Urethane Formation with an Excess of Isocyanate or Alcohol: Experimental and Ab Initio Study

 ,
,  , and
, and
Abstract
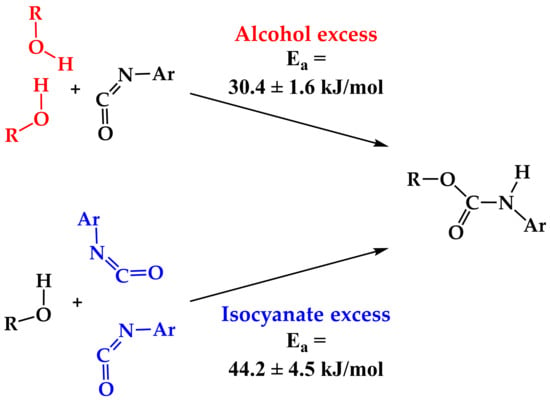
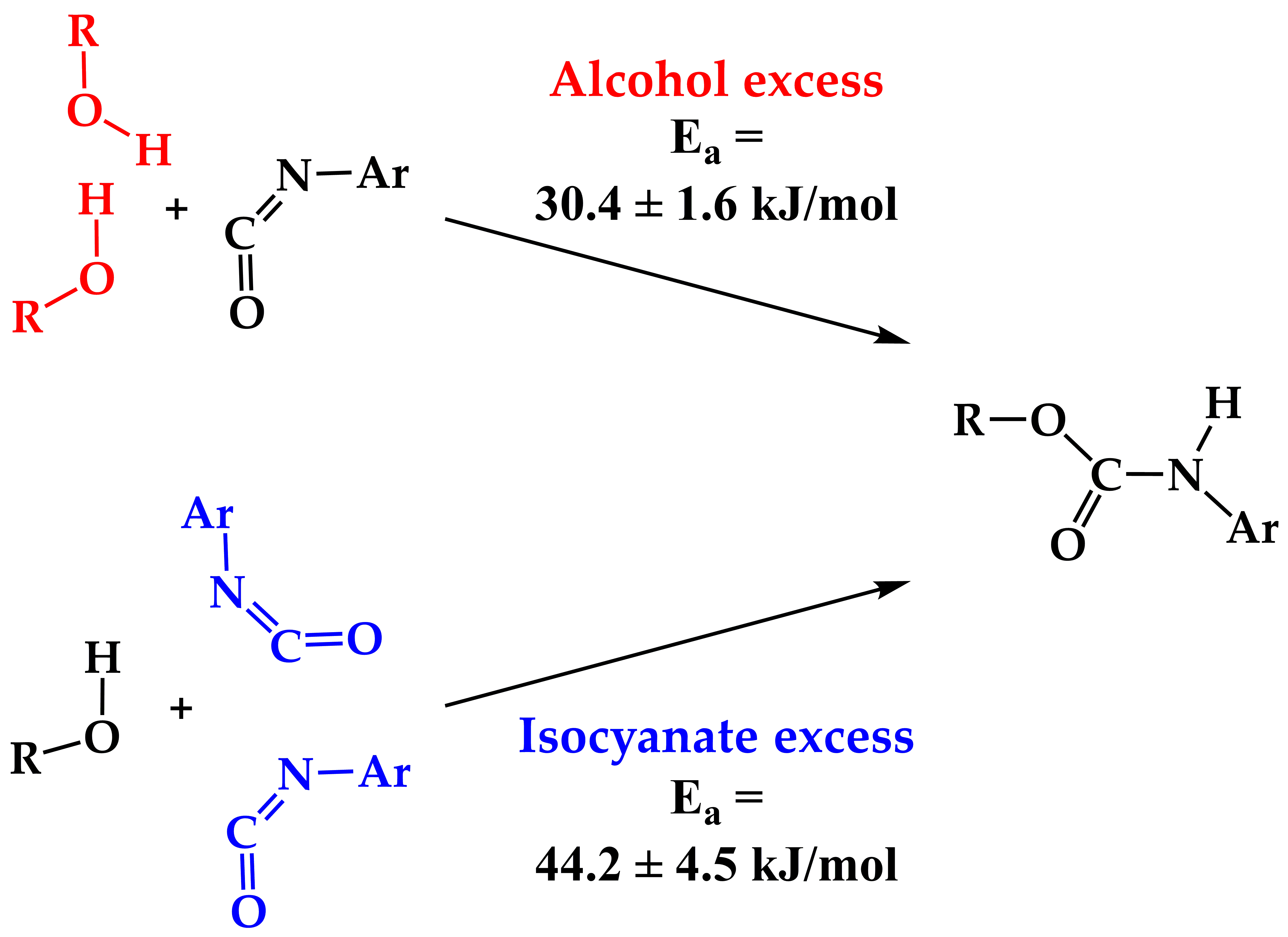
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Kinetic Experiments
2.3. Analysis Method
2.4. Theoretical Method
3. Results and Discussion
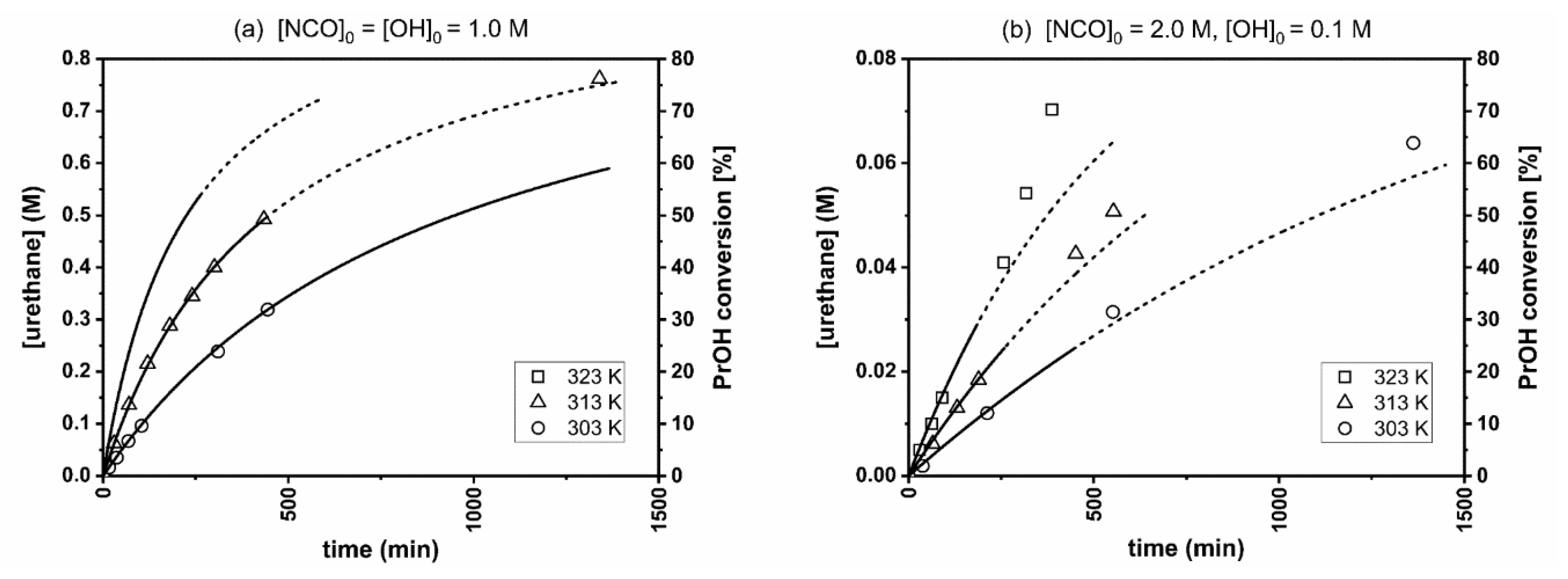
3.1. Results of the Kinetic Experiments
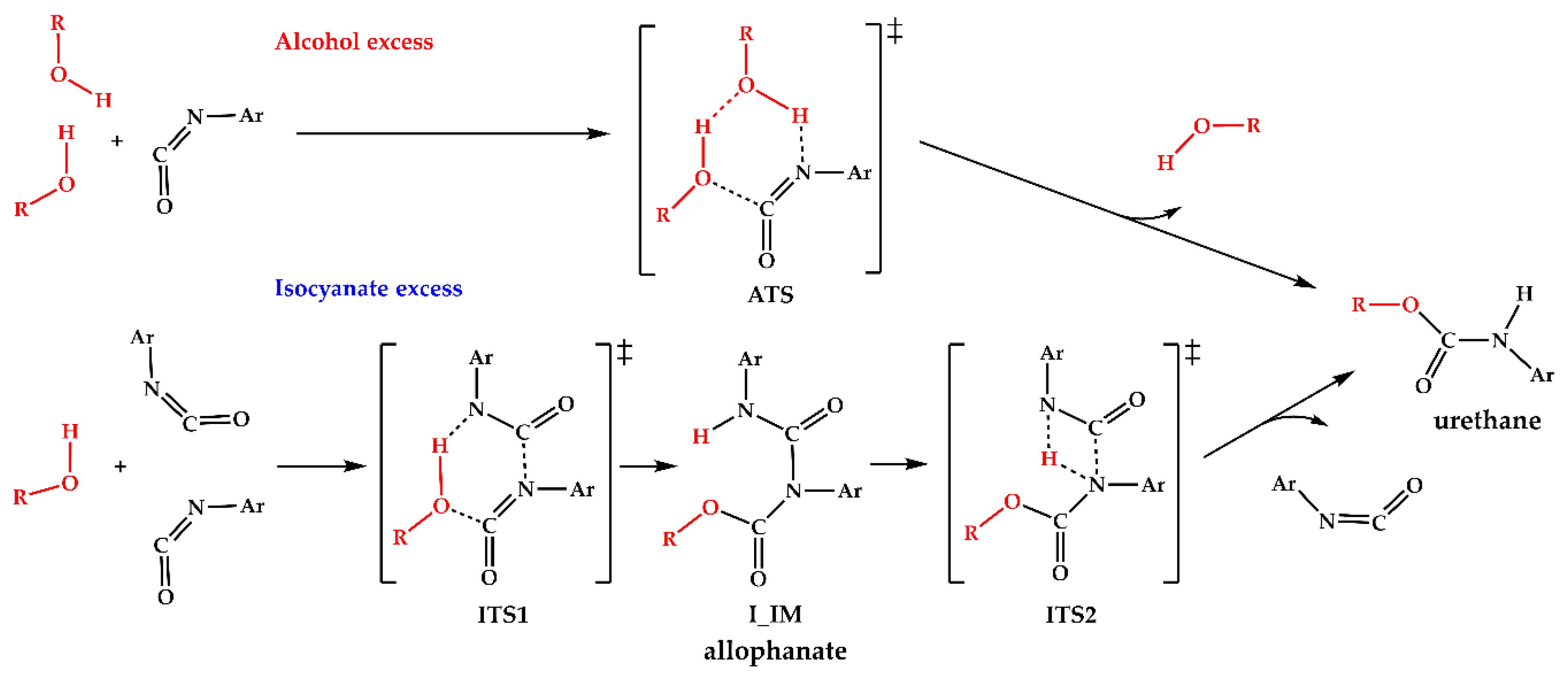
3.2. Results of the Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boros, R.Z.; Farkas, L.; Nehéz, K.; Viskolcz, B.; Szőri, M. An Ab Initio Investigation of the 4,4′-Methlylene Diphenyl Diamine (4,4′-MDA) Formation from the Reaction of Aniline with Formaldehyde. Polymers 2019, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.L.; Farnum, J.M. Relative Velocities of Reaction of Alcohols with Phenyl Isocyanate. J. Am. Chem. Soc. 1934, 56, 883–885. [Google Scholar] [CrossRef]
- Baker, J.W.; Holdsworth, J.B. The Mechanism of Aromatic Side-chain Reactions with Special Reference to the Polar Effects of Substituents. Part XIII. Kinetic Examination of the Reaction of Aryl Isocyanates with Methyl Alcohol. J. Chem. Soc. 1947, 713–726. [Google Scholar] [CrossRef]
- Baker, J.W.; Gaunt, J. The Mechanism of the Reaction of Aryl Isocyanates with Alcohols and Amines. Part III. The “Spontaneous” Reaction of Phenyl Isocyanate with Various Alcohols. Further Evidence Relating to the Anomalous Effect of Dialkylanilines in the Base-catalysed Reaction. J. Chem. Soc. 1949, 19–24. [Google Scholar] [CrossRef]
- Baker, J.W.; Gaunt, J. The Mechanism of the Reaction of Aryl Isocyanates with Alcohols and Amines. Part, V. Kinetic Investigations of the Reaction between Phenyl Isocyanate and Methyl and Ethyl Alcohols in Benzene Solution. J. Chem. Soc. 1949, 27–31. [Google Scholar] [CrossRef]
- Ephraim, S.; Woodward, A.E.; Mesrobian, R.B. Kinetic Studies of the Reaction of Phenyl Isocyanate with Alcohols in Various Solvents. J. Am. Chem. Soc. 1958, 80, 1326–1328. [Google Scholar] [CrossRef]
- Lammiman, S.A.; Satchell, R.S. The Kinetics and Mechanism of the Spontaneous Alcoholysis of p-Chlorophenyl Isocyanate in Diethyl Ether. The Association of Alcohols in Diethyl Ether. J. Chem. Soc. Perkin Trans. 1972, 2, 2300–2305. [Google Scholar] [CrossRef]
- Raspoet, G.; Nguyen, M.T. The Alcoholysis Reaction of Isocyanates Giving Urethanes: Evidence for a Multimolecular Mechanism. J. Org. Chem. 1998, 63, 6878–6885. [Google Scholar] [CrossRef]
- Makitra, R.G.; Midyana, G.G.; Pal’chikova, E.Ya.; Romanyuk, A.V. Solvent Effect on the Kinetics of Carbamoylation of Alcohols. Russ. J. Org. Chem. 2012, 48, 25–31. [Google Scholar] [CrossRef]
- Król, P.; Wojturska, J. Kinetic Study on the Reaction of 2,4-and 2,6-Tolylene Diisocyanate with 1-Butanol in the Presence of Styrene, as a Model Reaction for the Process that Yields Interpenetrating Polyurethane–Polyester Networks. J. Appl. Polym. Sci. 2003, 88, 327–336. [Google Scholar] [CrossRef]
- López, C.O.C.; Fejes, Z.; Viskolcz, B. Microreactor Assisted Method for Studying Isocyanate–Alcohol Reaction Kinetics. J. Flow Chem. 2019, 9, 199–204. [Google Scholar] [CrossRef]
- Çoban, M.; Aylin, F.; Konuklar, S. A Computational Study on the Mechanism and the Kinetics of Urethane Formation. Comput. Theor. Chem. 2011, 963, 168–175. [Google Scholar] [CrossRef]
- Kössl, F.; Lisaj, M.; Kozich, V.; Heyne, K.; Kühn, O. Monitoring the Alcoholysis of Isocyanates with Infrared Spectroscopy. Chem. Phys. Lett. 2015, 621, 41–45. [Google Scholar] [CrossRef]
- Wang, X.; Hu, W.; Gui, D.; Chi, X.; Wang, M.; Tian, D.; Liu, J.; Ma, X.; Pang, A. DFT Study of the Proton Transfer in the Urethane Formation between 2,4-Diisocyanatotoluene and Methanol. Bull. Chem. Soc. Jpn. 2013, 86, 255–265. [Google Scholar] [CrossRef]
- Somekawa, K.; Mitsushio, M.; Ueda, T. Molecular Simulation of Potential Energies, Steric Changes and Substituent Effects in Urethane Formation Reactions from Isocyanates. J. Comput. Chem. Jpn. 2016, 15, 32–40. [Google Scholar] [CrossRef]
- Akiyama, I.; Ogawa, M.; Takase, K.; Takamuku, T.; Yamaguchi, T.; Ohtori, N. Liquid Structure of 1-Propanol by Molecular Dynamics Simulations and X-Ray Scattering. J. Solution Chem. 2004, 33, 797–809. [Google Scholar] [CrossRef]
- Sun, W.; Silva, W.G.D.P.; van Wijngaarden, J. Rotational Spectra and Structures of Phenyl Isocyanate and Phenyl Isothiocyanate. J. Phys. Chem. A 2019, 123, 2351–2360. [Google Scholar] [CrossRef]
- Partington, J.R.; Cowley, E.G. Dipole Moments of Ethyl and Phenyl Isocyanates. Nature 1935, 135, 1038. [Google Scholar] [CrossRef]
- Lenzi, V.; Driest, P.J.; Dijkstra, D.J.; Ramos, M.M.D.; Marques, L.S.A. Investigation on the Intermolecular Interactions in Aliphatic Isocyanurate Liquids: Revealing the Importance of Dispersion. J. Mol. Liquids 2019, 280, 25–33. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; von Lilienfeld, O.A. Quantum Chemistry Structures and Properties of 134 Kilo Molecules. Sci. Data 2015, 1, 140022. [Google Scholar] [CrossRef]
- Boros, R.Zs.; Koós, T.; Cheikh, W.; Nehéz, K.; Farkas, L.; Viskolcz, B.; Szőri, M. A Theoretical Study on the Phosgenation of Methylene Diphenyl Diamine (MDA). Chem. Phys. Lett. 2018, 706, 568–576. [Google Scholar] [CrossRef]
- Schalk, O.; Townsend, D.; Wolf, T.J.A.; Holland, D.M.P.; Boguslavskiy, A.E.; Szőri, M. Albert Stolow Time-Resolved Photoelectron Spectroscopy of Nitrobenzene and its Aldehydes. Chem. Phys. Lett. 2018, 691, 379–387. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 Theory Using Reduced Order Perturbation Theory. J. Chem. Phys. 2007, 127, 124105. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.B.G.; Lawton, M. Drying of Organic Solvents: Quantitative Evaluation of the Efficiency of Several Desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView05; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. An Improved Algorithm for Reaction Path Following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on the Generalized Born Approximation with Asymmetric Descreening. J. Chem. Theory Comput. 2009, 5, 2447–2464. [Google Scholar] [CrossRef]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 90th ed.; CRC Press/Taylor and Francis: Boca Raton, FL, USA, 2010. [Google Scholar]
- Baev, A.K. Specific Intermolecular Interactions of Nitrogenated and Bioorganic Compounds; Springer: Heidelberg, Germany, 2013; p. 318. [Google Scholar]
- Majer, V.; Svoboda, V. Enthalpies of Vaporization of Organic Compounds: A Critical Review and Data Compilation; Blackwell Scientific Publications: Oxford, UK, 1985; p. 300. [Google Scholar]
- Lenzi, V.; Driest, P.J.; Dijkstra, D.J.; Ramos, M.M.; Marques, L.S. GAFF-IC: Realistic Viscosities for Isocyanate Molecules with a GAFF-based Force Field. Mol. Simulat. 2019, 45, 207–214. [Google Scholar] [CrossRef]
- Al Nabulsi, A.; Cozzula, D.; Hagen, T.; Leitner, W.; Müller, T.E. Isocyanurate Formation during Rigid Polyurethane Foam Assembly: A Mechanistic Study Based on in situ IR and NMR Spectroscopy. Polym. Chem. 2018, 9, 4891–4899. [Google Scholar] [CrossRef]
- Stern, T. Hierarchical fractal-structured allophanate-derived network formation in bulk polyurethane synthesis. Polym. Adv. Technol. 2017, 29, 1–12. [Google Scholar] [CrossRef]
- Delebecq, E.; Pascault, J.-P.; Boutevin, B.; Ganachaud, F. On the Versatility of Urethane/Urea Bonds: Reversibility, Blocked Isocyanate, and Non-isocyanate Polyurethane. Chem. Rev. 2013, 113, 80–118. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
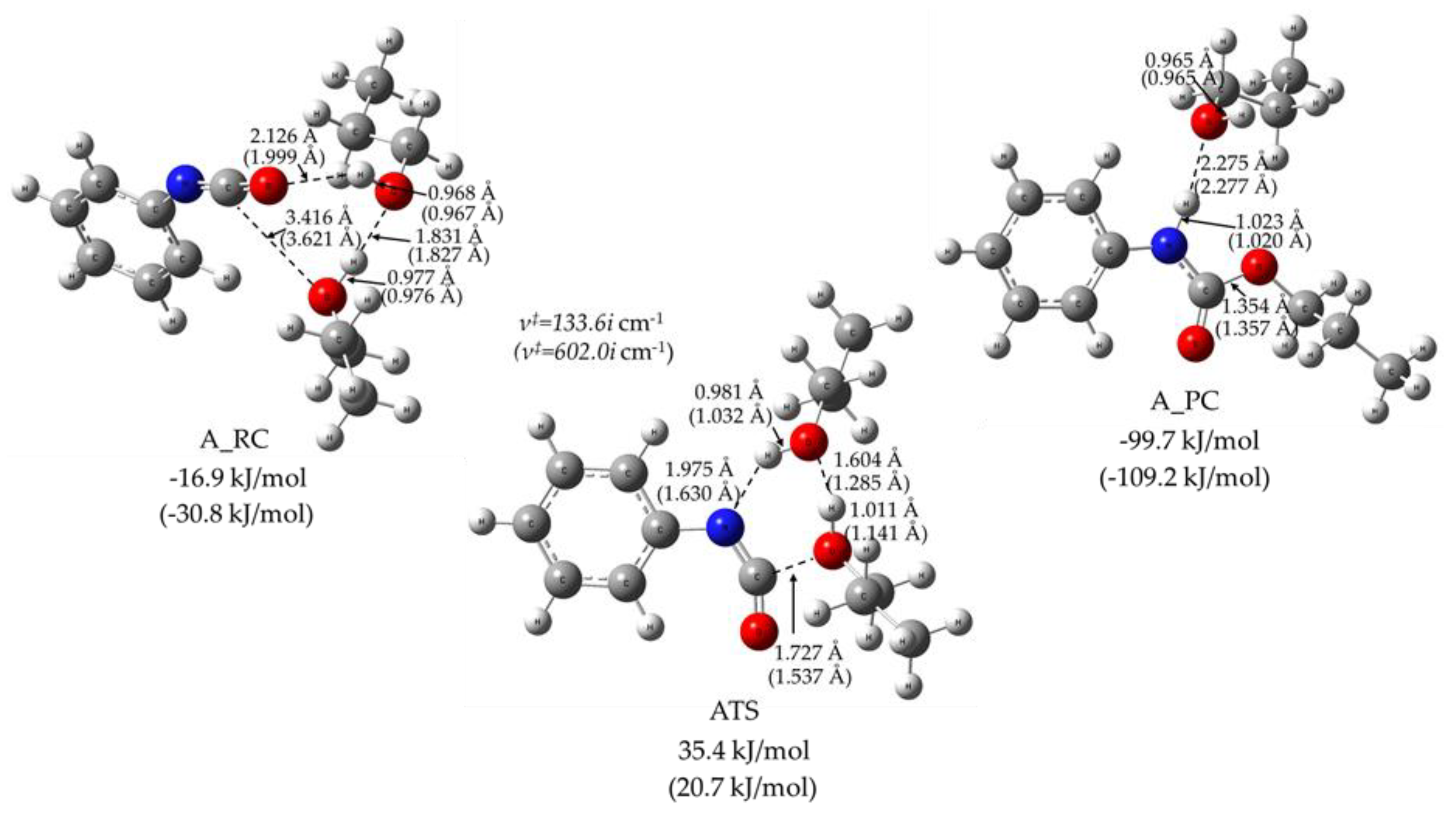{kind=link}
{kind=link}
| Temperature, K | Alcohol Excess [NCO]0/[OH]0 = 0.005 | Stoichiometric Ratio [NCO]0/[OH]0 = 1 | Isocyanate Excess [NCO]0/[OH]0 = 20 |
|---|---|---|---|
| kA × 105, M–1 s–1 | kS × 105, M–1 s–1 | kI × 105, M–1 s–1 | |
| 303 | n.m. | 1.76 ± 0.18 | 0.52 ± 0.04 |
| 313 | 0.16 ± 0.01 | 3.72 ± 0.32 | 0.91 ± 0.07 |
| 323 | 0.23 ± 0.01 | 7.41 ± 0.60 | 1.55 ± 0.11 |
| 333 | 0.33 ± 0.02 | n.m. | n.m. |
| Ea, kJ mol–1 | 30.4 ± 1.6 | 58.6 ± 6.0 | 44.2 ± 4.5 |
| A, M–1 s–1 | 18.8 ± 1.0 | 234113 ± 23971 | 214.9 ± 21.9 |
| Pathway | Species | ΔE0 | ΔH(T) | ΔG(T,P) | |||
|---|---|---|---|---|---|---|---|
| PrOH | THF | PrOH | THF | PrOH | THF | ||
| Alcohol Excess (A) | PhNCO + 2 PrOH | 0 | 0 | 0 | 0 | 0 | 0 |
| A_RC | −17.0 | −30.8 | −14.0 | −27.2 | 25.1 | 50.5 | |
| ATS | 35.4 | 20.7 | 32.7 | 17.0 | 91.4 | 119.2 | |
| A_PC | −99.7 | −109.3 | −100.9 | −109.8 | −47.4 | −17.2 | |
| Isocyanate Excess (I) | 2 PhNCO + PrOH | 0 | 0 | 0 | 0 | 0 | 0 |
| I_RC | −34.6 | −12.7 | −33.0 | −11.1 | −12.7 | 36.9 | |
| ITS1 | 51.1 | 62.6 | 44.0 | 55.7 | 62.6 | 141.3 | |
| I_IM | −152.3 | −139.3 | −160.1 | −147.2 | −139.3 | −59.0 | |
| ITS2 | 39.4 | 49.0 | 31.5 | 41.2 | 49.0 | 129.4 | |
| I_PC | −105.6 | −103.1 | −109.2 | −106.4 | −103.1 | −38.4 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheikh, W.; Rózsa, Z.B.; Camacho López, C.O.; Mizsey, P.; Viskolcz, B.; Szőri, M.; Fejes, Z. Urethane Formation with an Excess of Isocyanate or Alcohol: Experimental and Ab Initio Study. Polymers 2019, 11, 1543. https://doi.org/10.3390/polym11101543
Cheikh W, Rózsa ZB, Camacho López CO, Mizsey P, Viskolcz B, Szőri M, Fejes Z. Urethane Formation with an Excess of Isocyanate or Alcohol: Experimental and Ab Initio Study. Polymers. 2019; 11(10):1543. https://doi.org/10.3390/polym11101543
Chicago/Turabian StyleCheikh, Wafaa, Zsófia Borbála Rózsa, Christian Orlando Camacho López, Péter Mizsey, Béla Viskolcz, Milán Szőri, and Zsolt Fejes. 2019. "Urethane Formation with an Excess of Isocyanate or Alcohol: Experimental and Ab Initio Study" Polymers 11, no. 10: 1543. https://doi.org/10.3390/polym11101543