3.2.1. Steady-State Structural and Topological Properties
The steady-state microstructural and topological properties of a C
1000H
2002 melt undergoing simple shear flow are qualitatively very similar to those of the C
400H
802 and C
700H
1402 liquids, which were discussed in detail in prior publications [
2,
3,
4,
18]. These results are presented concisely herein; interested readers can refer to the cited references for more comprehensive discussions. Overall, steady-state shear properties of the C
1000H
2002 melt exhibit four distinct regions of behavior (
,
,
,
), as noted previously for the C
700H
1402 liquid [
2].
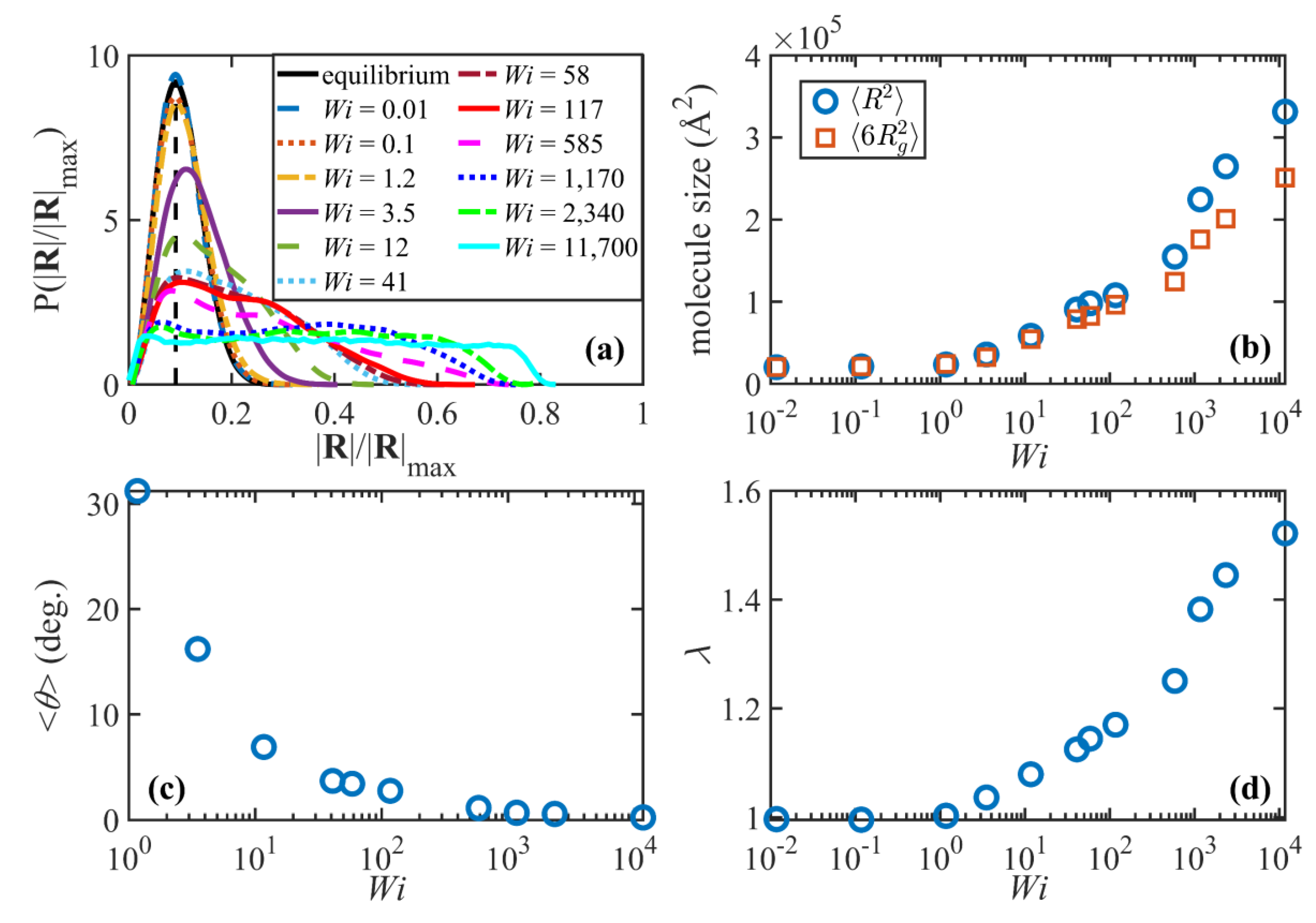
The probability distribution functions (PDFs) of the normalized end-to-end distance and the chain size (measured in terms of ensemble averages of chain end-to-end distance and six times the radius of gyration, respectively) are displayed for various values of
in
Figure 3a,b. In the linear viscoelastic regime (
), the PDFs are Gaussian and remain essentially unchanged from the quiescent state. The ensemble averages of the squared end-to-end distance and (6 times the) radius of gyration also remain constant and almost equal to each other in this regime. This suggests that the flow is too weak to significantly perturb the global molecular sizes. Keep in mind that the timescale of the flow is larger than the disengagement time (i.e.,
), implying that the constituent macromolecules have ample time for diffusive action to maintain their quiescent configurational properties even though the overall tube network begins to orient along a preferred direction in the shear plane relative to the direction of flow. Note that the ratio
approaches the theoretical value of 6 for long flexible Gaussian chains.
Figure 3c displays the ensemble average orientation angle,
, as a function of
.
is calculated as the angle between the principal eigenvector of the ensemble average of the unit end-to-end vector dyadic product,
, and the flow (
) direction. The orientation angle decreases from the zero-shear-rate limit of 45° (not shown in the figure) to about 30° at
. Finally, the tube stretch is shown as a function of
in
Figure 3d. The tube stretch is defined as the ratio
, where
is the quiescent primitive path length. Both
and
are calculated using the Z1 code. No chain stretch is observed in the linear viscoelastic region, as expected.
As the flow enters the weakly nonlinear regime,
(or equivalantly
), the orientation angle drops dramatically to values smaller than 5° and plateaus around 1–2° at higher
. The PDF of the end-to-end distance begins deviating from the equilibrium Gaussian distribution by developing a tail at higher values of
, indicating that a portion of the macromolecules have become partially extended by the applied flow. Notably, the PDF peak is still approximately at the same location as the equilibrium distribution, which suggests that the overall conformation of a significant number of chains has not yet been perturbed. The growth of molecular size and the deviation from Gaussian behavior can also be inferred from
Figure 3b, especially for
where
and
begin to diverge. (Note that there is no theory which indicates these two quantities are equivalent under flow conditions.) Interestingly, the tube network also begins to extend moderately in in this shear-rate region (
Figure 3d). This is an important observation because it contradicts the common notion of tube-based models that no stretching occurs for
. Quantitatively,
Figure 3d indicates that tubes are stretched about 16% at
, which is not negligible although just a fraction of the maximum theoretical tube stretch,
.
The third shear-rate regime of dynamical behavior is the range
(approximately
). Within this region, vorticity excursions start playing an important role in the system properties. Brownian fluctuations caused by the vorticity of the shear field lead to random excursions of the chain ends outside of the confining tubes; some of these excursions, especially those with shear-plane projections that possess negative orientation angles relative to the flow direction, induce rotation and retraction quasi-periodic tumbling cycles of the individual molecules at moderate and high shear rates similar to those observed in previous work [
2,
3,
8,
14,
18,
22]. A typical cycle begins as a chain molecule stretches and aligns in the flow direction (see
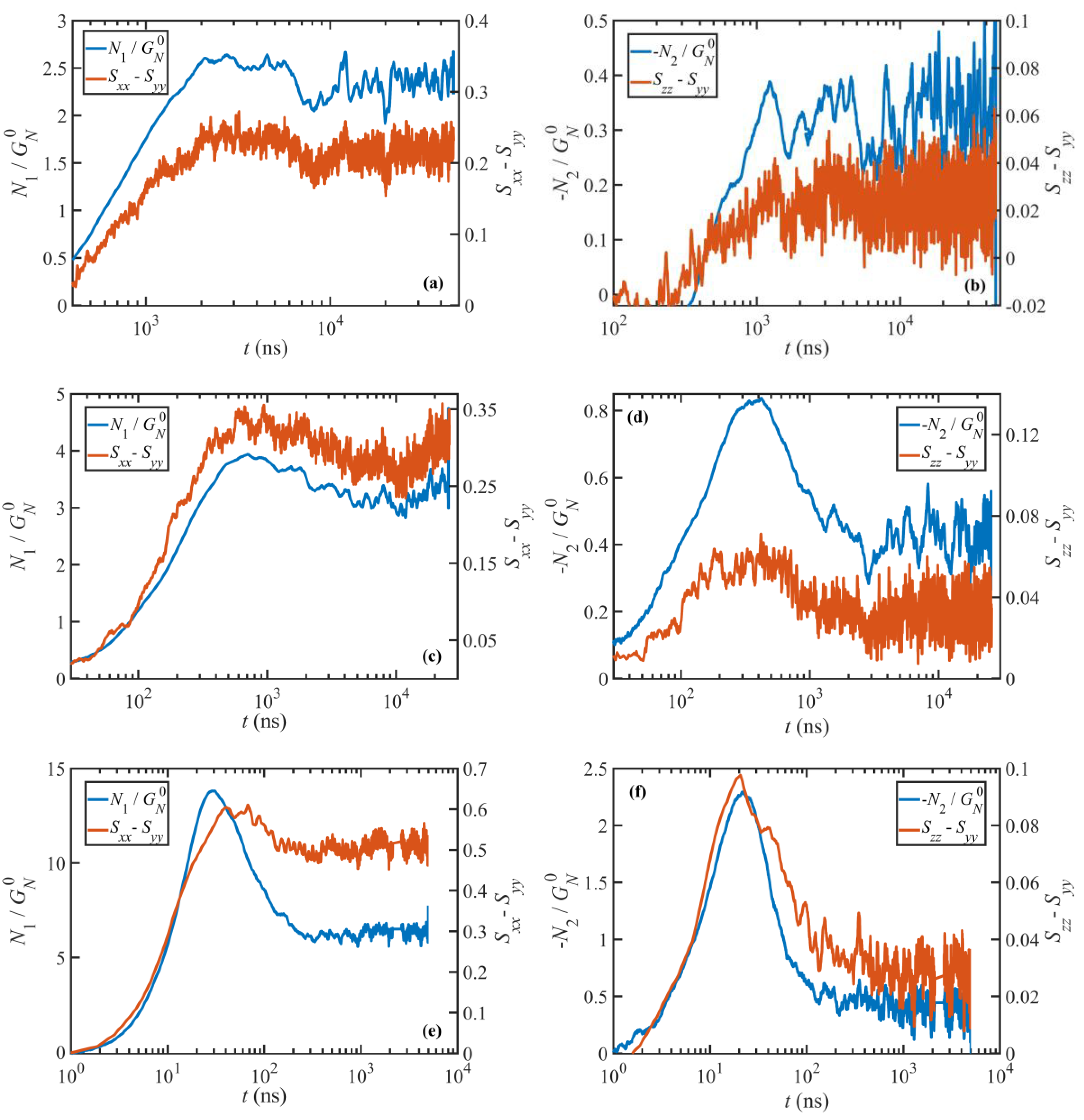
Figure 4c). At this point, due to the flow vorticity, chain ends fold backward along the spine of the molecule and slide toward the middle of the chain until the molecule collapses into a compressed configuration. Then the orientation of the chain flips as the chain ends cross and the molecule unravels until it adopts a stretched conformation again that concludes a half cycle. At the lower end of this range (
to 100), the cycle is very irregular, almost chaotic. Here, the macromolecules will reside in the compressed state for a long period of time (see
Figure 4a). Under this condition, the chain ends, which are typically very close to each other, exhibit a wagging behavior due to Brownian motion, passing each other back and forth multiple times before the molecule begins to reextend. As a consequence, the orientation angle of the chain end-to-end vector,
, oscillates haphazardly between
and
as evident in
Figure 4a. The orientation angle of the chain primary axis,
, however, does not oscillate as much as
suggesting that the body of the molecule does not wag like a solid object. (The primary axis of the molecule is defined as the eigenvector corresponding to the largest eigenvalue of the molecule gyration tensor.) Yet,
changes rapidly between positive and negative values when the molecule is in a collapsed and highly compressed state, indicating that the coiled chains wag for some indefinite period of time before they begin to unravel.
In the uppper portion of the range
(i.e.,
), the dynamical behavior of the macromolecules is much more regular and resembles the tumbling behavior observed in prior work [
2,
3,
8,
14,
18,
22]. During a typical cycle, the chain end-to-end distance varies dramatically from high values associated with the stretched configurations to values that are even smaller than the average equilibrium end-to-end distance. This is manifested in the wide non-Gaussian bimodal probability distribution function at this flow regime, as displayed in
Figure 3a. Specifically, the peak at low values of
shifts to the left as
increases and occurs at extensions smaller than the equilibrium peak, indicating the increasing population of the collapsed configurations during the course of the tumbling cycle. At the same time, the ensemble average molecule size (
Figure 3b) and tube stretch (
Figure 3d) increase with
in this flow region. Based on theoretical arguments, this is the region wherein tube stretch becomes significant. As mentioned earlier,
Figure 3d shows that tube stretching begins at lower flow strength than theoretically expected; however, tube stretch in the third flow region is apparently of a different nature than within the second flow regime. In the third region,
scales as
while this power-law exponent is 0.03 in the second range,
. This suggests that the tube stretch is influenced by the tumbling dynamics of the individual macromolecules, and that it has an influential contribution to the shear stress and constitutes a major relaxation mechanism in this intermediate flow strength regime. Note that the time-average orientation angle of the molecules is very close to its plateau value in the third region and does not change significantly, indicating that the chain end-to-end vectors are almost completely aligned in the flow direction on the molecular length scale, although not necessarily on the tube segment length scale.
The fourth and final flow regime is the strong flow region where
, approximately
. Although the molecules continue to stretch in this region (
Figure 3b,d), the molecular size and the tube stretch ultimately attain plateau values, which are significantly smaller than their corresponding maximum theoretical values. The tube stretch profile has an inflection point around
where the curvature changes from positive to negative. This signals a new regime where the tube stretch becomes saturated as chain rotation becomes the more dominant dynamic mechanism. The shape of the end-to-end distance distribution curve is also very different in this high
region compared to that at lower
regimes. Specifically, the distributions become relatively flat with a characteristic rotational peak at low
and a stretch peak that emerges at very high
. (See
Figure 3a. The stretch peaks can also be easily recognized in C
400H
802 and C
700H
1402 systems [
18].) These flat distributions, which become wider as
increases, are attributed to the more regular molecular rotation cycles at very high shear rates, as discussed by Nafar Sefiddashti, et al. [
3]. The skewed distributions within the intermediate
regime suggest that during a rotation cycle individual molecules spend on average a longer time at collapsed (or less stretched) configurations than they do at relatively stretched configurations [
18], or that some of the chains have not yet stretched enough to begin their rotation cycles (see
Figure 4a). Both cases lead to unbalanced lifetimes for various configurations, and consequently irregular rotation cycles. Within the high
regime, on the other hand, molecules undergo more regular periodic cycles. Hence various configurations between a highly stretched chain and a tightly packed coil have fairly similar lifetimes or probabilities (see
Figure 4b), which manifest in the flat probability distribution of the end-to-end distance [
18].
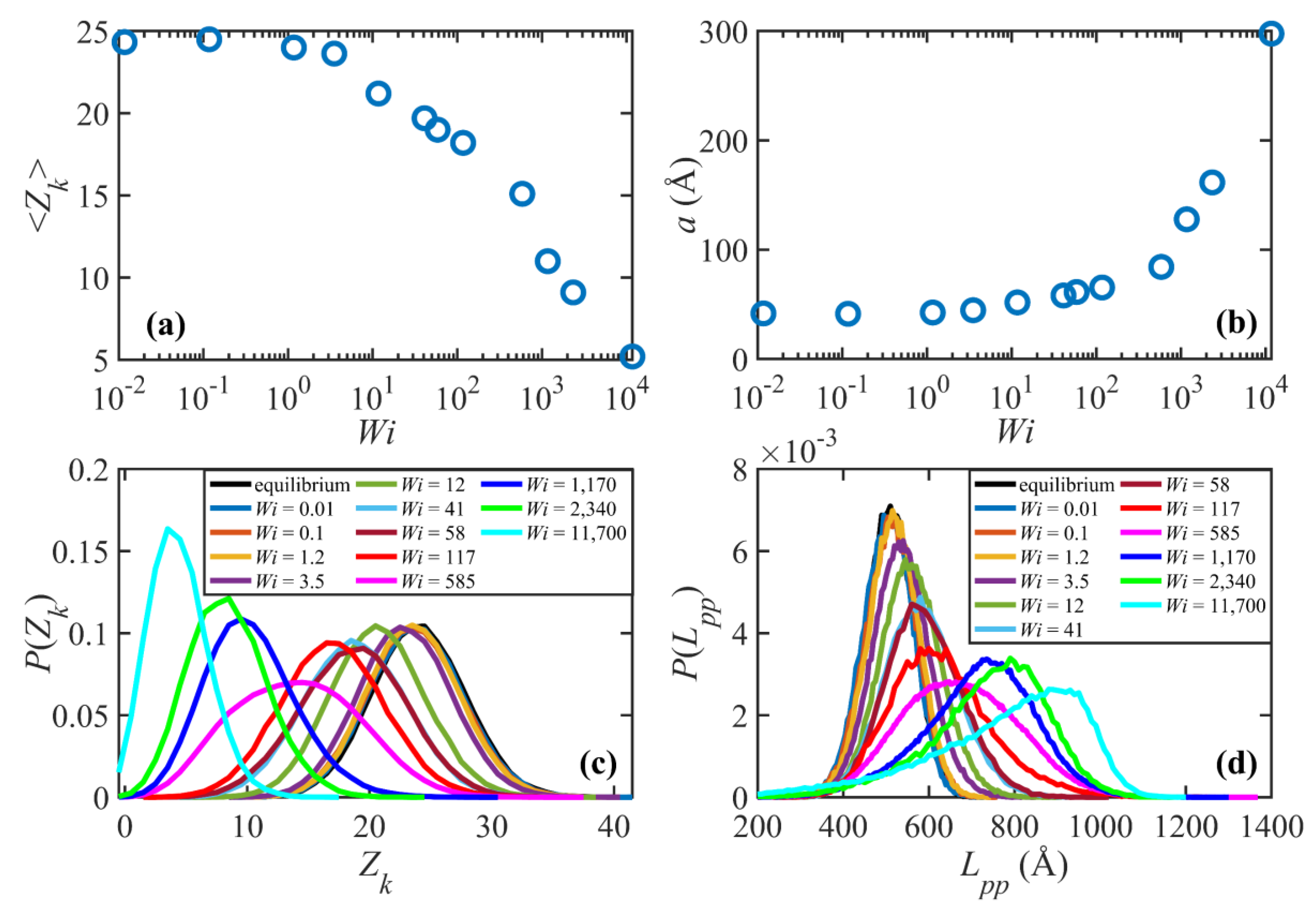
Figure 5 displays the entanglement network properties of the C
1000H
2002 melt at various
. The ensemble average entanglement density and the probability distribution function for the entanglement density are displayed in
Figure 5a,c.
Figure 5b shows the tube diameter, determined as the step length of the primitive path
[
2,
3]. The probability distribution function of the primitive path contour length is also shown in
Figure 5d for various values of
. Note that the primitive path contour length,
, is essentially commensurate with the tube stretch,
(see
Figure 3d), which is the normalized primitive path contour length. These plots show that, within the linear viscoelastic regime, the entanglement network is practically unperturbed as compared to quiescent conditions. Specifically, the entanglement density and tube diameter do not change as the flow strength increases. The probability distribution function for the entanglement density,
, follows a Poisson distribution and is independent of
in this regime.
exhibits a similar behavior, except that it follows a Gaussian distribution. As
increases and shear rate enters into the nonlinear viscoelastic regime, the tube network begins to lose entanglements. Notably, there is no sharp boundary between the second (
) and the third
) flow regimes, as discussed for the structural properties of the system. Rather, there is an initial stage of convective constraint release wherein the chains disentangle at a moderate rate in the region
such that
. Accordingly, the tube diameter increases moderately in this region. The probability distribution function for the entanglement density,
, shifts to the left with increasing
as the chains disentangle. The shape of the distribution, however, remains approximately similar to that of the linear viscoelastic regime and still follows a Poisson distribution. On the other hand,
shifts to the right and becomes wider (i.e., with a higher standard deviation); nevertheless, the distribution continues to follow a Gaussian distribution. These results suggest that, although by the end of this flow regime the system loses about 30% of its entanglements, the nature of the entanglement network does not change radically. Note that even at the highest shear rate within this regime, none of the chains has lost all of its entanglements. For instance, the curve for
in
Figure 5c shows that all molecules possess 5 or more kinks.
At higher flow strength, (i.e.,
), the entanglement density begins to drop dramatically as
increases with a power-law exponent of
. The tube diameter also increases substantially in this region, such that at
the tube diameter grows almost as large as the molecular radius of gyration. This means that a molecule could effectively diffuse as far as its size without feeling the confining tube. This interpretation essentially questions the existence of the tube concept and of an entangled system. These subtleties can be understood by examining the probability distribution of the entanglement density.
Figure 5c shows that the distributions begin to deviate somewhat from Poisson distributions. More importantly, these distributions suggest that, unlike before, in this flow region some of the molecules have lost all their entanglements and have become virtually unentangled. The distribution of the primitive path contour length also deviates considerably from the Gaussian distribution in this region. All of these observations suggest that the entanglement network is effectively destroyed due to the strong flow. This also might explain why the system behavior at such high shear rates resembles that of a dilute solution, as has been argued for shorter chain C
400H
802 and C
700H
1402 liquids [
3,
18]. In this regime, the tumbling cycles are comparatively more regular, similar to those of dilute solutions. Tube stretch approaches its plateau value as macromolecular tumbling becomes the dominant dynamic mechanism.
An important characteristic of the entanglement network is the tube orientation tensor,
, that is one of the principal variables of many tube-based constitutive models.
Figure 6 displays the nonzero components of
as functions of
at steady state obtained from the NEMD data for the C
1000H
2002 melt. The average orientation tensor of the tube segments in this figure is defined as
, where
is the unit end-to-end vector of an entanglement strand: knowing the positions of the entanglements (kinks) along the chain from the Z1 code, the end-to-end vectors of the entanglement strands can be easily identified and the appropriate ensemble averages of the components of the orientation tensor can then be readily calculated from the NEMD data. The component,
, begins to increase at very low
within the linear viscoelastic regime. This segmental orientation leads to an increase in the shear stress in this region, in agreement with theory.
passes through a maximum in the range
, which is somewhat higher than the theoretical prediction of
. At higher shear rates,
decreases almost monotonically. Such behavior can lead to excessive shear thinning, as observed in vesrsions of the tube model that do not incorporate CCR, especially within the shear rate range
or the plateau region where the tube stretch is insignificant. In fact, models that incorporate CCR predict a nearly constant
, and consequently constant shear stress, in the plateau region in agreement with typical experiments. Hence, the decrease in
observed in the NEMD data calls into question the theoretical mechanism of CCR in some tube-based models like MLD. The diagonal components of
remain nearly constant in the linear viscoelastic regime and then diverge from their equilibrium value (~0.33) as
increases. At very high shear rates, i.e.,
, the rate of change in these components increases significantly. This is the shear rate range wherein the entanglement network begins to disintegrate. Generally, the features of these plots are qualitatively similar to those of
and
(see
Figure 3d and
Figure 5a), which could be indicative of an inherent connection between
and
with the normal components of the tube segmental orientation tensor. This is specifically important from a modeling perspective as it suggests that the evolution equations for the tube stretch and entanglement density should be expressed in terms of the diagonal components of
rather than the shear component.
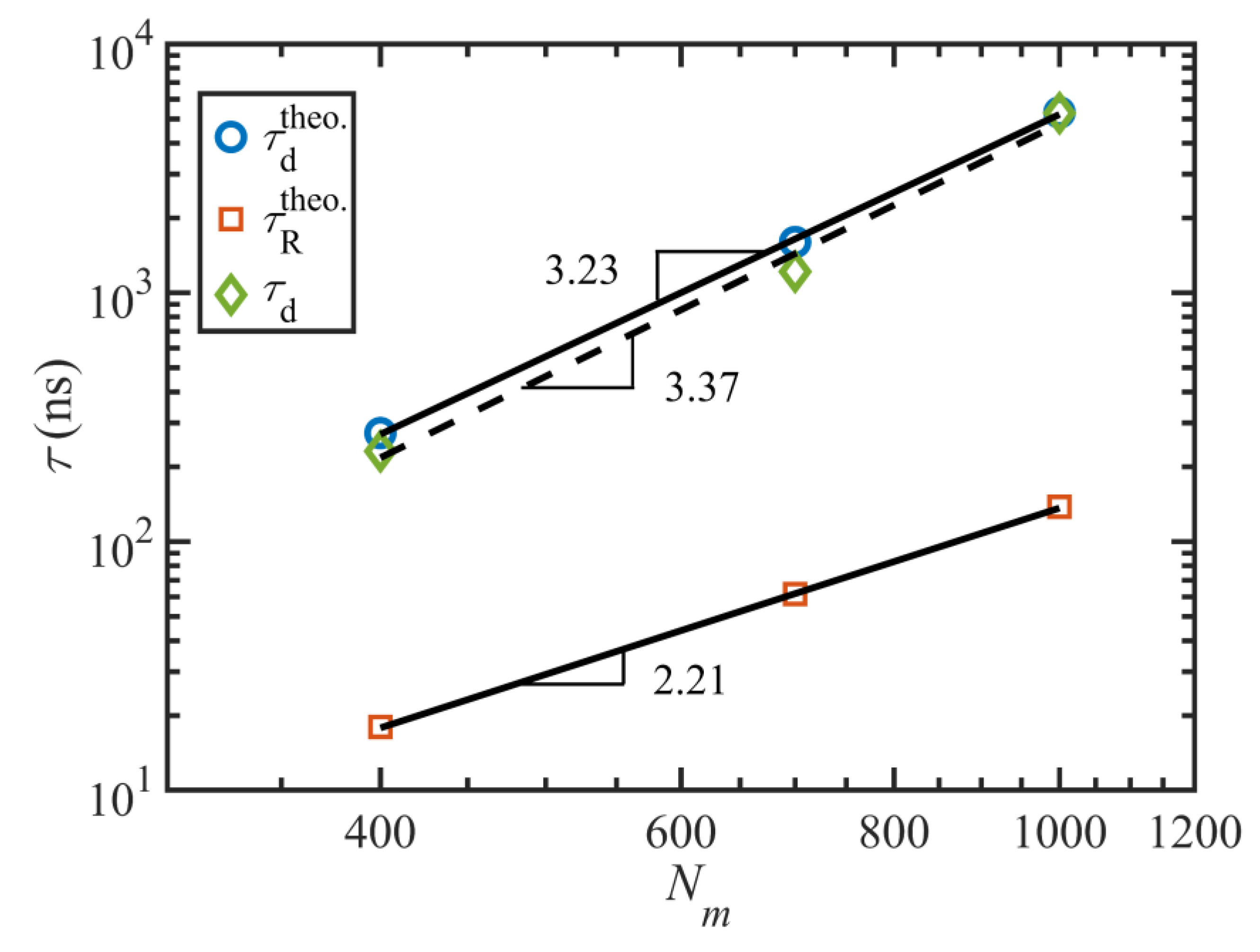
Figure 7 displays the important characteristic timescales of the C
1000H
2002 liquid as functions of
. These timescales are calculated based on fitting the autocorrelation function data of the end-to-end vector with the functional form
. Hence,
is the decorrelation time of the end-to-end vector, which is equal to the longest relaxation time (i.e., the disengagement time under quiescent conditions and within the linear viscoelastic regime).
quantifies the period of the rotation and retraction cycle of the macromolecules, assuming that the cycles are quasi-periodic. A characteristic time for the tumbling period can be defined conceptually as
[
2], displayed as diamonds in
Figure 7.
Figure 7 indicates that
does not change significantly within not only the linear viscoelastic region (
) but also in the nonlinear regime for
. At higher shear rates, the longest relaxation time decreases with a power-law exponent of
; this is consistent with the scaling exponents of the C
400H
802 and C
700H
1402 liquids at high shear rates [
2,
3]. Unlike for the C
1000H
2002 liquid, the relaxation times of C
400H
802 and C
700H
1402 decreased with shear rate at all
, and hence a separate power-law exponent for the
regime was reported in prior work [
2,
3,
18]. Nevertheless, a separate scaling factor for low
looks to be irrelevant here. This is perhaps caused by the higher entanglement density of the C
1000H
2002 melt, which possibly delays any meaningful change in the relaxation time until approximately
when
begins to decrease—see
Figure 5a.
also exhibits a power-law behavior that scales as
with flow strength. Although this value is slightly smaller than those of the C
400H
802 and C
700H
1402 melts (−0.78 and −0.75 respectively), they are all in reasonable agreement within statistical bounds. The ratio
averages about 7.3 over all
, which is reasonably close to
, similarly to the prior cases [
2,
3]. This suggests that a single timescale, one associated with the period of the molecular tumbling cycles, is the sole configurational relaxation mechanism of the C
1000H
2002 chains for
.
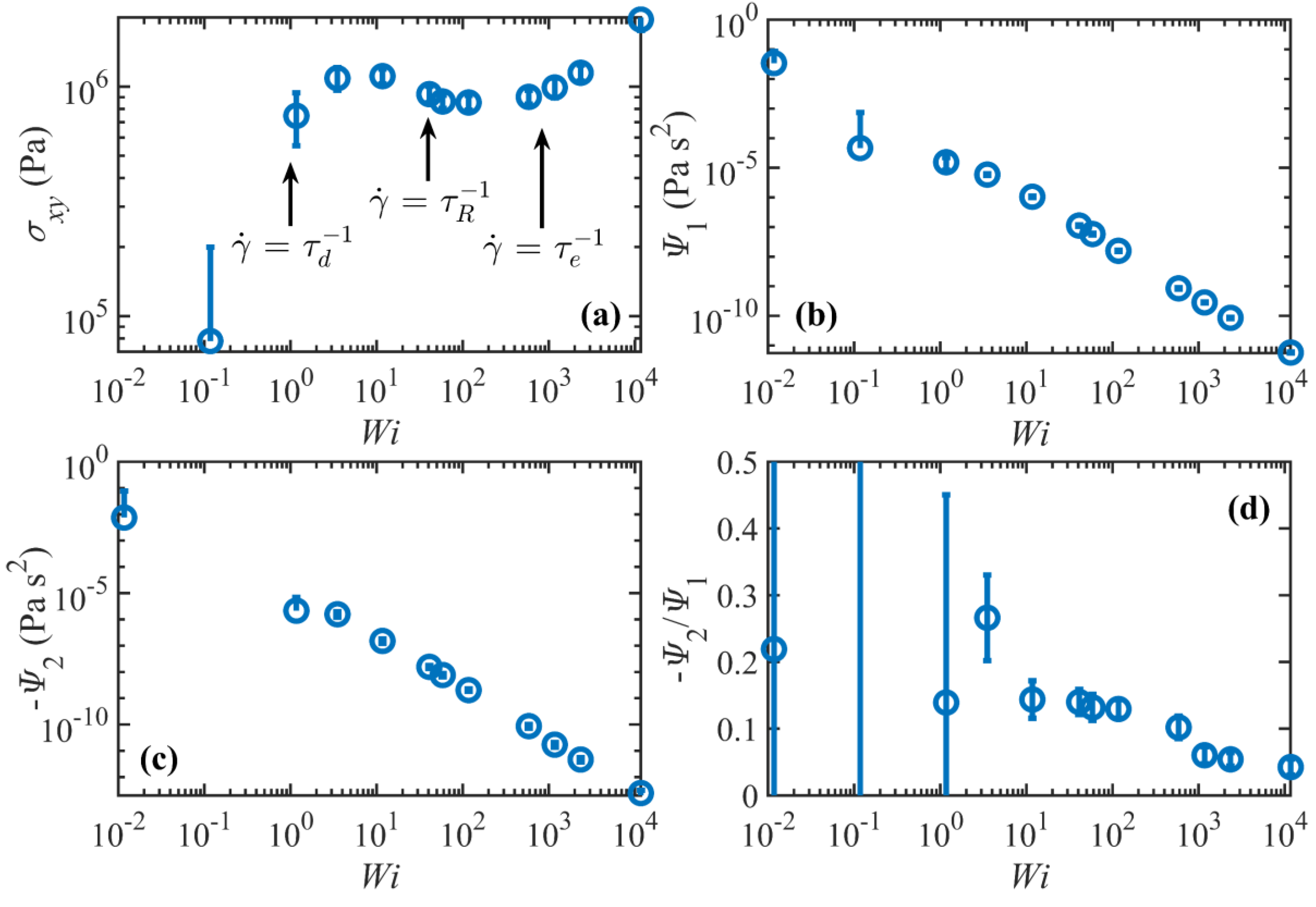
3.2.2. Rheological Response
Figure 8 displays the steady-state rheological properties of the C
1000H
2002 liquid as functions of
. As expected, the shear stress scales as
in the linear viscoelastic regime; however, at higher shear rates, the system’s response is quite different from typical experimental observations, as evident from
Figure 8a. Specifically, the shear stress passes through a maximum in the shear rate range
and a subsequent minimum in the range
, in contradiction with the experimentally observed plateau region where the shear stress remains approximately constant or increases slightly as shear rate increases, usually within the shear rate ranges
or
. Considering the uncertainties of the calculations, it appears that the local maximum and minimum in the shear stress profile occur roughly at about
and
, respectively, and the shear stress surpasses the local maximum value at a shear rate of approximately
. This possibly implies that the flow is unstable over a fairly wide range of shear rates. Such behavior is enticingly consistent with the discussion of Doi and Edwards (see Figure 7.22 of Reference [
34]) concerning the DE model predictions at high shear rates, who argued that the power-law exponent of the shear stress is very sensitive to the relaxation spectrum of the linear relaxation modulus. They argued that the absolute value of the exponent becomes smaller (closer to zero) as the relaxation spectrum becomes broader. Therefore, the shear stress should be approximately independent of the shear rate for polydisperse samples that are commonly used in experiments (hence the plateau), whereas a maximum in the shear stress profile could result from a completely monodisperse sample. Nevertheless, even for monodisperse samples, multiple relaxation processes tend to broaden the relaxation spectrum and weaken the shear rate dependence of the stress. However, as evident from
Figure 7, the number of timescales becomes effectively unity for
. Based on the DE model,
for
and as
becomes close to unity, the shear stress increases due to tube stretching [
34]. This implies that if the number of entanglements is not large enough (i.e.,
is not high enough), the shear rate dependence weakens. In
Figure 8a,
for
, which is consistent with this argument.
The plateau region in the shear stress profile has also been postulated to result from the onset of the molecular tumbling cycles that begin to manifest in this shear rate regime [
2,
3]. This hypothesis led to further investigations which indicated the possibility that shear banding, caused by the molecular periodicity, was a possible cause of the experimentally observed plateau in the shear stress profile [
36,
37,
38]; however, it is unlikely that shear banding occurs in the present simulations since the p-SLLOD equations of motion impose a homogeneous linear velocity profile throughout the simulation cell in the NEMD simulations. That being said, however, recent DPD simulations have demonstrated shear banding in monodisperse polymers in the same range of molecular weight where the flow curve is non-monotonic [
36,
37,
38,
39].
For
, the shear stress scales as
. The power-law exponents for the C
400H
802 and C
700H
1402 melts over the same range of shear rates are approximately
and
, respectively [
2,
3], which suggest a molecular weight dependence for the shear stress at these high shear rates.
Figure 8b,c show the first normal stress coefficient
, and the second normal stress coefficient
, where
and
. Both coefficients exhibit strong shear thinning behavior in the nonlinear regime with power-law exponents of –1.7 and –1.8, in agreement with those of the C
700H
1402 melt [
2]. The ratio
ranges over
in the nonlinear regime, again in reasonable agreement with C
700H
1402 melt [
2] and typical experimental values [
40].

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}