Makromol. Chem. 1988, 189, 2443–2458. [Google Scholar] [CrossRef]
- Laumer, T.; Stichel, T.; Amend, P.; Schmidt, M. Simultaneous laser beam melting of multimaterial polymer parts. J. Laser Appl. 2015, 27, S29204. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Milling Time/h | Vsheath/m3/h | x50,3/µm | Span/- |
|---|---|---|---|
| 10 | not rounded | 56.5 | 4.20 |
| 10 | 3 | 92.1 | 2.73 |
| 10 | 7 | 34.4 | 4.73 |
| 15 | not rounded | 51.5 | 3.76 |
| 15 | 3 | 126.2 | 3.54 |
| 15 | 7 | 21.5 | 6.29 |
| Sample | ΧPBT,SLS profile/% | ΧPBT,melt anneal/% |
|---|---|---|
| 1:1 PBT–PC comminuted | 23 | 13 |
| 1:1 PBT–PC rounded 3 m3/h | 28 | 8 |
| 1:1 PBT–PC rounded 7 m3/h | 26 | 4 |
| 1:1 PBT–PC comminuted (+MgO) | 20 | 4 |
| 1:1 PBT–PC rounded 3 m3/h (+MgO) | 26 | 0 |
| 1:1 PBT–PC rounded 7 m3/h (+MgO) | 29 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dechet, M.A.; Gómez Bonilla, J.S.; Lanzl, L.; Drummer, D.; Bück, A.; Schmidt, J.; Peukert, W. Spherical Polybutylene Terephthalate (PBT)—Polycarbonate (PC) Blend Particles by Mechanical Alloying and Thermal Rounding. Polymers 2018, 10, 1373. https://doi.org/10.3390/polym10121373
Dechet MA, Gómez Bonilla JS, Lanzl L, Drummer D, Bück A, Schmidt J, Peukert W. Spherical Polybutylene Terephthalate (PBT)—Polycarbonate (PC) Blend Particles by Mechanical Alloying and Thermal Rounding. Polymers. 2018; 10(12):1373. https://doi.org/10.3390/polym10121373
Chicago/Turabian StyleDechet, Maximilian A., Juan S. Gómez Bonilla, Lydia Lanzl, Dietmar Drummer, Andreas Bück, Jochen Schmidt, and Wolfgang Peukert. 2018. "Spherical Polybutylene Terephthalate (PBT)—Polycarbonate (PC) Blend Particles by Mechanical Alloying and Thermal Rounding" Polymers 10, no. 12: 1373. https://doi.org/10.3390/polym10121373