Theoretical Modeling of Polymer Translocation: From the Electrohydrodynamics of Short Polymers to the Fluctuating Long Polymers



Abstract
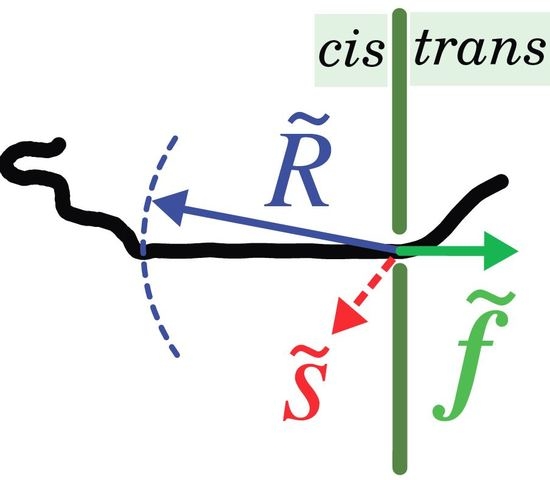
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Electrohydrodynamic Approach to the Translocation of Short Polymers
2.1. Theory
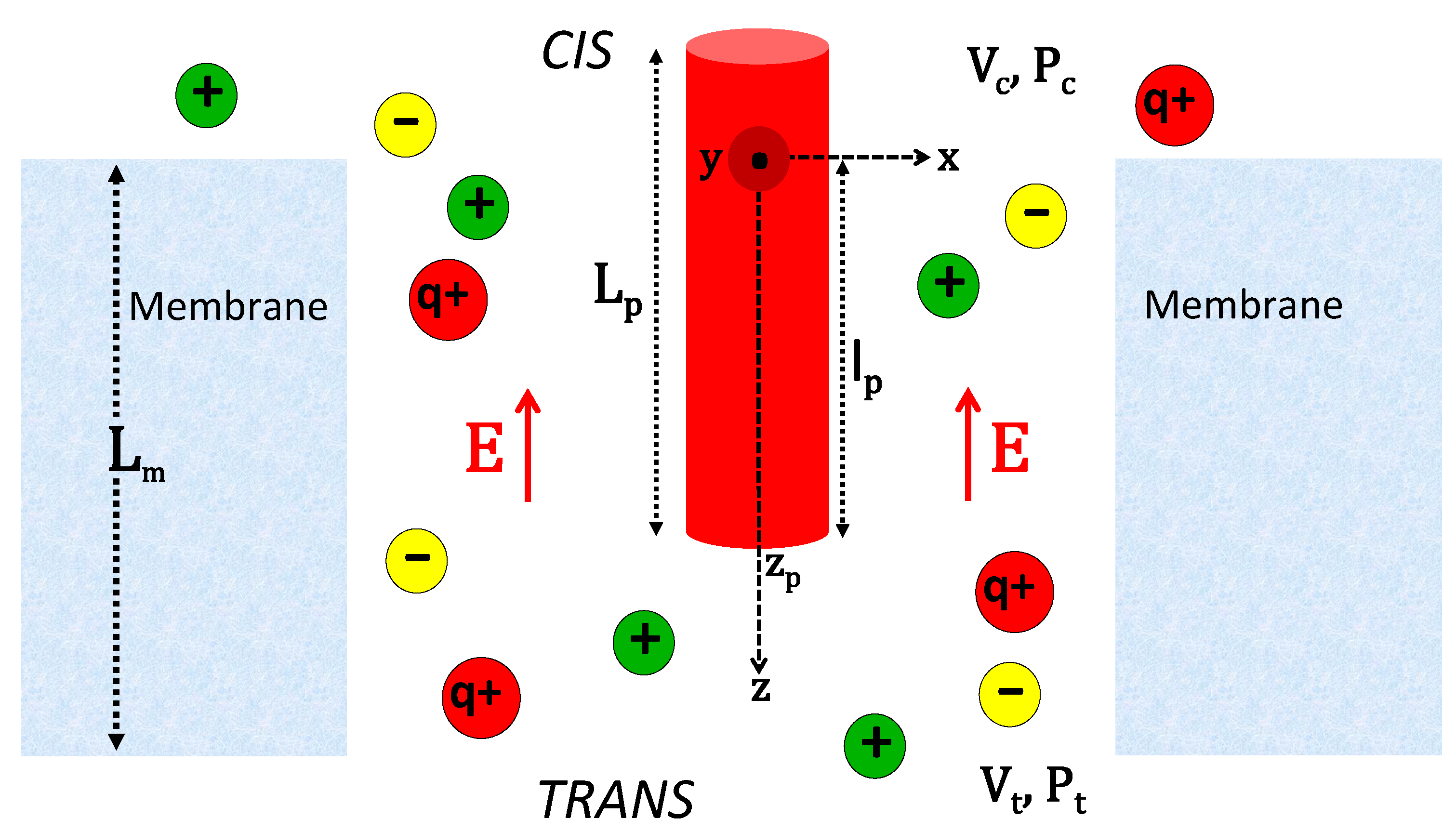
2.1.1. Electrohydrodynamic Formalism of Polymer Translocation
2.1.2. Derivation of the Polymer Velocity
2.1.3. Derivation of the Interaction Potential
2.2. Polymer Conductivity of Solid-State Pores: MF Electrohydrodynamics with Monovalent Salt
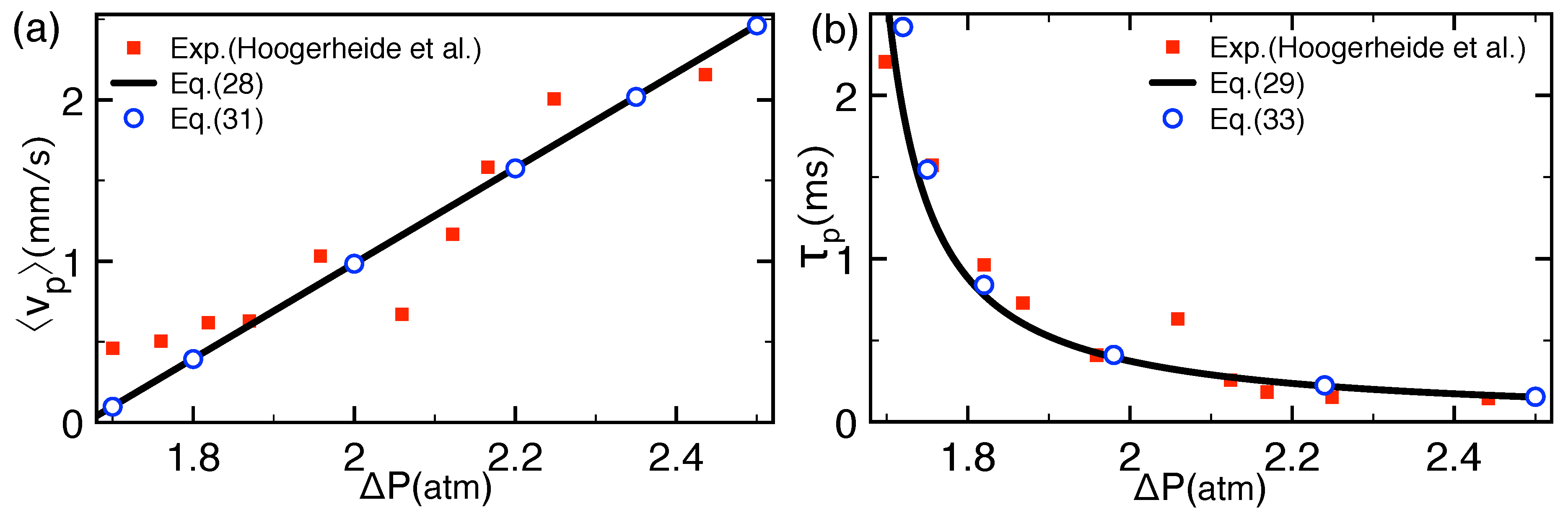
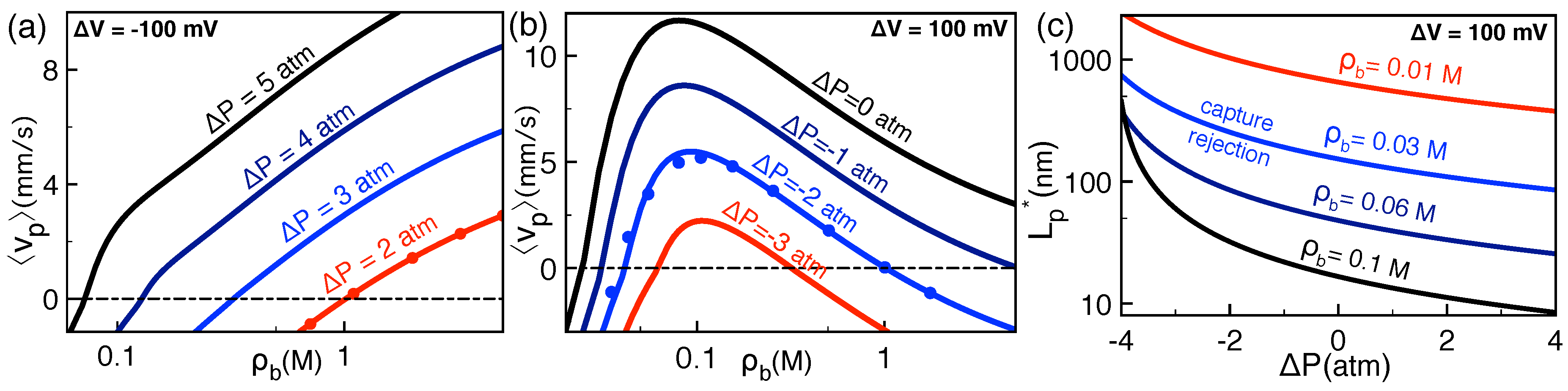
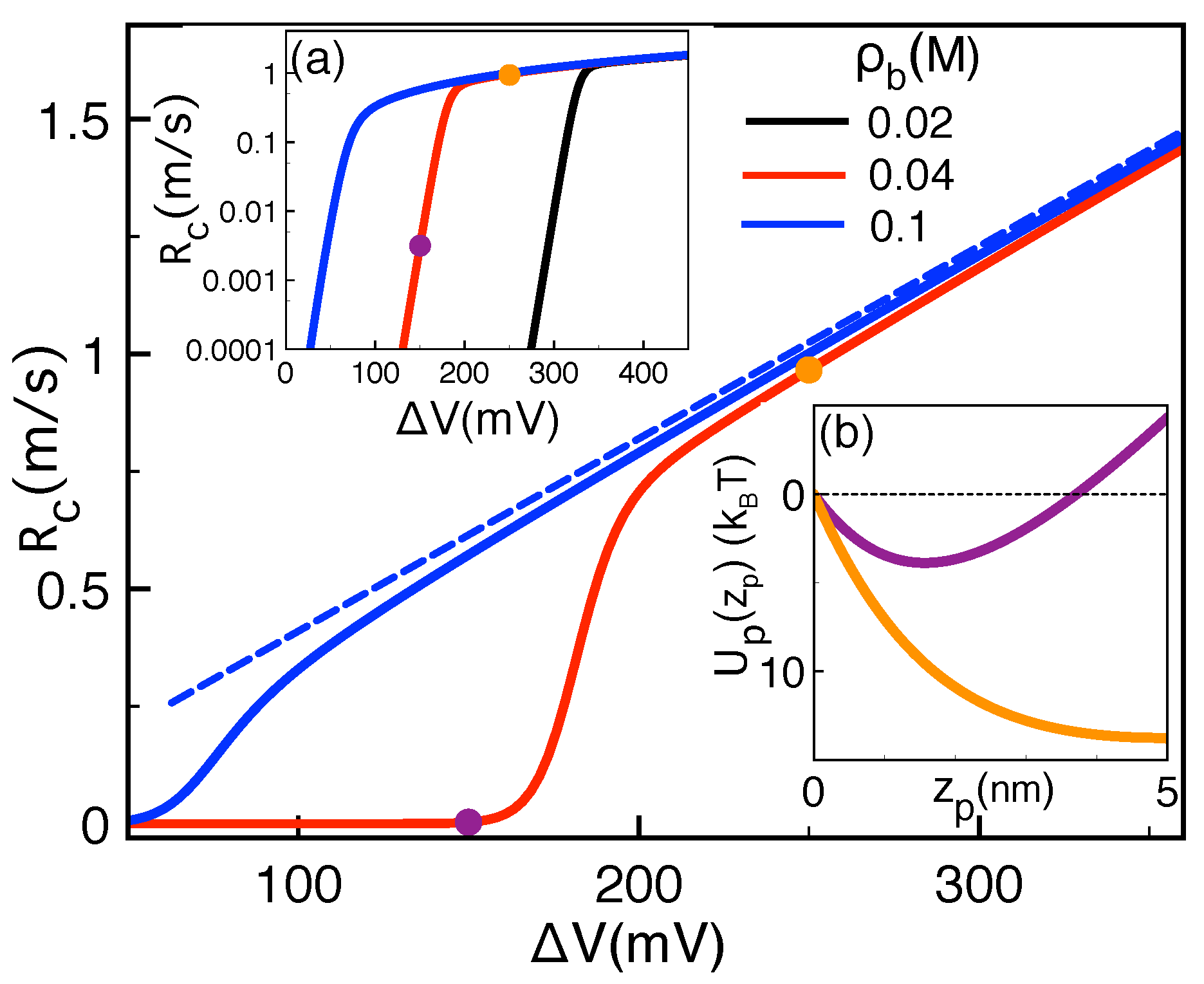
2.2.1. Comparison with Pressure-Voltage Trapping Experiments
2.2.2. Salt and Polymer Length Dependence of Pressure-Voltage-Driven Translocation Events
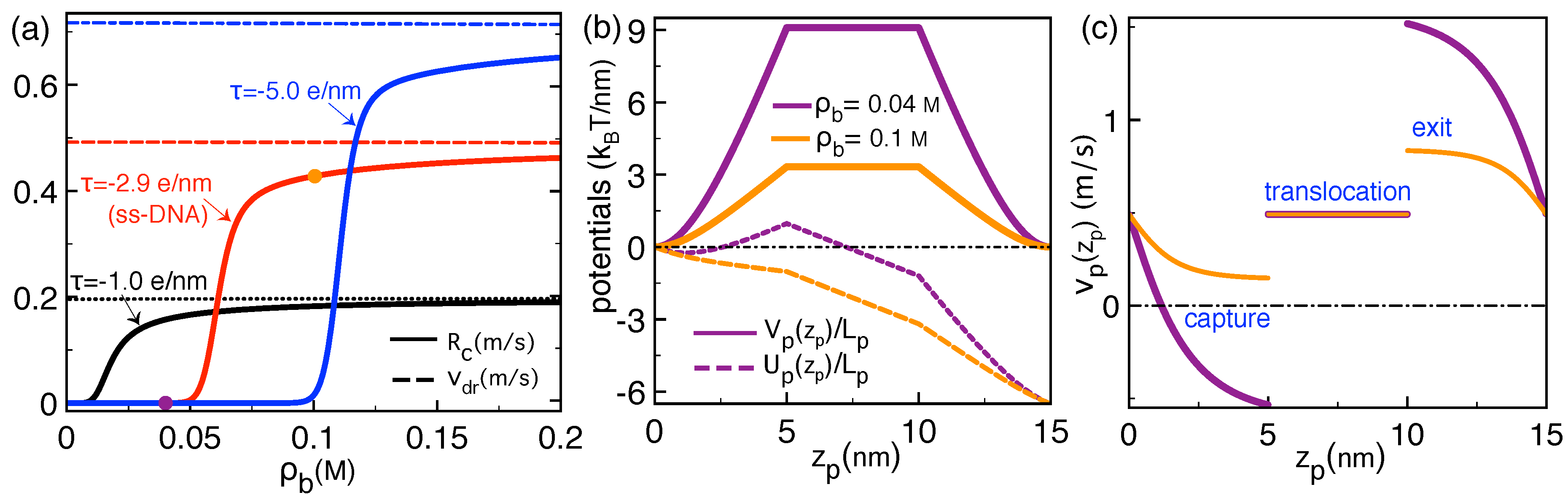
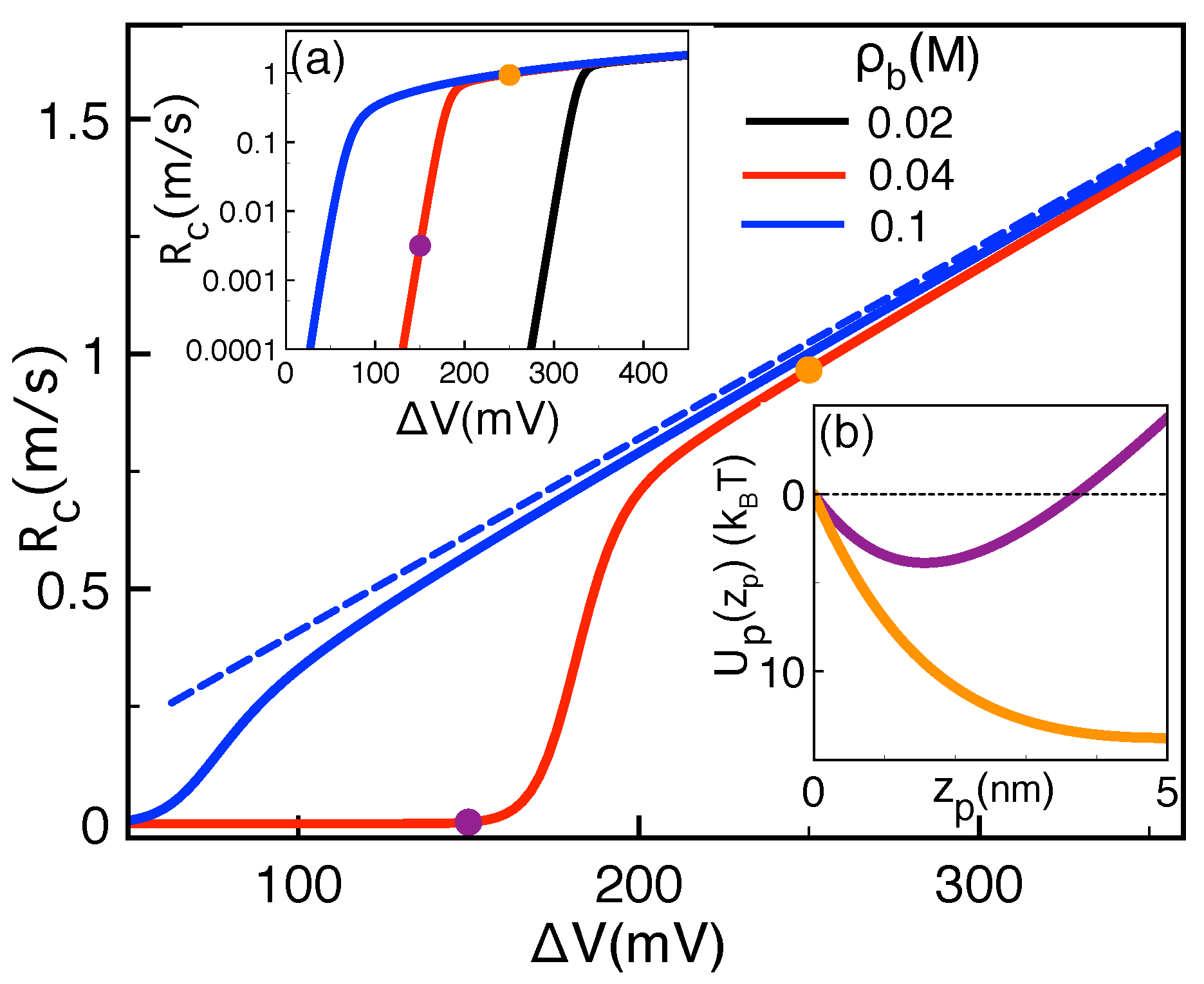
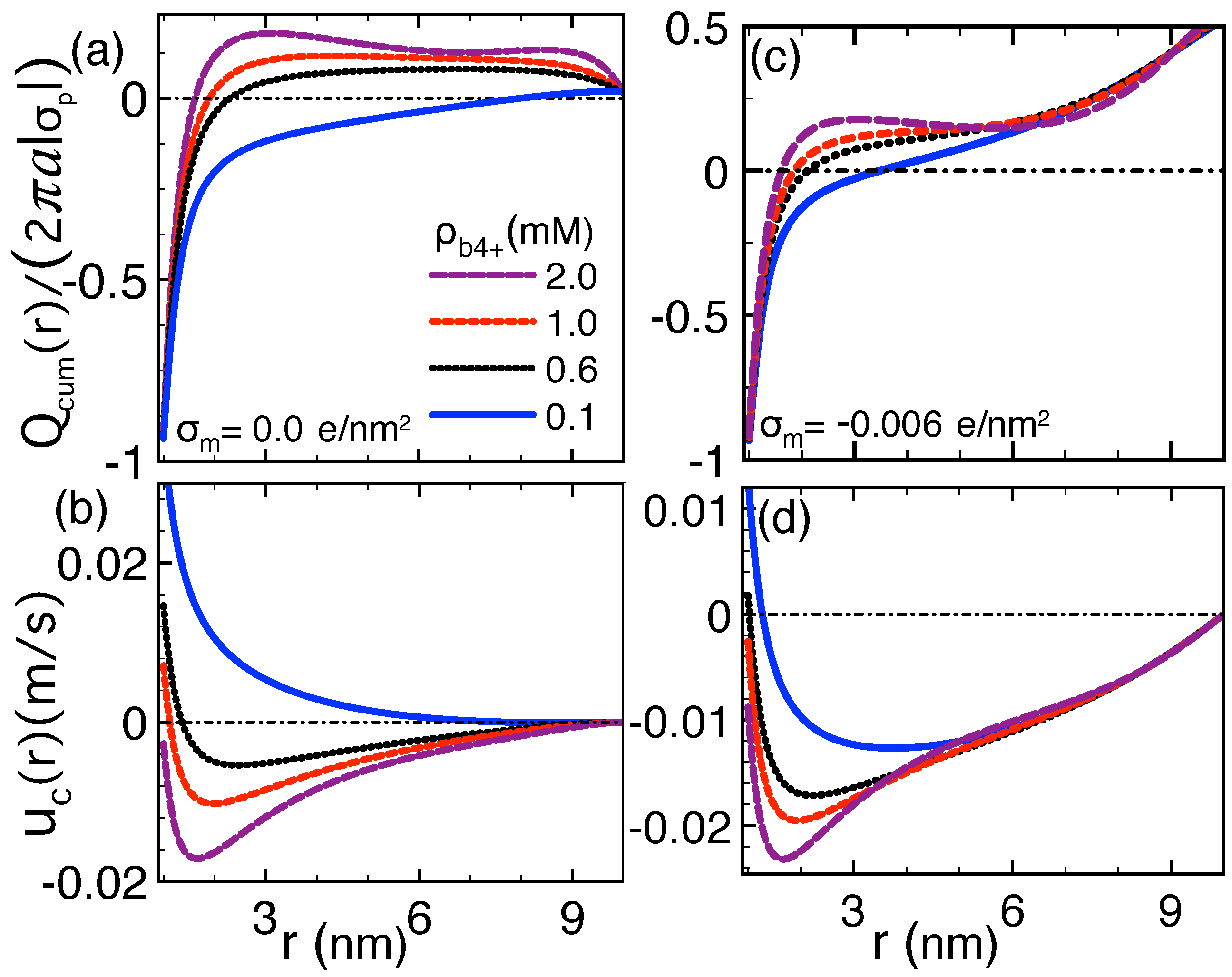
2.3. Correlation-Induced DNA Mobility Inversion by Polyvalent Counterions in Solid-State Pores
2.4. Polymer Conductivity of Biological Pores: Image-Charge Barrier against Drift Force
2.5. Limitation of the Stiff Polymer Approximation
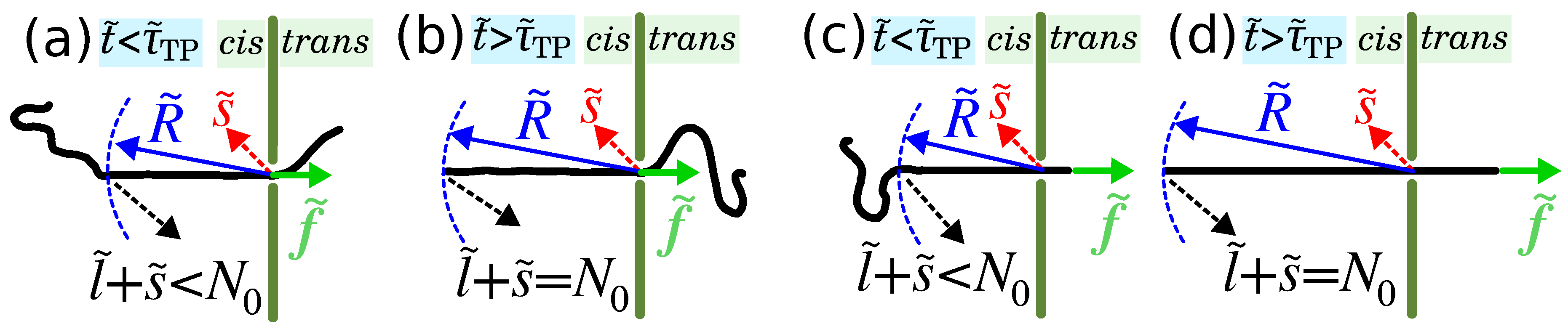
3. Iso-Flux Tension Propagation (IFTP) Theory for the Translocation of Long Polymers
3.1. Coarse-Grained Polymer Model
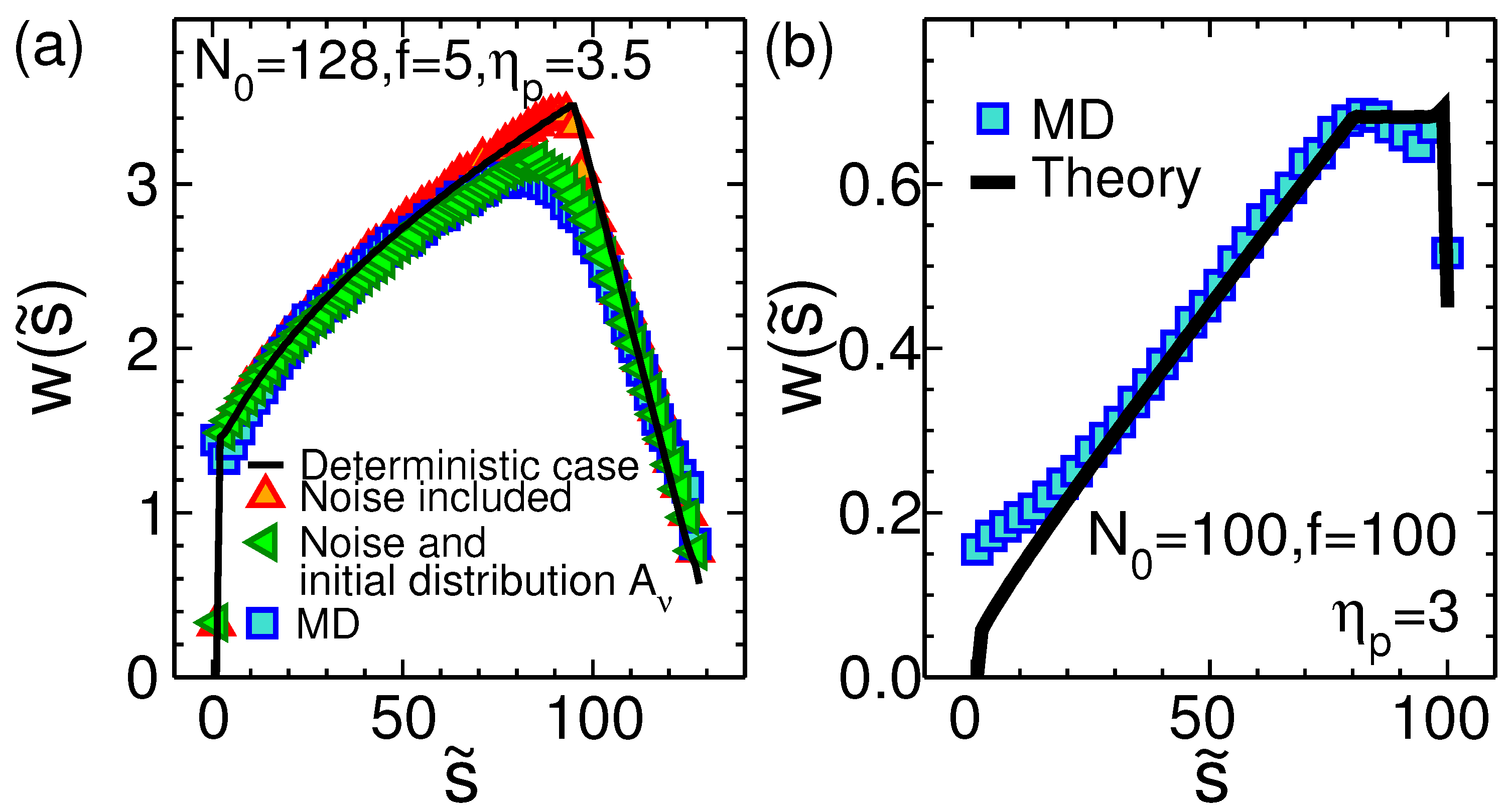
3.2. Waiting Time Distribution
3.3. Scaling of the Translocation Time for a Flexible Polymer
3.4. Scaling of the Translocation Time for a Stiff Polymer
4. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A. Coefficients of the Average Velocity 〈vp〉 in Equations (28) and (31)
Appendix B. Components of the Polymer Translocation Time τ in Equation (29)
References
- Schoch, R.B.; Han, J.; Renaud, P. Transport phenomena in nanofluidics. Rev. Mod. Phys. 2008, 80, 839–883. [Google Scholar] [CrossRef] [Green Version]
- Wanunu, W. Nanopores: A journey towards DNA sequencing. Phys. Life Rev. 2012, 9, 125–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palyulin, V.V.; Ala-Nissila, T.; Metzler, R. Polymer translocation: The first two decades and the recent diversification. Soft Matter 2014, 10, 9016–9037. [Google Scholar] [CrossRef] [PubMed]
- Kasianowicz, J.J.; Brandin, E.; Branton, D.; Deamer, D.W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. USA 1996, 93, 13770–13773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrickson, S.E.; Misakian, M.; Robertson, B.; Kasianowicz, J.J. Driven DNA transport into an asymmetric nanometer-scale pore. Phys. Rev. Lett. 2000, 14, 3057–3060. [Google Scholar] [CrossRef] [PubMed]
- Meller, A.; Nivon, L.; Branton, D. Voltage-Driven DNA Translocations through a Nanopore. Phys. Rev. Lett. 2001, 86, 3435–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonthuis, D.J.; Zhang, J.; Hornblower, B.; Mathé, J.; Shklovskii, B.I.; Meller, A. Self-Energy-Limited Ion Transport in Subnanometer Channels. Phys. Rev. Lett. 2006, 97, 128104. [Google Scholar] [CrossRef]
- Smeets, R.M.M.; Keyser, U.F.; Krapf, D.; Wue, M.-Y.; Dekker, N.H.; Dekker, C. Salt dependence of ion transport and DNA translocation through solid-state nanopores. Nano Lett. 2006, 6, 89–95. [Google Scholar] [CrossRef]
- Clarke, J.; Wu, H.C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef]
- Wanunu, M.; Morrison, W.; Rabin, Y.; Grosberg, A.Y.; Meller, A. Electrostatic focusing of unlabelled DNA into nanoscale pores using a salt gradient. Nat. Nanotechnol. 2010, 5, 160–165. [Google Scholar] [CrossRef]
- Sung, W.; Park, P.J. Polymer Translocation through a Pore in a Membrane. Phys. Rev. Lett. 1996, 77, 783. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Huopaniemi, I.; Ala-Nissila, T.; Ying, S.C. Polymer translocation through a nanopore under an applied external field. J. Chem. Phys. 2006, 124, 1–7. [Google Scholar] [CrossRef]
- Ikonen, T.; Bhattacharya, A.; Ala-Nissila, T.; Sung, W. Unifying model of driven polymer translocation. Phys. Rev. E 2012, 85, 051803. [Google Scholar] [CrossRef] [Green Version]
- Farahpour, F.; Maleknejad, A.; Varnikc, F.; Ejtehadi, M.R. Chain deformation in translocation phenomena. Soft Matter 2013, 9, 2750–2759. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Morrison, W.H.; Luo, K.; Ala-Nissila, T.; Ying, S.-C.; Milchev, A.; Binder, K. Scaling exponents of forced polymer translocation through a nanopore. Eur. Phys. J. E 2009, 29, 423. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, T. Nonequilibrium dynamics of polymer translocation and straightening. Phys. Rev. E 2007, 76, 021803. [Google Scholar] [CrossRef]
- Saito, T.; Sakaue, T. Dynamical diagram and scaling in polymer driven translocation. Eur. Phys. J. E 2011, 34, 135. [Google Scholar] [CrossRef]
- Sakaue, T. Dynamics of Polymer Translocation: A Short Review with an Introduction of Weakly-Driven Regime. Polymers 2016, 8, 424. [Google Scholar] [CrossRef]
- Sarabadani, J.; Ikonen, T.; Ala-Nissila, T. Iso-flux tension propagation theory of driven polymer translocation: The role of initial configurations. J. Chem. Phys. 2014, 141, 214907. [Google Scholar] [CrossRef] [Green Version]
- Sarabadani, J.; Ghosh, B.; Chaudhury, S.; Ala-Nissila, T. Dynamics of end-pulled polymer translocation through a nanopore. Europhys. Lett. 2017, 120, 38004. [Google Scholar] [CrossRef] [Green Version]
- Sarabadani, J.; Ikonen, T.; Mokkonen, H.; Ala-Nissila, T.; Coarson, S.; Wanunu, M. Driven translocation of a semi-flexible polymer through a nanopore. Sci. Rep. 2017, 7, 7423. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S. Effect of Salt Concentration on the Electrophoretic Speed of a Polyelectrolyte through a Nanopore. Phys. Rev. Lett. 2007, 98, 238104. [Google Scholar] [CrossRef]
- Zhang, J.; Shklovskii, B.I. Effective charge and free energy of DNA inside an ion channel. Phys. Rev. E 2007, 75, 021906. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.T.A.; Muthukumar, M. Polymer capture by electro-osmotic flow of oppositely charged nanopores. J. Chem. Phys. 2007, 126, 164903. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, M. Theory of capture rate in polymer translocation. J. Chem. Phys. 2010, 132, 195101. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, M. Communication: Charge, diffusion, and mobility of proteins through nanopores. J. Chem. Phys. 2014, 141, 081104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, N.A.W.; Muthukumar, M.; Keyser, U.F. Translocation frequency of double-stranded DNA through a solid-state nanopore. Phys. Rev. E 2016, 93, 022401. [Google Scholar] [CrossRef] [PubMed]
- Buyukdagli, S.; Ala-Nissila, T. Controlling Polymer Capture and Translocation by Electrostatic Polymer-pore interactions. J. Chem. Phys. 2017, 147, 114904. [Google Scholar] [CrossRef]
- Buyukdagli, S. Facilitated polymer capture by charge inverted electroosmotic flow in voltage-driven polymer translocation. Soft Matter 2018, 14, 3541–3549. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Wang, Y.; Cao, B.; Guo, Z.; Chen, Y.; Yang, G. The suppression and promotion of DNA charge inversion by mixing counterions. Soft Matter 2015, 11, 4099–4105. [Google Scholar] [CrossRef]
- Buyukdagli, S. Enhanced polymer capture speed and extended translocation time in pressure-solvation traps. Phys. Rev. E 2018, 97, 062406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buyukdagli, S.; Sarabadani, J.; Ala-Nissila, T. Dielectric trapping of biopolymers translocating through insulating membranes. Polymers 2018, 10, 1242. [Google Scholar] [CrossRef]
- Hoogerheide, D.P.; Lu, B.; Golovchenko, J.A. Pressure-voltage trap for DNA near a solid-state nanopore. ACS Nano 2014, 8, 738. [Google Scholar] [CrossRef] [PubMed]
- Avalos, J.B.; Rubi, J.M.; Bedeaux, D. Dynamics of rodlike polymers in dilute solution. Macromolecules 1993, 26, 2550–2561. [Google Scholar] [CrossRef]
- Buyukdagli, S.; Ala-Nissila, T. Controlling Polymer Translocation and Ion Transport via Charge Correlations. Langmuir 2014, 30, 12907. [Google Scholar] [CrossRef]
- Wanunu, M.; Sutin, J.; Mcnally, B.; Chow, A.; Meller, A. DNA Translocation Governed by Interactions with Solid-State Nanopores. Biophys. J. 2008, 95, 4716. [Google Scholar] [CrossRef]
- Matysiak, S.; Montesi, A.; Pasquali, M.; Kolomeisky, A.B.; Clementi, C. Dynamics of polymer translocation through nanopores: Theory meets experiment. Phys. Rev. Lett. 2006, 96, 118103. [Google Scholar] [CrossRef]
- Wong, C.T.A.; Muhtukumar, M. Polymer translocation through alpha-hemolysin pore with tunable polymer-pore electrostatic interaction. J. Chem. Phys. 2010, 133, 045101. [Google Scholar] [CrossRef]
- Ansalone, P.; Chinappi, M.; Rondoni, L.; Cecconi, F. Driven diffusion against electrostatic or effective energy barrier across α-hemolysin. J. Chem. Phys. 2017, 143, 154109. [Google Scholar] [CrossRef]
- Abramowitz, M.; Stegun, I.A. Handbook of Mathematical Functions; Dover Publications: New York, NY, USA, 1972. [Google Scholar]
- Meller, A.; Branton, D. Single molecule measurements of DNA transport through a nanopore. Electrophoresis 2002, 23, 2583–2591. [Google Scholar] [CrossRef]
- Rowghanian, P.; Grosberg, A.Y. Force-driven polymer translocation through a nanopore: An old problem revisited. J. Phys. Chem. B 2011, 115, 14127. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, T.; Bhattacharya, A.; Ala-Nissila, T.; Sung, W. Influence of non-universal effects on dynamical scaling in driven polymer translocation. J. Chem. Phys. 2012, 137, 085101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikonen, T.; Bhattacharya, A.; Ala-Nissila, T.; Sung, W. Influence of pore friction on the universal aspects of driven polymer translocation. Europhys. Lett. 2013, 103, 38001. [Google Scholar] [CrossRef] [Green Version]
- Sarabadani, J.; Ikonen, T.; Ala-Nissila, T. Theory of polymer translocation through a flickering nanopore under an alternating driving force. J. Chem. Phys. 2015, 143, 074905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarabadani, J.; Ala-Nissila, T. Theory of pore-driven and end-pulled polymer translocation dynamics through a nanopore: An overview. J. Phys. Condens. Matter 2018, 30, 274002. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buyukdagli, S.; Sarabadani, J.; Ala-Nissila, T. Theoretical Modeling of Polymer Translocation: From the Electrohydrodynamics of Short Polymers to the Fluctuating Long Polymers. Polymers 2019, 11, 118. https://doi.org/10.3390/polym11010118
Buyukdagli S, Sarabadani J, Ala-Nissila T. Theoretical Modeling of Polymer Translocation: From the Electrohydrodynamics of Short Polymers to the Fluctuating Long Polymers. Polymers. 2019; 11(1):118. https://doi.org/10.3390/polym11010118
Chicago/Turabian StyleBuyukdagli, Sahin, Jalal Sarabadani, and Tapio Ala-Nissila. 2019. "Theoretical Modeling of Polymer Translocation: From the Electrohydrodynamics of Short Polymers to the Fluctuating Long Polymers" Polymers 11, no. 1: 118. https://doi.org/10.3390/polym11010118
APA StyleBuyukdagli, S., Sarabadani, J., & Ala-Nissila, T. (2019). Theoretical Modeling of Polymer Translocation: From the Electrohydrodynamics of Short Polymers to the Fluctuating Long Polymers. Polymers, 11(1), 118. https://doi.org/10.3390/polym11010118