Synthesis and Structural Characterization of a Series of One-Dimensional Heteronuclear Dirhodium-Silver Coordination Polymers

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Physical Measurements
2.3. Crystallography
2.4. Synthesis
2.4.1. Synthesis of [Rh2(μ-O2CPh)4(OEt2)2] (1a) and [Rh2(μ-O2CPh)4(OCMe2)2] (1b)
2.4.2. Synthesis of [Rh2(μ-O2CCH2OEt)4(HO2CCH2OEt)2] (2a) and [Rh2(μ-O2CCH2OEt)4(THF)2] (2b and 2b′)
2.4.3. Synthesis of (PPh4)n[Rh2(μ-O2CMe)4Ag(CN)2]n (3a) and {(PPh4)[Rh2(μ-O2CMe)4Ag(CN)2]·2CH2Cl2}n (3b)
2.4.4. Synthesis of (PPh4)n[Rh2(μ-O2CPh)4Ag(CN)2]n (4)
2.4.5. Synthesis of (PPh4)n[Rh2(μ-O2CCH2OEt)4Ag(CN)2]n (5)
2.4.6. Synthesis of (PPh4)2{Rh2(μ-O2CCMe3)4[Ag(CN)2]2} (6)
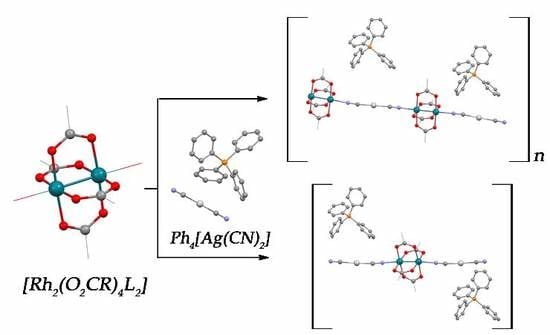
3. Results and Discussion
3.1. Synthesis and Spectroscopic Characterization
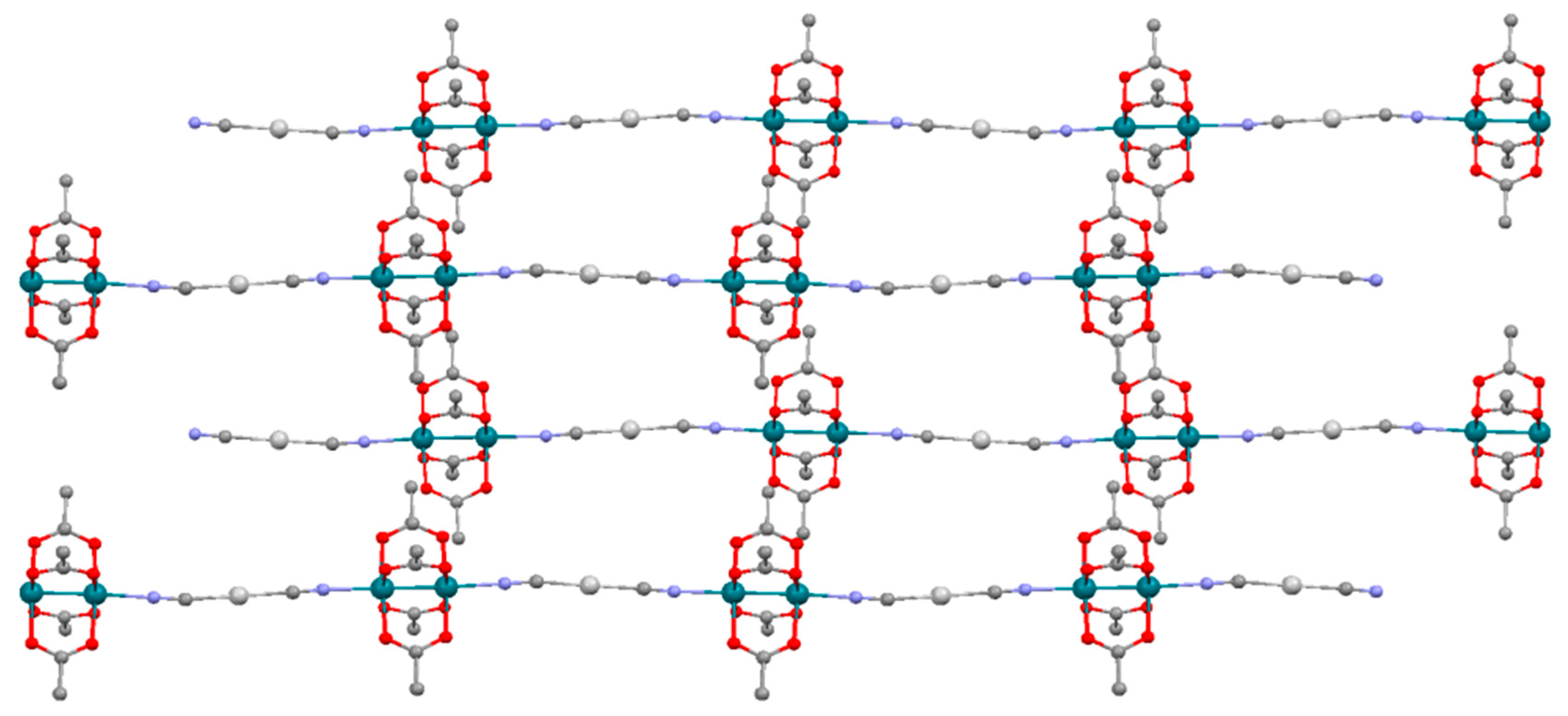
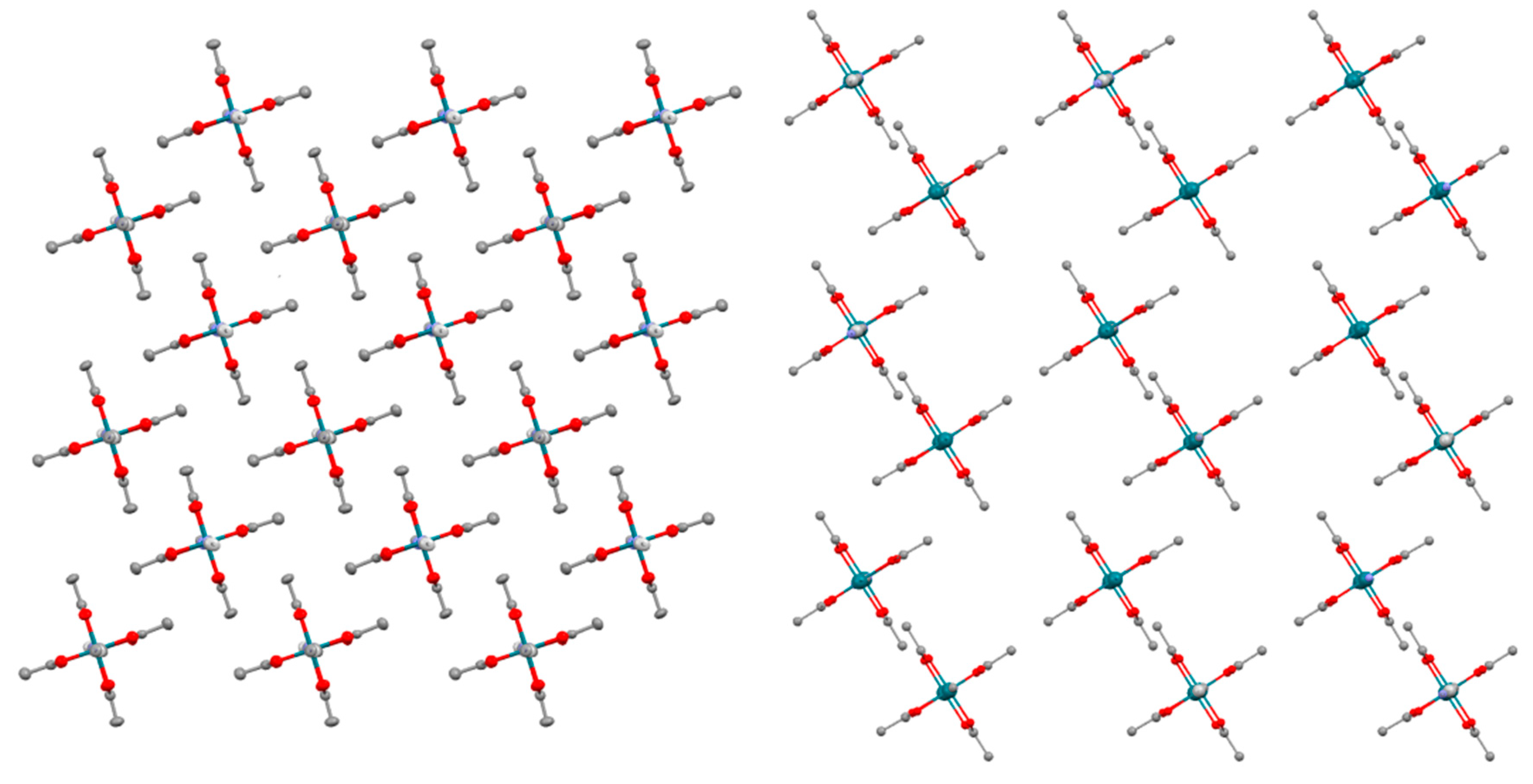
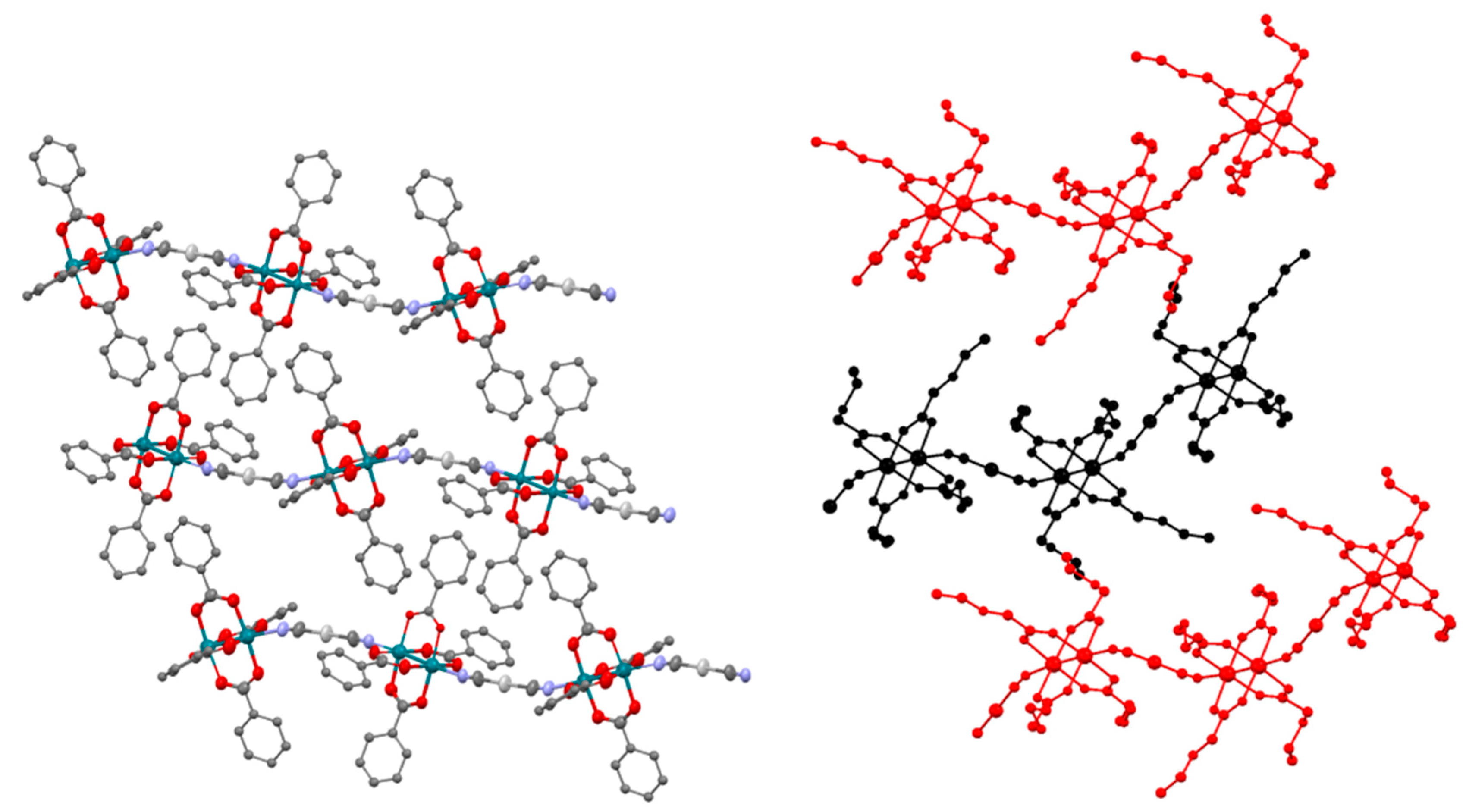
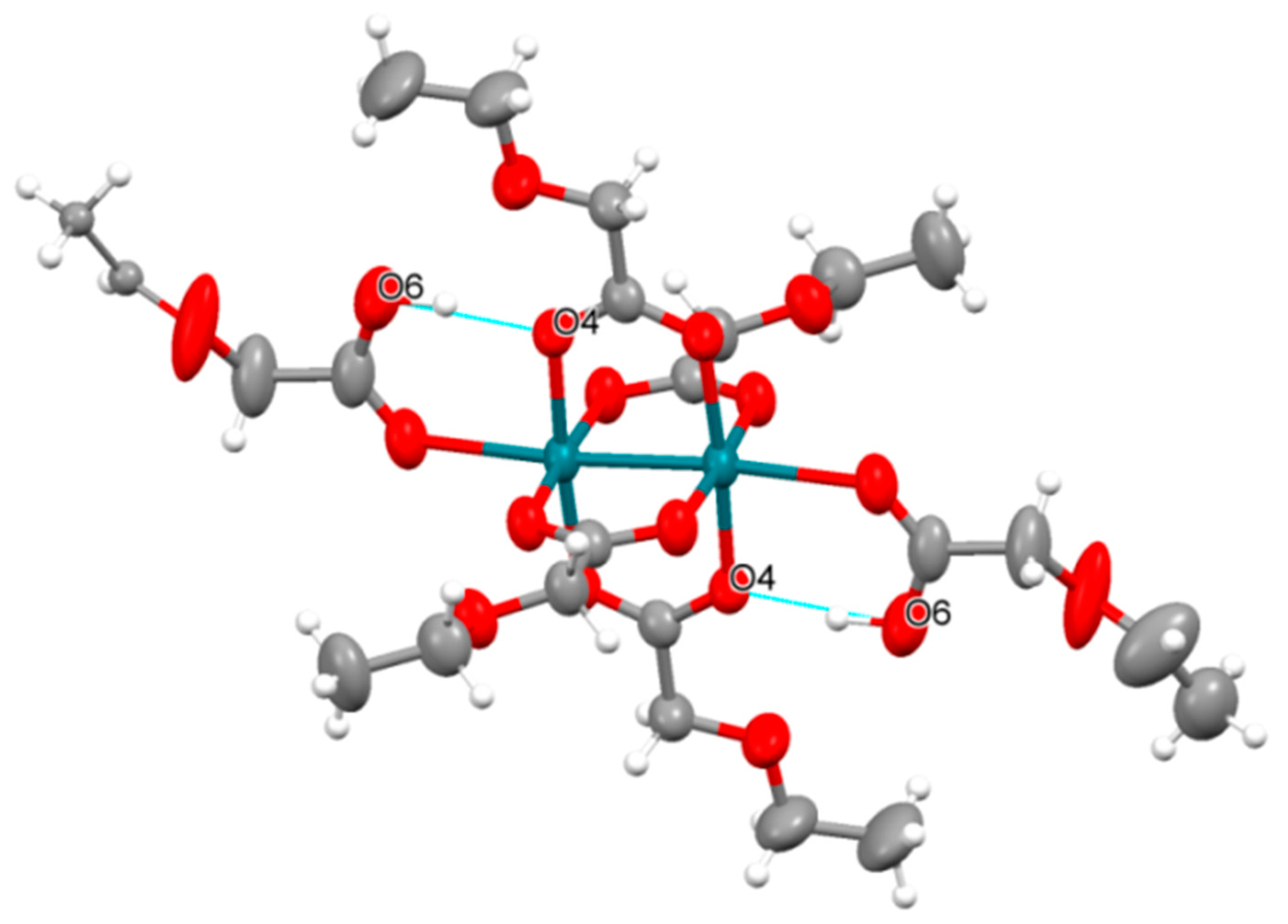
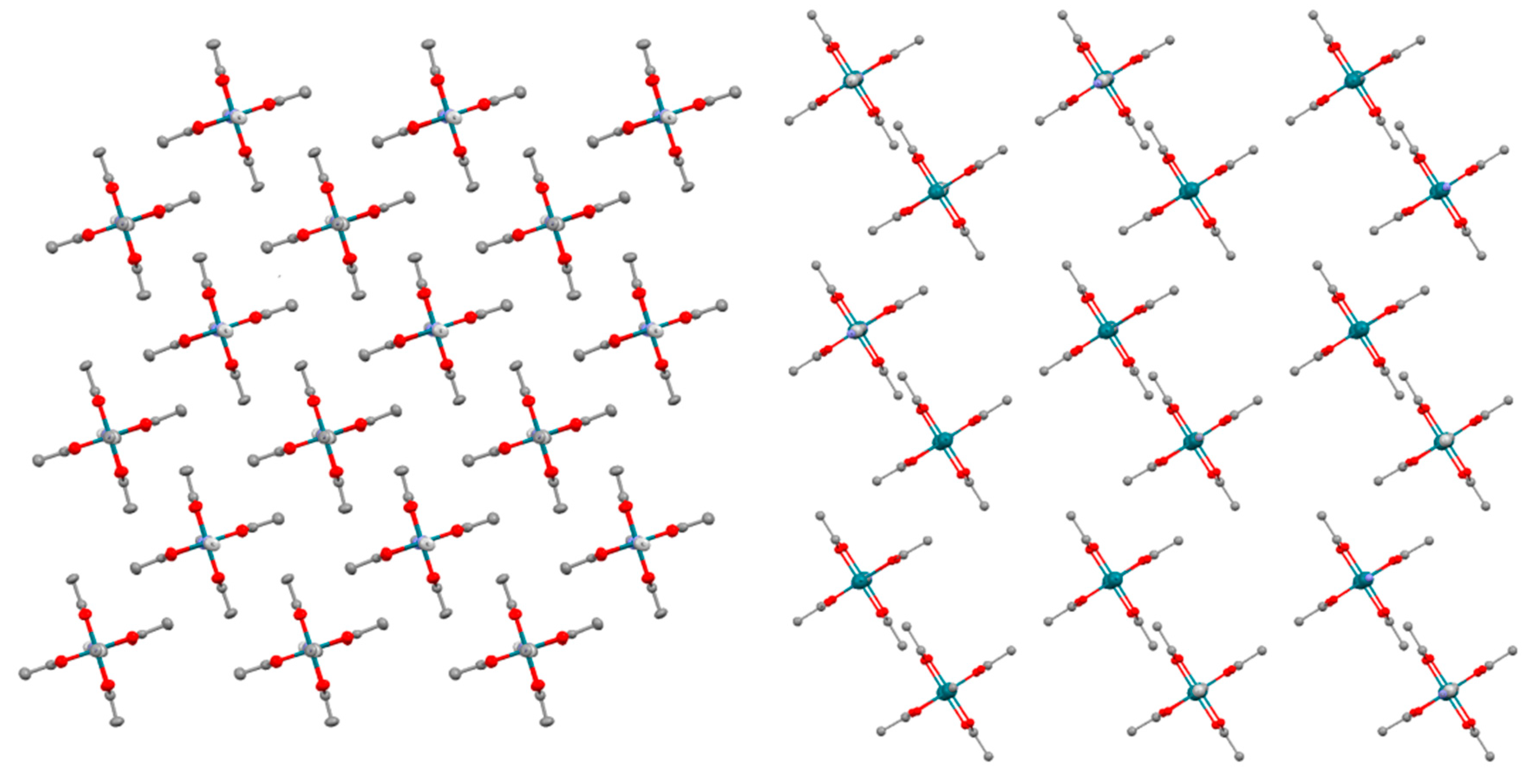
3.2. Crystal Structures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cotton, F.A.; Walton, R.A. Multiple Bonds between Metal Atoms, 2nd ed.; Wiley: New York, NY, USA, 1982. [Google Scholar]
- Cotton, F.A.; Murillo, C.; Walton, R.A. Multiple Bonds between Metal Atoms, 3rd ed.; Springer: New York, NY, USA, 2005. [Google Scholar]
- Liddle, S.T. (Ed.) Molecular Metal-Metal Bonds: Compounds, Synthesis, Properties; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Liao, K.; Liu, W.; Niemeyer, Z.L.; Ren, Z.; Bacsa, J.; Musaev, D.G.; Sigman, M.S.; Davies, H.M.L. Site-Selective Carbene-Induced C–H Functionalization Catalyzed by DirhodiumTetrakis(triarylcyclopropanecarboxylate) Complexes. ACS Catal. 2018, 8, 678–682. [Google Scholar] [CrossRef]
- Adly, F.G. On the Structure of Chiral Dirhodium(II) Carboxylate Catalysts: Stereoselectivity Relevance and Insights. Catalysts 2017, 7, 347. [Google Scholar] [CrossRef]
- Berry, J.F. Metal–metal multiple bonded intermediates in catalysis. J. Chem. Sci. 2015, 127, 209–214. [Google Scholar] [CrossRef]
- Hansen, J.; Davies, H.M.L. High symmetry dirhodium(II) paddlewheel complexes as chiral catalysts. Coord. Chem. Rev. 2008, 252, 545–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohata, J.; Ball, T.Z. Rhodium at the chemistry–biology interface. Dalton Trans. 2018, 47, 14855–14860. [Google Scholar] [CrossRef] [PubMed]
- Jalilehvand, F.; Enriquez Garcia, A.; Niksirat, P. Reactions of Antitumor Active Dirhodium(II) Tetraacetate Rh2(CH3COO)4 with Cysteine and Its Derivatives. ACS Omega 2017, 2, 6174–6186. [Google Scholar] [CrossRef]
- Knoll, J.D.; Turro, C. Control and utilization of ruthenium and rhodium metal complex excited states for photoactivated cancer therapy. Coord. Chem. Rev. 2015, 282–283, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Peña, B.; Barhoumi, R.; Burghardt, R.C.; Turro, C.; Dunbar, K.R. Confocal Fluorescence Microscopy Studies of a Fluorophore-Labeled Dirhodium Compound: Visualizing Metal–Metal Bonded Molecules in Lung Cancer (A549) Cells. J. Am. Chem. Soc. 2014, 136, 7861–7864. [Google Scholar] [CrossRef]
- Leung, C.-H.; Zhong, H.J.; Chan, D.S.H.; Ma, D.L. Bioactive iridium and rhodium complexes as therapeutic agents. Coord. Chem. Rev. 2013, 257, 1764–1776. [Google Scholar] [CrossRef]
- Sarkar, M.; Daw, P.; Ghatak, T.; Bera, J.K. Amide-Functionalized Naphthyridines on a RhII–RhII Platform: Effect of Steric Crowding, Hemilability, and Hydrogen-Bonding Interactions on the Structural Diversity and Catalytic Activity of Dirhodium(II) Complexes. Chem. Eur. J. 2014, 20, 16537–16549. [Google Scholar] [CrossRef]
- Amo-Ochoa, P.; Jiménez-Aparicio, R.; Perles, J.; Torres, M.R.; Gennari, M.; Zamora, F. Structural Diversity in Paddlewheel Dirhodium(II) Compounds through Ionic Interactions: Electronic and Redox Properties. Cryst. Growth Des. 2013, 13, 4977–4985. [Google Scholar] [CrossRef]
- Cotton, F.A.; Dikarev, E.V.; Petrukhina, M.A.; Schmitz, M.; Stang, P.J. Supramolecular Assemblies of Dimetal Complexes with Polydentate N-Donor Ligands: From a Discrete Pyramid to a 3D Channel Network. Inorg. Chem. 2002, 41, 2903–2908. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yano, N.; Shimodaira, T.; Yan, Y.-N.; Yamasaki, M.; Tanaka, H.; Omata, K.; Kawamoto, T.; Handa, M. Paddlewheel-Type Dirhodium Tetrapivalate Based Coordination Polymer: Synthesis, Characterization, and Self-Assembly and Disassembly Transformation Properties. Eur. J. Inorg. Chem. 2016, 2810–2815. [Google Scholar] [CrossRef]
- Fritsch, N.; Wick, C.R.; Waidmann, T.; Dral, P.O.; Tucher, J.; Heinemann, F.W.; Shubina, T.E.; Clark, T.; Burzlaff, N. Multiply Bonded Metal(II) Acetate (Rhodium, Ruthenium, and Molybdenum) Complexes with the trans-1,2-Bis(N-methylimidazol-2-yl)ethylene Ligand. Inorg. Chem. 2014, 53, 12305–12314. [Google Scholar] [CrossRef] [PubMed]
- Dikarev, E.V.; Shpanchenko, R.V.; Andreini, K.W.; Block, E.; Jin, J.; Petrukhina, M.A. Powder Diffraction Study of a Coordination Polymer Comprised of Rigid Building Blocks: [Rh2(O2CCH3)4‚µ2-Se2C5H8-Se,Se′]∞. Inorg. Chem. 2004, 43, 5558–5563. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, S.-J.; Lough, A.J. New dirhodium(II,II) carboxylates with 2,6-bis(N-1,2,4-triazolyl)pyridinato ligand (btp). Polyhedron 2001, 20, 3073–3078. [Google Scholar] [CrossRef]
- Gonzalez-Belman, O.F.; Yazmín Varela, Y.; Flores-Álamo, M.; Wrobel, K.; Gutierrez-Granados, S.; Peralta-Hernández, J.M.; Jiménez-Halla, J.O.C.; Serrano, O. Microwave-Assisted Synthesis and Characterization of [Rh2(OAc)4(L)2] Paddlewheel Complexes: A Joint Experimental and Computational Study. Int. J. Inorg. Chem. 2017, 2017, 5435436. [Google Scholar] [CrossRef]
- Ye, Q.-S.; Li, X.-N.; Jin, Y.; Yu, J.; Chang, Q.-W.; Jiang, J.; Yan, C.-X.; Li, J.; Liu, W.-P. Synthesis, crystal structures and catalytic activity of tetrakis(acetato)dirhodium(II) complexes with axial picoline ligands. Inorg. Chim. Acta 2015, 434, 113–120. [Google Scholar] [CrossRef]
- Cmoch, P.; Głaszczka, R.; Jaźwiński, J.; Kamieńskia, B.; Senkara, E. Adducts of nitrogenous ligands with rhodium(II) tetracarboxylates and tetraformamidinate: NMR spectroscopy and density functional theory calculations. Magn. Reson. Chem. 2014, 52, 61–68. [Google Scholar] [CrossRef]
- Heyduk, A.F.; Krodel, D.J.; Meyer, E.E.; Nocera, D.G. A Luminescent Heterometallic Dirhodium−Silver Chain. Inorg. Chem. 2002, 41, 634–636. [Google Scholar] [CrossRef]
- Uemura, K. One-dimensional complexes extended by unbridged metal–metal bonds based on a HOMO–LUMO interaction at the dz2 orbital between platinum and heterometal atoms. Dalton Trans. 2017, 46, 5474–5492. [Google Scholar] [CrossRef]
- Dikarev, E.V.; Andreini, K.W.; Petrukhina, M.A. On the Road to a Termolecular Complex with Acetone: A Heterometallic Supramolecular Network {[Rh2(O2CCF3)4]·μ2-OCMe2·[Cu4(O2CCF3)4]}. Inorg. Chem. 2004, 43, 3219–3224. [Google Scholar] [CrossRef] [PubMed]
- Uemura, K.; Ebihara, M. One-Dimensionally Extended Paddlewheel Dirhodium Complexes from Metal–Metal Bonds with Diplatinum Complexes. Inorg. Chem. 2011, 50, 7919–7921. [Google Scholar] [CrossRef] [PubMed]
- Uemura, K.; Ebihara, M. Paramagnetic One-Dimensional Chains Comprised of Trinuclear Pt–Cu–Pt and Paddlewheel Dirhodium Complexes with Metal–Metal Bonds. Inorg. Chem. 2013, 52, 5535–5550. [Google Scholar] [CrossRef] [PubMed]
- Uemura, K.; Kanbara, T.; Ebihara, M. Two Types of Heterometallic One-Dimensional Alignment Composed of Acetamidate-Bridged Dirhodium and Pivalamidate-Bridged Diplatinum Complexes. Inorg. Chem. 2014, 53, 4621–4628. [Google Scholar] [CrossRef]
- Uemura, K.; Yamada, T.; Kanbara, T.; Ebihara, M. Acetamidate-bridged paddlewheel dirhodium complex sandwiched by mononuclear platinum complexes with axial metal–metal bonds affording neutral heterometallic one-dimensional alignments. Inorg. Chim. Acta 2015, 424, 194–201. [Google Scholar] [CrossRef]
- Yamada, T.; Ebihara, M.; Uemura, K. Heterometallic one-dimensional chain with tetradeca metal repetition constructed by amidate bridged dirhodium and pivalate bridged diplatinum complexes influenced by hydrogen bonding. Dalton Trans. 2016, 45, 12322–12328. [Google Scholar] [CrossRef]
- Uemura, K. Magnetic behavior in heterometallic one-dimensional chains or octanuclear complex regularly aligned with metal-metal bonds as –Rh-Rh-Pt-Cu-Pt. J. Mol. Struct. 2018, 1162, 31–36. [Google Scholar] [CrossRef]
- Amo-Ochoa, P.; Delgado, S.; Gallego, A.; Gómez-García, C.J.; Jiménez-Aparicio, R.; Martínez, G.; Perles, J.; Rosario Torres, M.R. Structure and Properties of One-Dimensional Heterobimetallic Polymers Containing Dicyanoaurate and Dirhodium(II) Fragments. Inorg. Chem. 2012, 51, 5844–5849. [Google Scholar] [CrossRef]
- Dance, I.; Scudder, M. Supramolecular Motifs: Concerted Multiple Phenyl Embraces between Ph4P+ Cations Are Attractive and Ubiquitous. Chem. Eur. J. 1996, 2, 481–486. [Google Scholar] [CrossRef]
- Dance, I.; Scudder, M. Concerted supromolecular motifs: Linear columns and zigzag chains of multiple phenyl embraces involving PPh4P+ cations in crystals. J. Chem. Soc. Dalton Trans. 1996, 19, 3755–3769. [Google Scholar] [CrossRef]
- Scudder, M.; Dance, I. Sixfold phenyl embraces with substituted phenyl in PPh3. J. Chem. Soc. Dalton Trans. 2000, 17, 2909–2915. [Google Scholar] [CrossRef]
- Dance, I.; Scudder, M. Crystal supramolecularity: Elaborate six-, eight- and twelve-fold concerted phenyl embraces in compounds [M(PPh3)3]z and [M(PPh3)4]z. New J. Chem. 1998, 22, 481–492. [Google Scholar] [CrossRef]
- Ali, B.; Dance, I.; Scudder, M.; Craig, D. Dimorphs of (Ph4P)2[Cd2(SPh)6]: Crystal Packing Analyses and the Interplay of Intermolecular and Intramolecular Energies. Cryst. Growth Des. 2002, 2, 601–607. [Google Scholar] [CrossRef]
- Janssen, F.F.B.J.; de Gelder, R.; Rowan, A.E. 2. The Multiple Phenyl Embrace as a Synthon in Cu(I)/PPh3/N-Donor Ligand Coordination Polymers. Cryst. Growth Des. 2011, 11, 4326–4333. [Google Scholar] [CrossRef]
- Zaręba, J.K.; Białek, M.J.; Janczak, J.; Zoń, J.; Dobosz, A. Extending the Family of Tetrahedral Tectons: Phenyl Embraces in Supramolecular Polymers of Tetraphenylmethane-based Tetraphosphonic Acid Templated by Organic Bases. Cryst. Growth Des. 2014, 14, 6143–6153. [Google Scholar] [CrossRef]
- Cotton, F.A.; Felthouse, T.R. Structural studies of three tetrakis(carboxylato)dirhodium(II) adducts in which carboxylate groups and axial ligands are varied. Inorg. Chem. 1980, 19, 323–328. [Google Scholar] [CrossRef]
- Mikuriya, M.; Yamamoto, J.; Ishida, H.; Yoshioka, D.; Handa, M. Preparation and Crystal Structure of Tetrakis(μ-pivalato-O,O′)bis[(pivalic acid-O)rhodium(II)]. X-Ray Struct. Anal. Online 2011, 27, 7–8. [Google Scholar] [CrossRef]
- Barral, M.C.; Jiménez-Aparicio, R.; Priego, J.L.; Royer, E.C.; Saucedo, M.J.; Urbanos, F.A.; Amador, U. Non-polymeric diruthenium(II,III) carboxylates. Crystal structures of [Ru2Cl(µ-O2CCMe3)4(H2O)] and [Ru2Cl(µ-O2CCHMe2)4(thf)] (thf = tetrahydrofuran). J. Chem. Soc. Dalton Trans. 1995, 13, 2183–2187. [Google Scholar] [CrossRef]
- Masaaki, A.; Yoichi, S.; Tadashi, Y.; Tasuku, I. X-Ray Structure, Ligand Substitution, and Other Properties of Trinuclear Ruthenium Complex, [Ru3(μ3-O)(μ-C6H5COO)6(py)3](PF6) (py = pyridine), and X-Ray Structure of Dinuclear Ruthenium Complex, [Ru2(μ-C6H5COO)4Cl]. Bull. Chem. Soc. Jpn. 1992, 65, 1585–1590. [Google Scholar] [CrossRef]
- Bino, A.; Cotton, F.A.; Felthouse, T.R. Structural studies of some multiply bonded dirutheniumtetracarboxylate compounds. Inorg. Chem. 1979, 18, 2599–2604. [Google Scholar] [CrossRef]
- Barral, M.C.; Jiménez-Aparicio, R.; Priego, J.L.; Royer, E.C.; Urbanos, F.A.; Amador, U. Diruthenium(II,III) Carboxylate Compounds: Existence of both Polymeric and Ionic Forms in Solution and Solid State. Inorg. Chem. 1998, 37, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Naito, S.; Tanibe, T.; Saito, E.; Miyao, T.; Moriet, W. A Novel Reaction Pathway in Olefin-Deuterium Reaction inside the Microporous of Rh(II) Dicarboxylate Polymer Complexes. Chem. Lett. 2001, 30, 1178–1189. [Google Scholar] [CrossRef]
- Nukada, R.; Mori, W.; Takamizawa, S.; Mikuriya, M.; Handa, M.; Naono, H. Microporous Structure of a Chain Compound of Copper(II) Benzoate Bridged by Pyrazine. Chem. Lett. 1999, 28, 367–368. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1b | 2a | 2b | 2b′ | 3a | |
|---|---|---|---|---|---|
| Formula | C34H32 O10Rh2 | C24H42 O18Rh2 | C24H44O14Rh2 | C34H32AgN2 O8PRh2 | |
| fw | 806.41 | 824.40 | 762.41 | 941.28 | |
| Space group | C2/c | P21/c | P-1 | P21/c | P21/n |
| a/Å | 18.873(3) | 8.1426(19) | 8.687(5) | 13.5323(13) | 11.7168(8) |
| b/Å | 15.669(3) | 24.685(6) | 13.558(5) | 15.0682(15) | 26.3945(17) |
| c/Å | 23.988(4) | 8.4475(19) | 14.312(5) | 8.0473(8) | 11.7290(8) |
| α/° | 71.929(5) | ||||
| β/° | 103.999(15) | 99.906(4) | 84.838(5) | 98.481(2) | 93.595(1) |
| α/° | 86.272(5) | ||||
| V/Å3 | 6883(2) | 1672.7(7) | 1594.8(12) | 1623.0(3) | 3620.2(4) |
| Z | 8 | 2 | 2 | 2 | 4 |
| d calc/g·cm−3 | 1.556 | 1.637 | 1.588 | 1.560 | 1.727 |
| μ/mm−1 | 1.013 | 1.060 | 1.096 | 1.077 | 1.531 |
| R indices | R1 = 0.0491 wR2 = 0.1590 | R1 = 0.0402 wR2 = 0.1002 | R1 = 0.0393 wR2 = 0.1093 | R1 = 0.0335 wR2 = 0.0900 | R1 = 0.0494 wR2 = 0.1687 |
| GooF on F2 | 1.031 | 0.997 | 0.998 | 0.997 | 0.999 |
| 3b | 4 | 5 | 6 | |
|---|---|---|---|---|
| Formula | C36H36AgCl4N2O8PRh2 | C54H40Ag N2O8PRh2 | C42H48Ag N2O12PRh2 | C72H75Ag2 N4O8P2Rh2 |
| fw | 1111.13 | 1189.54 | 1117.48 | 1607.86 |
| Space group | P-1 | P-1 | P21/c | Pnma |
| a/Å | 13.1180(12) | 12.2802(9) | 13.1982(13) | 22.016(4) |
| b/Å | 13.6197(13) | 13.3534(9) | 22.201(2) | 33.450(2) |
| c/Å | 13.6949(13) | 17.3652(12) | 16.9090(17) | 11.691(6) |
| α/° | 87.033(2) | 83.105(1) | ||
| β/° | 78.278(2) | 83.373(1) | 111.788(2) | |
| α/° | 79.928(2) | 64.333(1) | ||
| V/Å3 | 2358.5(4) | 2541.6(3) | 4600.6(8) | 8610(5) |
| Z | 2 | 2 | 4 | 4 |
| d calc/g·cm−3 | 1.565 | 1.554 | 1.613 | 1.240 |
| μ/mm−1 | 1.407 | 1.109 | 1.225 | 0.907 |
| R indices | R1 = 0.0396 wR2 = 0.1187 | R1 = 0.0587 wR2 = 0.1881 | R1 = 0.0379 wR2 = 0.1040 | R1 = 0.0686 wR2 = 0.1648 |
| GooF on F2 | 0.995 | 1.008 | 1.042 | 0.993 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, P.; Fernandez-Bartolome, E.; Cortijo, M.; Delgado-Martínez, P.; González-Prieto, R.; Priego, J.L.; Torres, M.R.; Jiménez-Aparicio, R. Synthesis and Structural Characterization of a Series of One-Dimensional Heteronuclear Dirhodium-Silver Coordination Polymers. Polymers 2019, 11, 111. https://doi.org/10.3390/polym11010111
Cruz P, Fernandez-Bartolome E, Cortijo M, Delgado-Martínez P, González-Prieto R, Priego JL, Torres MR, Jiménez-Aparicio R. Synthesis and Structural Characterization of a Series of One-Dimensional Heteronuclear Dirhodium-Silver Coordination Polymers. Polymers. 2019; 11(1):111. https://doi.org/10.3390/polym11010111
Chicago/Turabian StyleCruz, Paula, Estefania Fernandez-Bartolome, Miguel Cortijo, Patricia Delgado-Martínez, Rodrigo González-Prieto, José L. Priego, M. Rosario Torres, and Reyes Jiménez-Aparicio. 2019. "Synthesis and Structural Characterization of a Series of One-Dimensional Heteronuclear Dirhodium-Silver Coordination Polymers" Polymers 11, no. 1: 111. https://doi.org/10.3390/polym11010111