Nanochannel-Confined TAMRA-Polypyrrole Stained DNA Stretching by Varying the Ionic Strength from Micromolar to Millimolar Concentrations


 ,
,  ,
,
Abstract
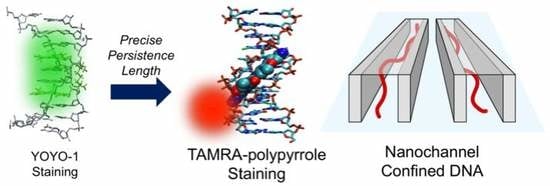
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of TAMRA-Polypyrrole
2.3. Flow Chamber
2.4. Preparation of T4 DNA for Nanochannel
2.5. DNA Loading into PDMS Nanochannels
2.6. Microscopy
2.7. Calculation of the Effective Diameter (w) of DNA
3. Results and Discussion
3.1. DNA Contour Length (L)
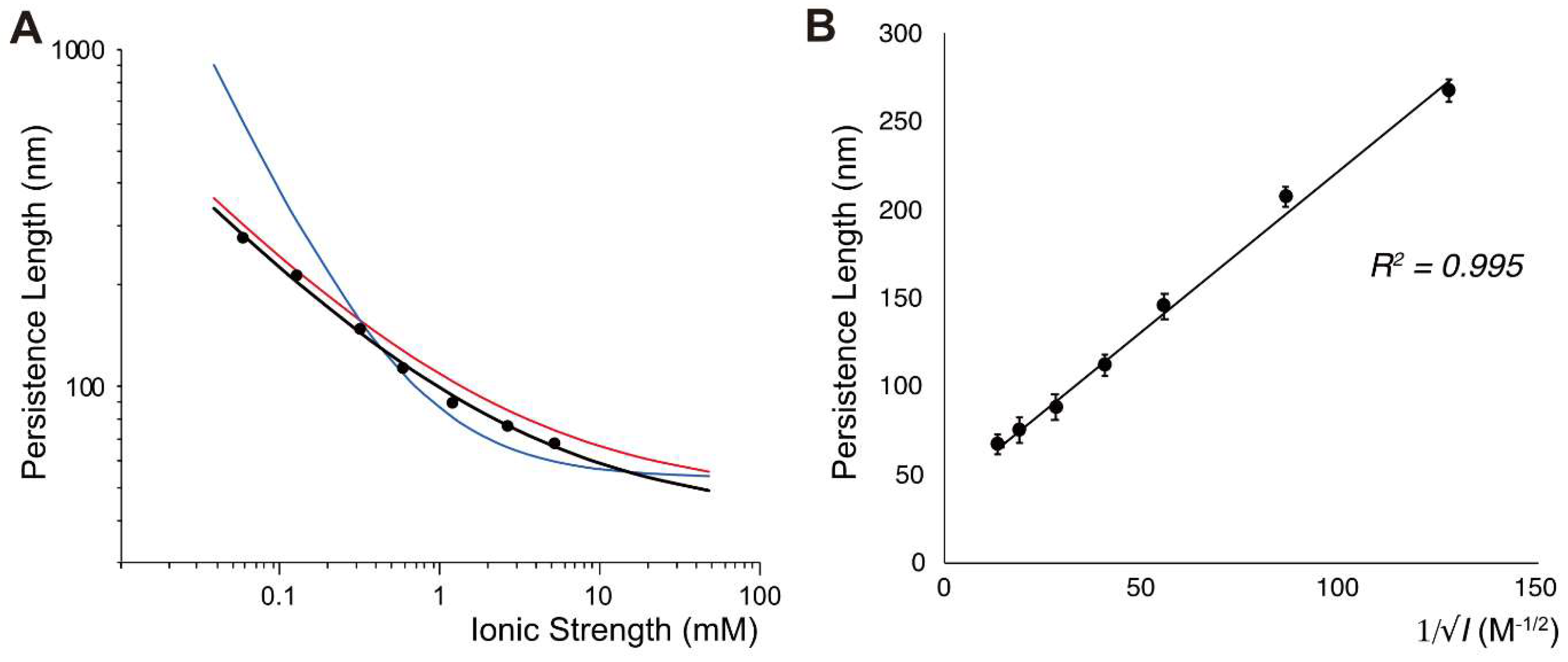
3.2. DNA Persistence Length (p)
3.3. TAMRA-Polypyrrole Stained DNA in Nanochannel
3.4. Weak-Confinement Regime
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Smith, S.B.; Cui, Y.J.; Bustamante, C. Overstretching B-DNA: The elastic response of individual double-stranded and single-stranded DNA molecules. Science 1996, 271, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.G.; Smith, S.B.; Bloomfield, V.A.; Bustamante, C. Ionic effects on the elasticity of single DNA molecules. Proc. Natl. Acad. Sci. USA 1997, 94, 6185–6190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, Y.; Lee, S.; Jo, K. Visualization of large elongated DNA molecules. Electrophoresis 2015, 36, 2057–2071. [Google Scholar] [CrossRef] [PubMed]
- Brochard, F.; de Gennes, P.G. Dynamics of confined polymer chains. J. Chem. Phys. 1977, 67, 52–56. [Google Scholar] [CrossRef]
- Odijk, T. On the Statistics and Dynamics of Confined or Entangled Stiff Polymers. Macromolecules 1983, 16, 1340–1344. [Google Scholar] [CrossRef]
- Tegenfeldt, J.O.; Prinz, C.; Cao, H.; Chou, S.; Reisner, W.W.; Riehn, R.; Wang, Y.M.; Cox, E.C.; Sturm, J.C.; Silberzan, P.; et al. The dynamics of genomic-length DNA molecules in 100-nm channels. Proc. Natl. Acad. Sci. USA 2004, 101, 10979–10983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisner, W.; Morton, K.J.; Riehn, R.; Wang, Y.M.; Yu, Z.N.; Rosen, M.; Sturm, J.C.; Chou, S.Y.; Frey, E.; Austin, R.H. Statics and dynamics of single DNA molecules confined in nanochannels. Phys. Rev. Lett. 2005, 94, 196101. [Google Scholar] [CrossRef]
- Jo, K.; Dhingra, D.M.; Odijk, T.; de Pablo, J.J.; Graham, M.D.; Runnheim, R.; Forrest, D.; Schwartz, D.C. A single-molecule barcoding system using nanoslits for DNA analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 2673–2678. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kim, K.S.; Kounovsky, K.L.; Chang, R.; Jung, G.Y.; Jo, K.; Schwartz, D.C. Nanochannel Confinement: DNA Stretch Approaching Full Contour Length. Lab Chip 2011, 11, 1721–1729. [Google Scholar] [CrossRef]
- Chang, R.; Jo, K. DNA conformation in nanochannels: Monte Carlo simulation studies using a primitive DNA model. J. Chem. Phys. 2012, 136, 095101. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.; Jeong, H.; Jung, G.Y.; Chang, R.; Chen, Y.L.; Jo, K. Nanoslit Confined DNA at Low Ionic Strengths. ACS Macro Lett. 2014, 3, 926–930. [Google Scholar] [CrossRef]
- Dorfman, K.D.; King, S.B.; Olson, D.W.; Thomas, J.D.P.; Tree, D.R. Beyond Gel Electrophoresis: Microfluidic Separations, Fluorescence Burst Analysis and DNA Stretching. Chem. Rev. 2013, 113, 2584–2667. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, S.M.; Zec, H.C.; Wang, T.H. Analysis of single nucleic acid molecules in micro- and nano-fluidics. Lab Chip 2016, 16, 790–811. [Google Scholar] [CrossRef] [Green Version]
- Rems, L.; Kawale, D.; Lee, L.J.; Boukany, P.E. Flow of DNA in micro/nanofluidics: From fundamentals to applications. Biomicrofluidics 2016, 10, 043403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rye, H.S.; Yue, S.; Wemmer, D.E.; Quesada, M.A.; Haugland, R.P.; Mathies, R.A.; Glazer, A.N. Stable fluorescent complexes of double-stranded DNA with bis-intercalating asymmetric cyanine dyes: Properties and applications. Nucleic Acids Res. 1992, 20, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Perkins, T.T.; Quake, S.R.; Smith, D.E.; Chu, S. Relaxation of a Single DNA Molecule Observed by Optical Microscopy. Science 1994, 264, 822–826. [Google Scholar] [CrossRef]
- Carlsson, C.; Jonsson, M.; Akerman, B. Double Bands in DNA Gel-Electrophoresis Caused by Bis-Intercalating Dyes. Nucleic Acids Res. 1995, 23, 2413–2420. [Google Scholar] [CrossRef]
- Gurrieri, S.; Wells, K.S.; Johnson, I.D.; Bustamante, C. Direct visualization of individual DNA molecules by fluorescence microscopy: Characterization of the factors affecting signal/background and optimization of imaging conditions using YOYO. Anal Biochem. 1997, 249, 44–53. [Google Scholar] [CrossRef]
- Murade, C.U.; Subramaniam, V.; Otto, C.; Bennink, M.L. Force spectroscopy and fluorescence microscopy of dsDNA-YOYO-1 complexes: Implications for the structure of dsDNA in the overstretching region. Nucleic Acids Res. 2010, 38, 3423–3431. [Google Scholar] [CrossRef]
- Tycon, M.A.; Dial, C.F.; Faison, K.; Melvin, W.; Fecko, C.J. Quantification of dye-mediated photodamage during single-molecule DNA imaging. Anal. Biochem. 2012, 426, 13–21. [Google Scholar] [CrossRef]
- Lee, S.; Oh, Y.; Lee, J.; Choe, S.; Lim, S.; Lee, H.S.; Jo, K.; Schwartz, D.C. DNA binding fluorescent proteins for the direct visualization of large DNA molecules. Nucleic Acids Res. 2016, 44, e6. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A.; Carlsson, C.; Jonsson, M.; Albinsson, B. Characterization of the binding of the fluorescent dyes YO and YOYO to DNA by polarized light spectroscopy. J. Am. Chem. Soc. 1994, 116, 8459–8465. [Google Scholar] [CrossRef]
- Akerman, B.; Tuite, E. Single- and double-strand photocleavage of DNA by YO, YOYO and TOTO. Nucleic Acids Res. 1996, 24, 1080–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundukad, B.; Yan, J.; Doyle, P.S. Effect of YOYO-1 on the mechanical properties of DNA. Soft Matter 2014, 10, 9721–9728. [Google Scholar] [CrossRef] [PubMed]
- Bakajin, O.B.; Duke, T.A.J.; Chou, C.F.; Chan, S.S.; Austin, R.H.; Cox, E.C. Electrohydrodynamic stretching of DNA in confined environments. Phys. Rev. Lett. 1998, 80, 2737–2740. [Google Scholar] [CrossRef]
- Günther, K.; Mertig, M.; Seidel, R. Mechanical and structural properties of YOYO-1 complexed DNA. Nucleic Acids Res. 2010, 38, 6526–6532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Wang, C.; Song, J.; Kim, D.; Oh, Y.; Ko, W.; Lee, J.; Park, J.; Lee, H.S.; Jo, K. Investigation of various fluorescent protein-DNA binding peptides for effectively visualizing large DNA molecules. RSC Adv. 2016, 6, 46291–46298. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.; Won, N.; Shin, E.; Kim, S.-H.; Chun, M.-Y.; Gu, J.; Jung, G.-Y.; Lim, K.-I.; Jo, K. Single-molecule DNA visualization using AT-specific red and non-specific green DNA-binding fluorescent proteins. Analyst 2019. [Google Scholar] [CrossRef]
- Lee, S.; Kawamoto, Y.; Vaijayanthi, T.; Park, J.; Bae, J.; Kim-Ha, J.; Sugiyama, H.; Jo, K. TAMRA-polypyrrole for A/T sequence visualization on DNA molecules. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Dobrynin, A.V. Effect of counterion condensation on rigidity of semiflexible polyelectrolytes. Macromolecules 2006, 39, 9519–9527. [Google Scholar] [CrossRef]
- Odijk, T. Scaling theory of DNA confined in nanochannels and nanoslits. Phys. Rev. E 2008, 77, 060901. [Google Scholar] [CrossRef] [PubMed]
- Daoud, M.; de Gennes, P.G. Statistics of Macromolecular Solutions Trapped in Small Pores. J. Phys. Paris 1977, 38, 85–93. [Google Scholar] [CrossRef]
- Lee, S.; Jo, K. Visualization of Surface-tethered Large DNA Molecules with a Fluorescent Protein DNA Binding Peptide. J. Vis. Exp. 2016, 112, e54141. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, H.S.; Lim, S.; Jo, K. Visualization of UV-induced damage on single DNA molecules. Chem. Commun. 2013, 49, 4740–4742. [Google Scholar] [CrossRef] [PubMed]
- Stigter, D. Interactions of highly charged colloidal cylinders with applications to double-stranded. Biopolymers 1977, 16, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Stigter, D. Charged Colloidal Cylinder with a Gouy Double Layer. J. Colloid Interface Sci. 1975, 53, 296–306. [Google Scholar] [CrossRef]
- Schellman, J.A.; Stigter, D. Electrical double layer, zeta potential and electrophoretic charge of double-stranded DNA. Biopolymers 1977, 16, 1415–1434. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.; Marko, J.; Siggia, E.; Smith, S. Entropic elasticity of lambda-phage DNA. Science 1994, 265, 1599–1600. [Google Scholar] [CrossRef] [Green Version]
- Odijk, T. Polyelectrolytes near Rod Limit. J. Polym. Sci. Part B Polym. Phys. 1977, 15, 477–483. [Google Scholar] [CrossRef]
- Skolnick, J.; Fixman, M. Electrostatic Persistence Length of a Wormlike Polyelectrolyte. Macromolecules 1977, 10, 944–948. [Google Scholar] [CrossRef]
- Dobrynin, A.V. Electrostatic persistence length of semiflexible and flexible polyelectrolytes. Macromolecules 2005, 38, 9304–9314. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Carrillo, J.M.Y. Swelling of biological and semiflexible polyelectrolytes. J. Phys. Condens. Matter 2009, 21, 424112. [Google Scholar] [CrossRef] [PubMed]
- Porschke, D. Persistence Length and Bending Dynamics of DNA from Electrooptical Measurements at High Salt Concentrations. Biophys. Chem. 1991, 40, 169–179. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Balducci, A.; Doyle, P.S. Ionic effects on the equilibrium dynamics of DNA confined in nanoslits. Nano Lett. 2008, 8, 1683–1688. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.K.; Hsieh, C.C.; Chen, Y.L.; Chou, C.F. Effects of Topology and Ionic Strength on Double-Stranded DNA Confined in Nanoslits. Macromolecules 2012, 45, 2920–2927. [Google Scholar] [CrossRef]
- Smith, S.; Finzi, L.; Bustamante, C. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 1992, 258, 1122–1126. [Google Scholar] [CrossRef]
- Reisner, W.; Pedersen, J.N.; Austin, R.H. DNA confinement in nanochannels: Physics and biological applications. Rep. Prog. Phys. 2012, 75, 106601. [Google Scholar] [CrossRef]
- Yang, Y.Z.; Burkhardt, T.W.; Gompper, G. Free energy and extension of a semiflexible polymer in cylindrical confining geometries. Phys. Rev. E 2007, 76, 011804. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, A.B.; Reifenberger, J.G.; Chuang, H.M.; Cao, H.; Dorfman, K.D. Measuring the wall depletion length of nanoconfined DNA. J. Chem. Phys. 2018, 149, 104901. [Google Scholar] [CrossRef]
- Chen, J.Z.Y. Self-Avoiding Wormlike Chain Confined in a Cylindrical Tube: Scaling Behavior. Phys. Rev. Lett. 2018, 121, 037801. [Google Scholar] [CrossRef]
- Muralidhar, A.; Tree, D.R.; Dorfman, K.D. Backfolding of Wormlike Chains Confined in Nanochannels. Macromolecules 2014, 47, 8446–8458. [Google Scholar] [CrossRef]
- Werner, E.; Mehlig, B. Scaling regimes of a semiflexible polymer in a rectangular channel. Phys. Rev. E 2015, 91. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Doyle, P.S. Comparisons of a Polymer in Confinement versus Applied Force. Macromolecules 2013, 46, 6336–6344. [Google Scholar] [CrossRef]
- Muralidhar, A.; Quevillon, M.J.; Dorfman, K.D. The Backfolded Odijk Regime for Wormlike Chains Confined in Rectangular Nanochannels. Polymers 2016, 8, 79. [Google Scholar] [CrossRef]
- Dai, L.; Ng, S.Y.; Doyle, P.S.; van der Maarel, J.R.C. Conformation Model of Back-Folding and Looping of a Single DNA Molecule Confined Inside a Nanochannel. ACS Macro Lett. 2012, 1, 1046–1050. [Google Scholar] [CrossRef]
- Werner, E.; Cheong, G.K.; Gupta, D.; Dorfman, K.D.; Mehlig, B. One-Parameter Scaling Theory for DNA Extension in a Nanochannel. Phys. Rev. Lett. 2017, 119, 268102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Z.Y. Conformational Properties of a Back-Folding Wormlike Chain Confined in a Cylindrical Tube. Phys. Rev. Lett. 2017, 118, 247802. [Google Scholar] [CrossRef]
- Chen, Y.L. Electro-entropic excluded volume effects on DNA looping and relaxation in nanochannels. Biomicrofluidics 2013, 7, 054119. [Google Scholar] [CrossRef]
- Muralidhar, A.; Dorfman, K.D. Backfolding of DNA Confined in Nanotubes: Flory Theory versus the Two-State Cooperativity Model. Macromolecules 2016, 49, 1120–1126. [Google Scholar] [CrossRef]
- Odijk, T. DNA confined in nanochannels: Hairpin tightening by entropic depletion. J. Chem. Phys. 2006, 125, 204904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, J.M.; Tremblett, A.F.; McLure, Z.R.N. Free Energy of a Folded Polymer under Cylindrical Confinement. Macromolecules 2017, 50, 9515–9524. [Google Scholar] [CrossRef] [Green Version]
- Micheletti, C.; Orlandini, E. Knotting and Unknotting Dynamics of DNA Strands in Nanochannels. ACS Macro Lett. 2014, 3, 876–880. [Google Scholar] [CrossRef]
- Amin, S.; Khorshid, A.; Zeng, L.; Zimny, P.; Reisner, W. A nanofluidic knot factory based on compression of single DNA in nanochannels. Nat. Commun. 2018, 9, 1506. [Google Scholar] [CrossRef] [PubMed]
- Krog, J.; Alizadehheidari, M.; Werner, E.; Bikkarolla, S.K.; Tegenfeldt, J.O.; Mehlig, B.; Lomholt, M.A.; Westerlund, F.; Ambjornsson, T. Stochastic unfolding of nanoconfined DNA: Experiments, model and bayesian analysis. J. Chem., Phys. 2018, 149, 215101. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Miller, J.J.; Muralidhar, A.; Mahshid, S.; Reisner, W.; Dorfman, K.D. Experimental Evidence of Weak Excluded Volume Effects for Nanochannel Confined DNA. ACS Macro Lett. 2015, 4, 759–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, F.; Utko, P.; Reisner, W.; Larsen, N.B.; Kristensen, A. Confinement Spectroscopy: Probing Single DNA Molecules with Tapered Nanochannels. Nano Lett. 2009, 9, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Utko, P.; Persson, F.; Kristensen, A.; Larsen, N.B. Injection molded nanofluidic chips: Fabrication method and functional tests using single-molecule DNA experiments. Lab Chip 2011, 11, 303–308. [Google Scholar] [CrossRef]
- Werner, E.; Mehlig, B. Confined polymers in the extended de Gennes regime. Phys. Rev. E 2014, 90. [Google Scholar] [CrossRef]
- Dai, L.; van der Maarel, J.R.C.; Doyle, P.S. Extended de Gennes Regime of DNA Confined in a Nanochannel. Macromolecules 2014, 47, 2445–2450. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IS (mM) | w (nm) | IS (mM) | w (nm) |
|---|---|---|---|
| 0.061 | 250 | 1.82 | 38 |
| 0.075 | 224 | 2.23 | 34 |
| 0.10 | 193 | 2.71 | 31 |
| 0.13 | 164 | 3.57 | 27 |
| 0.33 | 98 | 5.26 | 22 |
| 0.60 | 70 | 7.54 | 18 |
| 0.80 | 60 | 9.72 | 16 |
| 1.23 | 47 | 19.4 | 11 |
| 1.40 | 44 | 47.0 | 7.8 |
| 1.59 | 41 | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Lee, Y.; Kim, Y.; Wang, C.; Park, J.; Jung, G.Y.; Chen, Y.; Chang, R.; Ikeda, S.; Sugiyama, H.; et al. Nanochannel-Confined TAMRA-Polypyrrole Stained DNA Stretching by Varying the Ionic Strength from Micromolar to Millimolar Concentrations. Polymers 2019, 11, 15. https://doi.org/10.3390/polym11010015
Lee S, Lee Y, Kim Y, Wang C, Park J, Jung GY, Chen Y, Chang R, Ikeda S, Sugiyama H, et al. Nanochannel-Confined TAMRA-Polypyrrole Stained DNA Stretching by Varying the Ionic Strength from Micromolar to Millimolar Concentrations. Polymers. 2019; 11(1):15. https://doi.org/10.3390/polym11010015
Chicago/Turabian StyleLee, Seonghyun, Yelin Lee, Yongkyun Kim, Cong Wang, Jungyul Park, Gun Young Jung, Yenglong Chen, Rakwoo Chang, Shuji Ikeda, Hiroshi Sugiyama, and et al. 2019. "Nanochannel-Confined TAMRA-Polypyrrole Stained DNA Stretching by Varying the Ionic Strength from Micromolar to Millimolar Concentrations" Polymers 11, no. 1: 15. https://doi.org/10.3390/polym11010015